
Conformational Remodeling and Allosteric Regulation Underlying EGFR Mutant-Induced Activation: A Multi-Scale Analysis Using MD, MSMs, and NRI


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
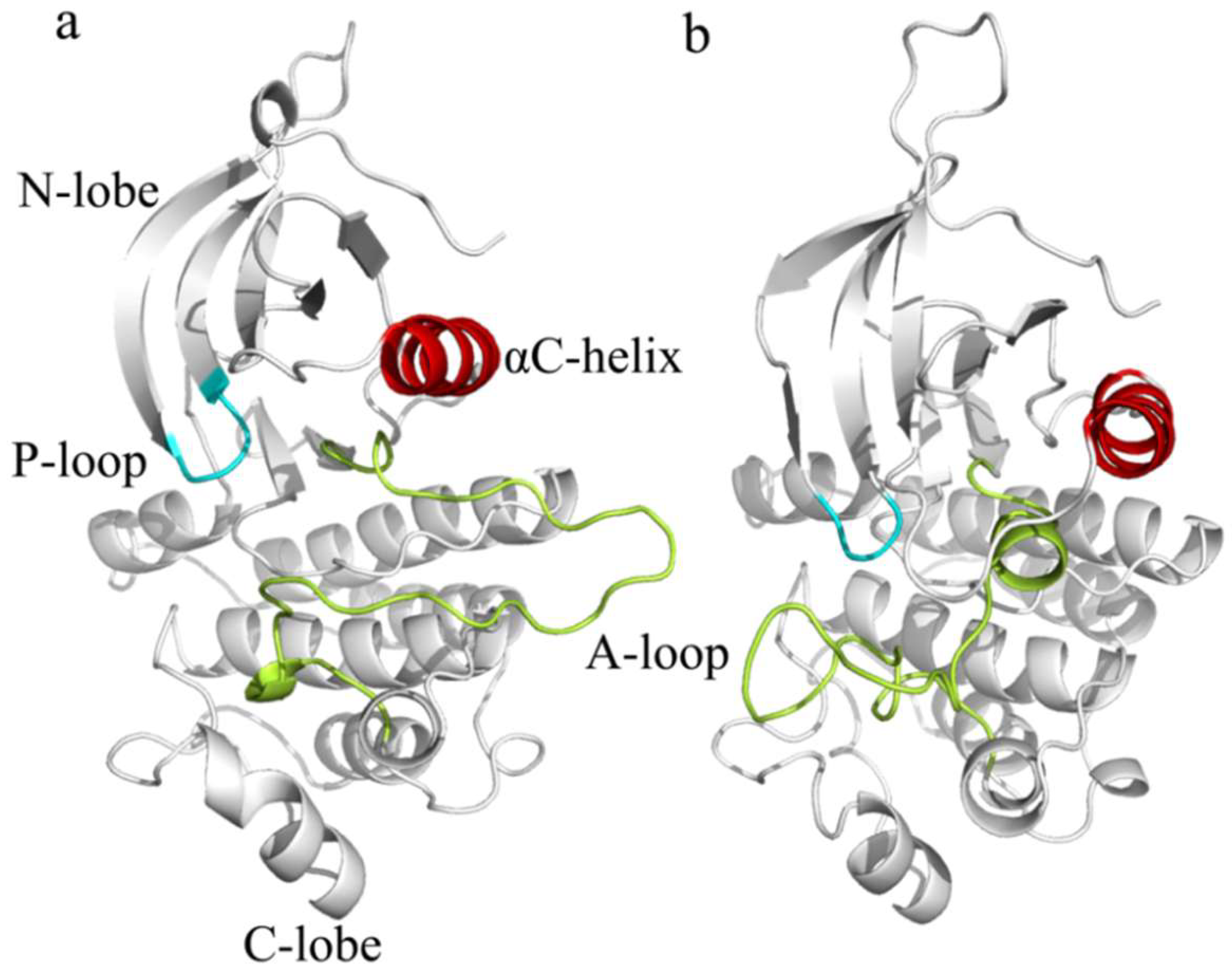
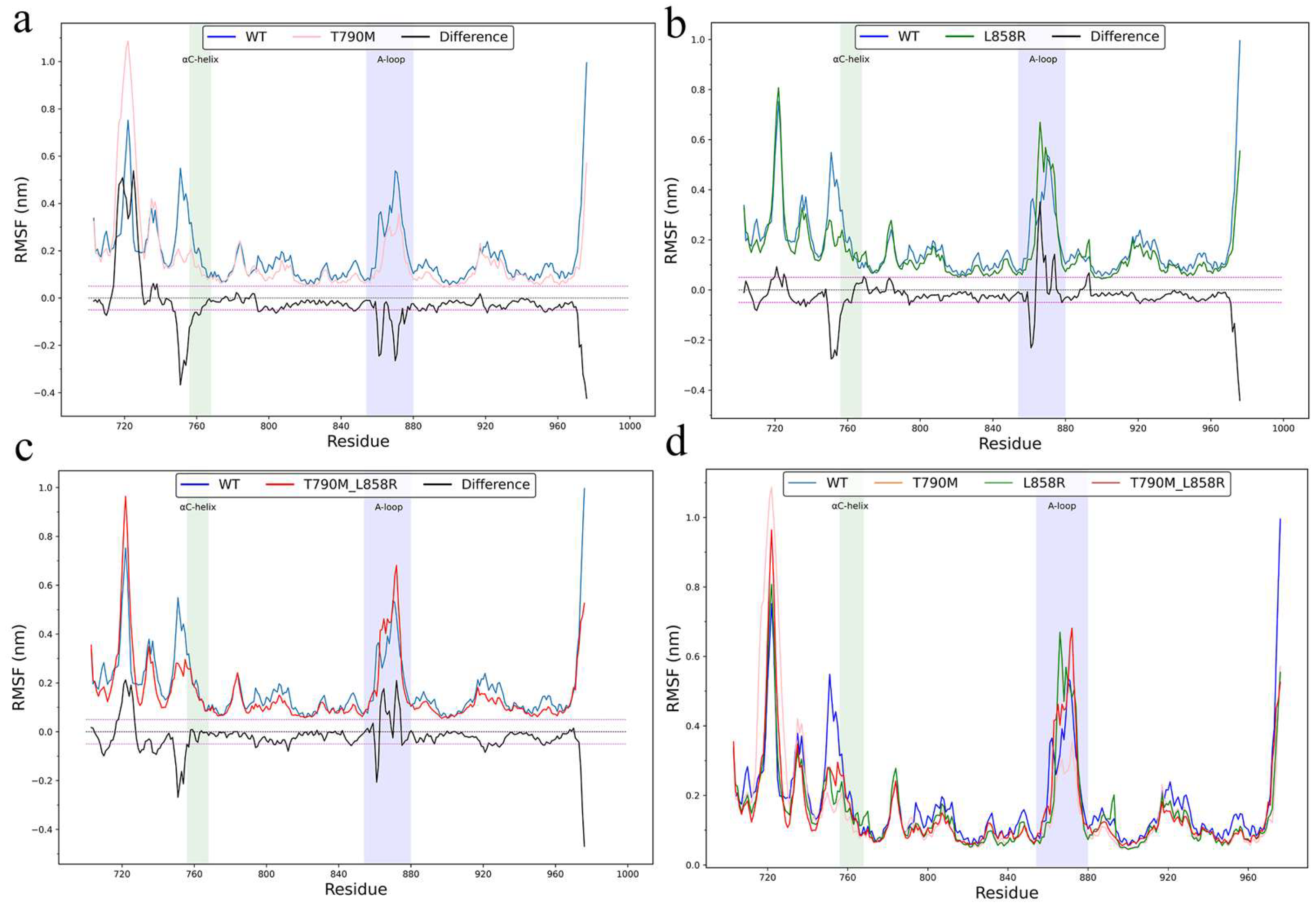
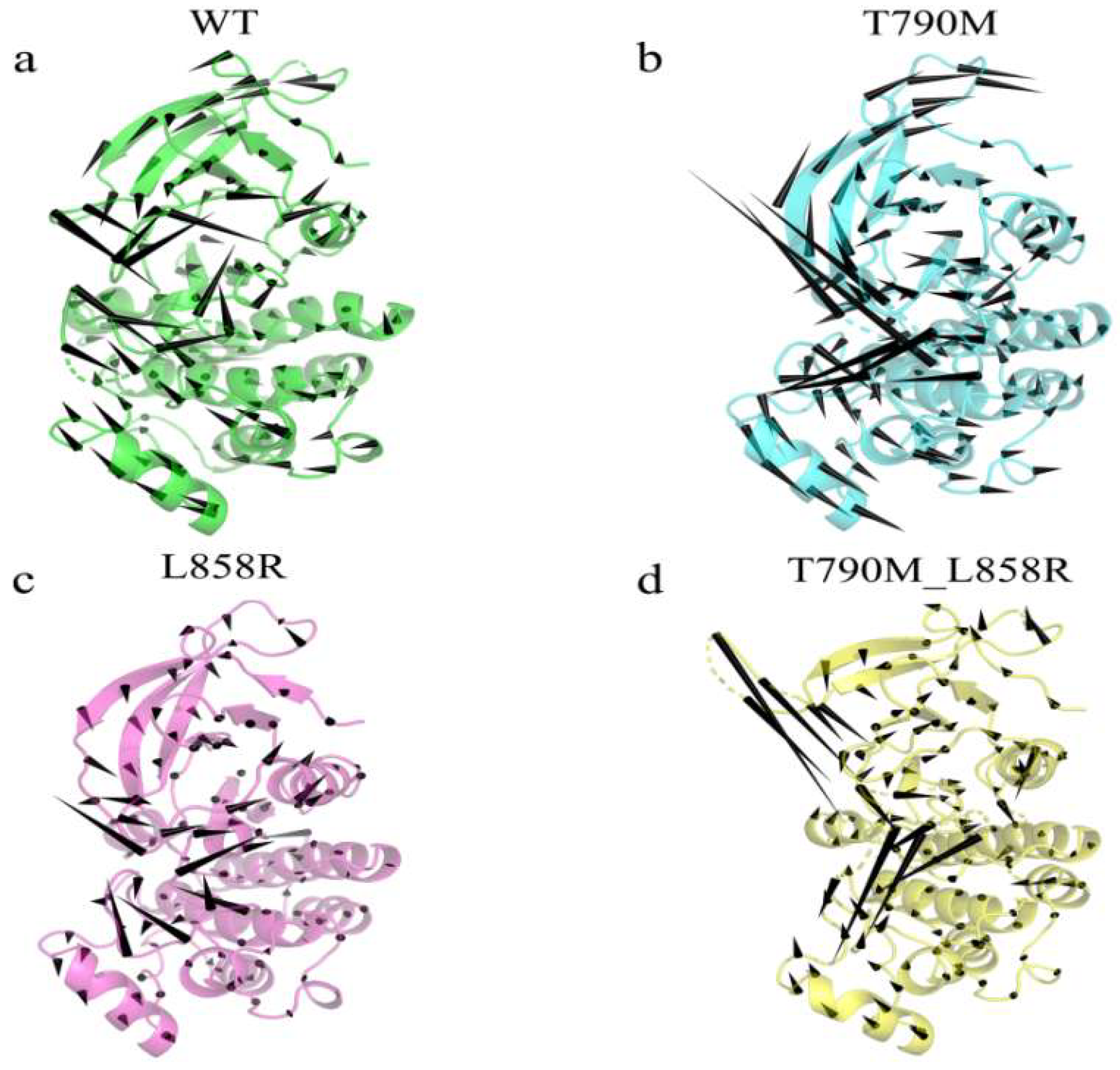
2.1. Structural Flexibility and Molecular Motion Analysis
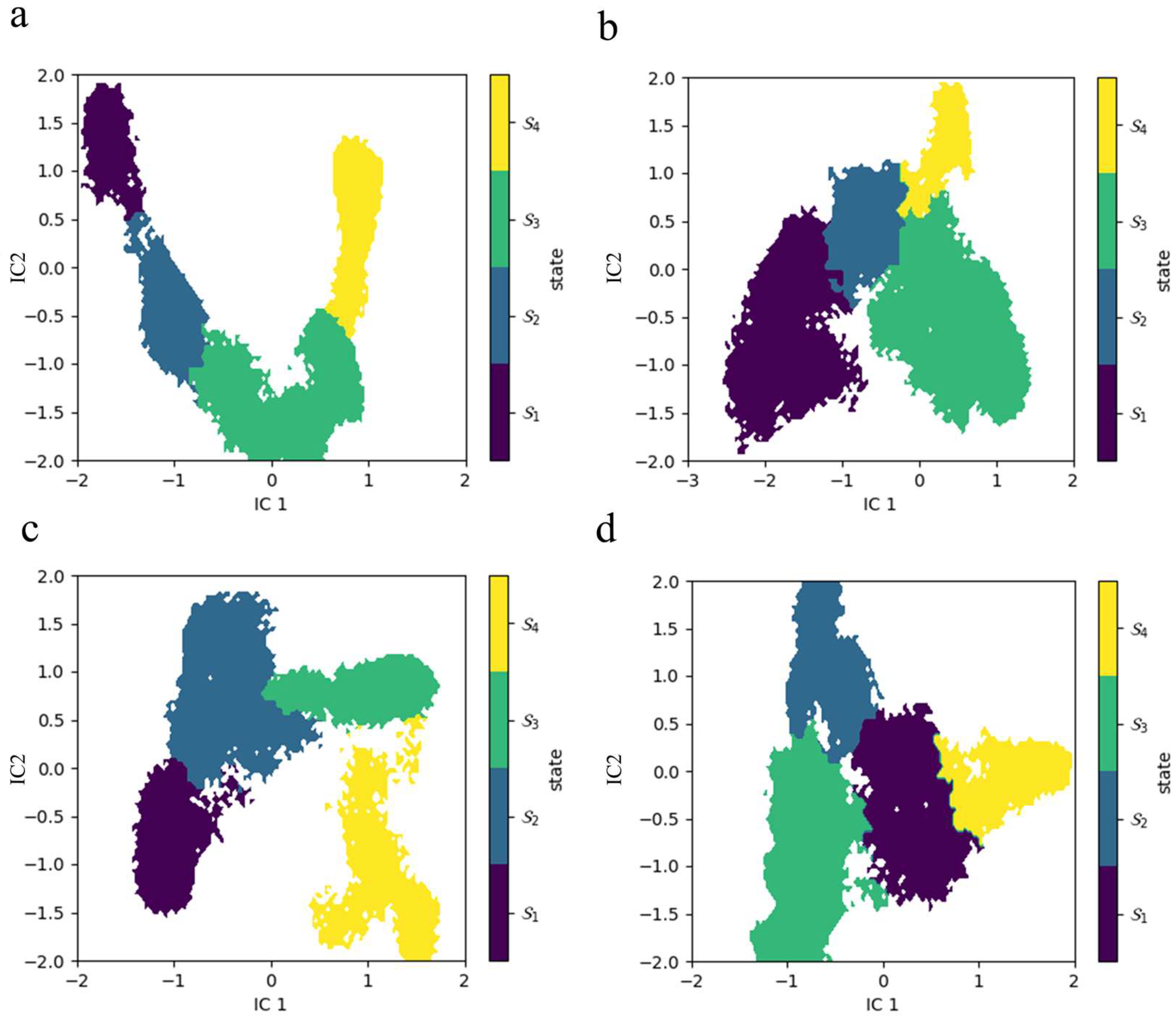
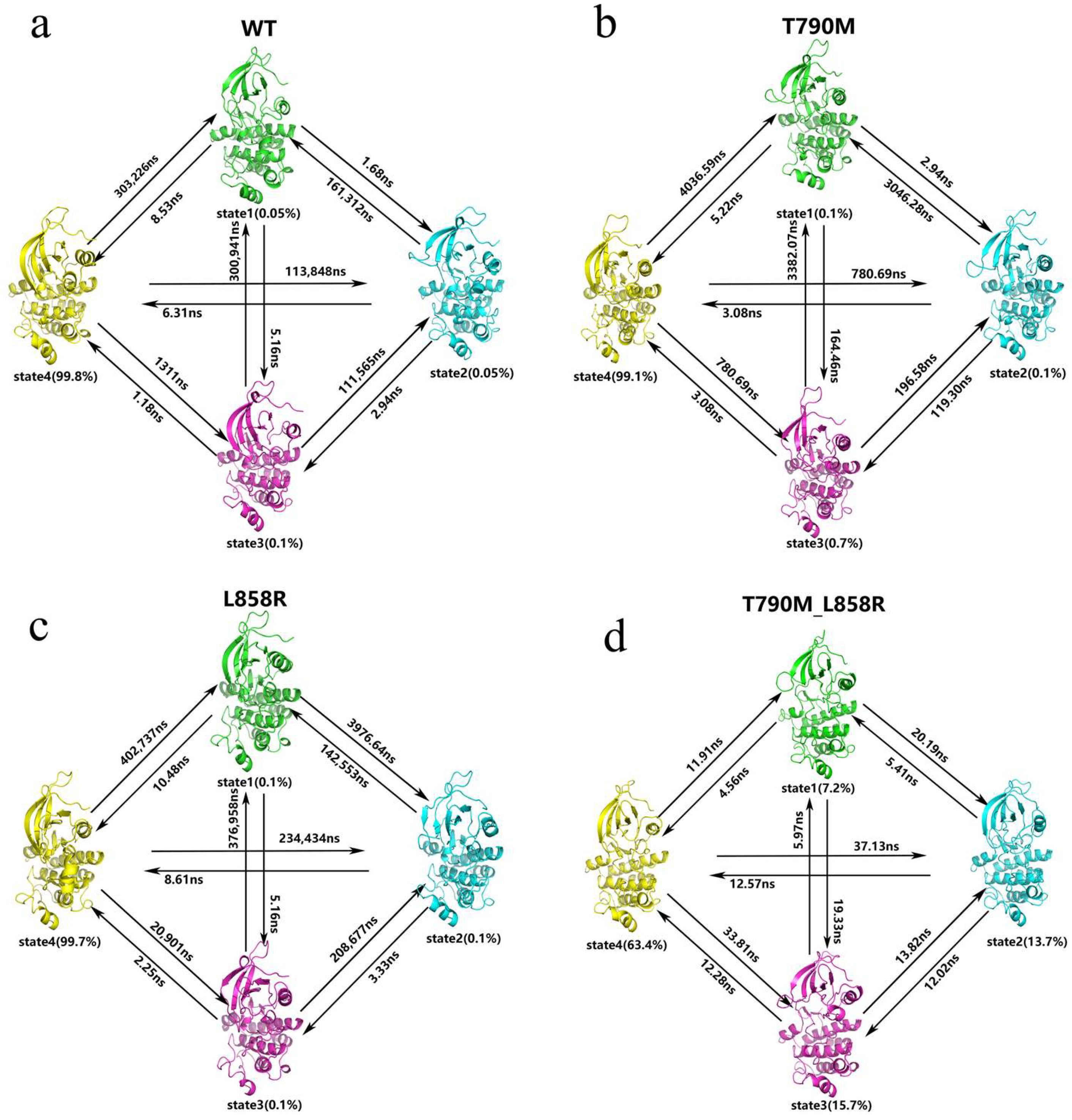
2.2. Thermodynamics and Kinetics of Conformational Transitions
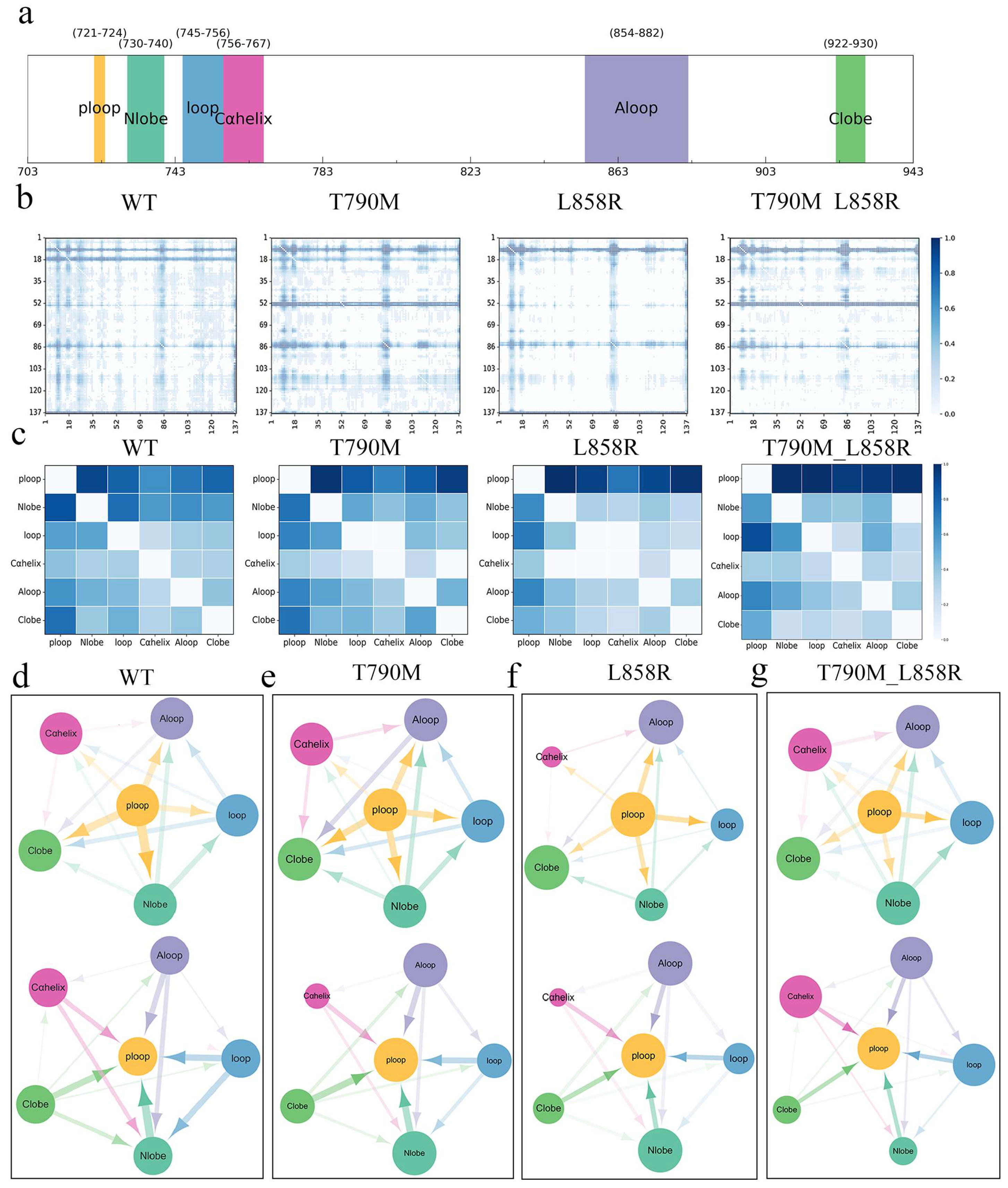
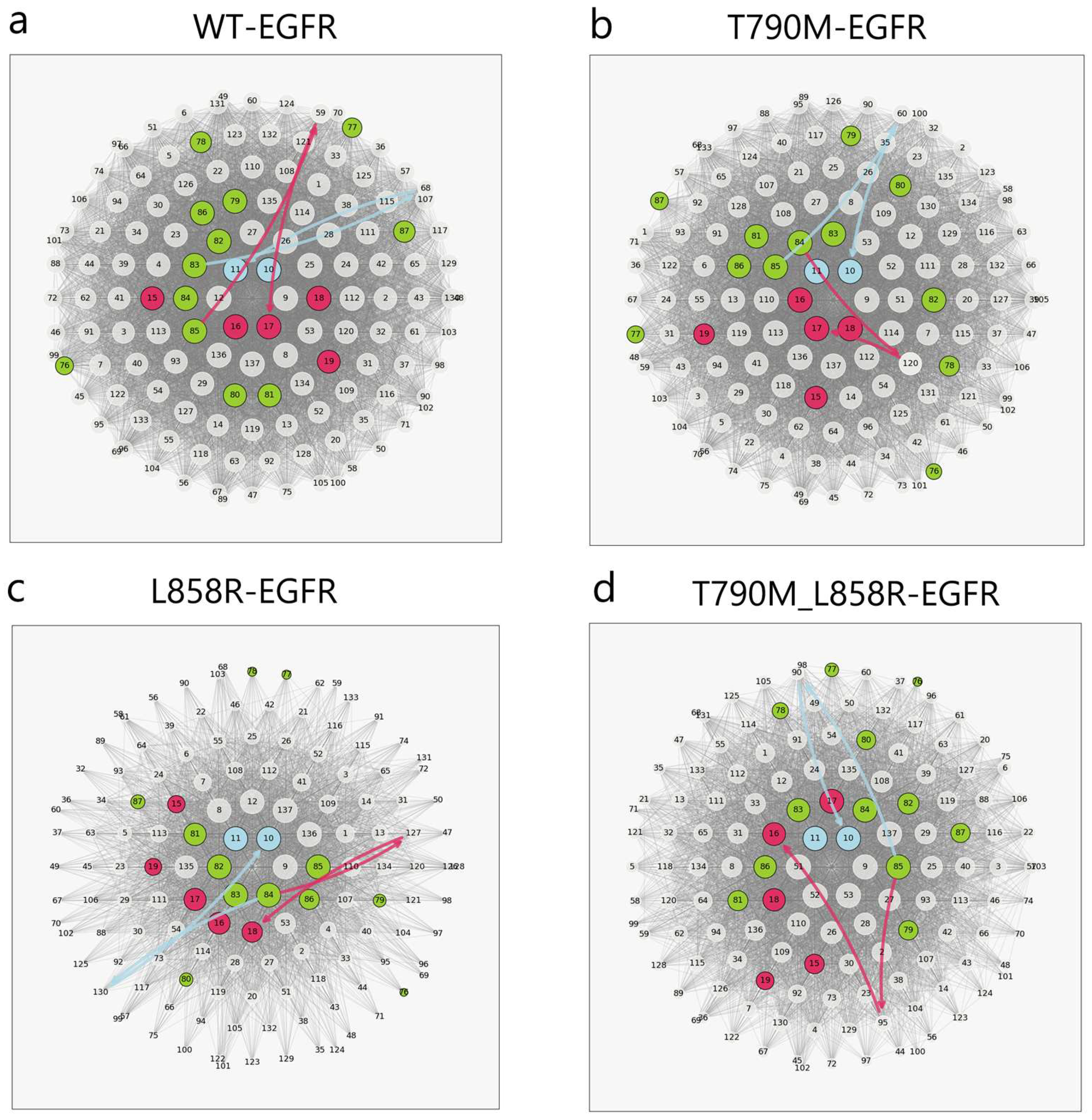
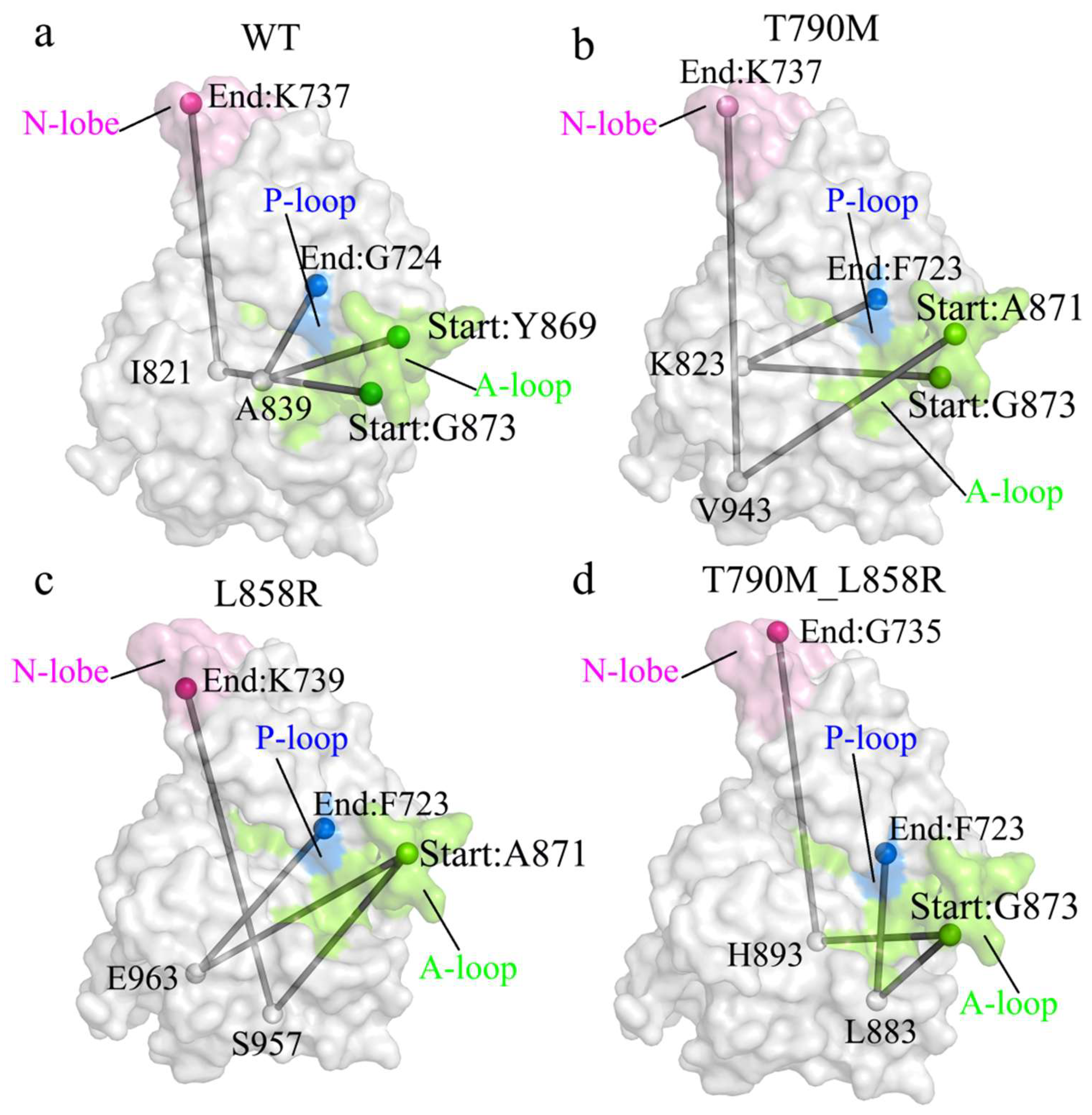
2.3. Interaction Network and Allosteric Pathway Analysis
3. Discussion
4. Materials and Methods
4.1. Protein Model Construction
4.2. Metadynamics Simulation
4.3. Conformational Flexibility and Molecular Motion Analysis
4.4. Markov State Models
4.5. Neural Relational Inference
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef] [PubMed]
- Tsim, S.; O’dowd, C.A.; Milroy, R.; Davidson, S. Staging of non-small cell lung cancer (NSCLC): A review. Respir. Med. 2010, 104, 1767–1774. [Google Scholar] [CrossRef]
- Chevallier, M.; Borgeaud, M.; Addeo, A.; Friedlaender, A. Oncogenic driver mutations in non-small cell lung cancer: Past, present and future. World J. Clin. Oncol. 2021, 12, 217. [Google Scholar] [CrossRef]
- Shan, Y.; Eastwood, M.P.; Zhang, X.; Kim, E.T.; Arkhipov, A.; Dror, R.O.; Jumper, J.; Kuriyan, J.; Shaw, D.E. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 2012, 149, 860–870. [Google Scholar] [CrossRef]
- Vani, B.P.; Aranganathan, A.; Tiwary, P. Exploring kinase Asp-Phe-Gly (DFG) loop conformational stability with AlphaFold2-RAVE. J. Chem. Inf. Model. 2023, 64, 2789–2797. [Google Scholar] [CrossRef]
- Reveguk, I.; Simonson, T. Classifying protein kinase conformations with machine learning. Protein Sci. 2024, 33, e4918. [Google Scholar] [CrossRef]
- Kanbour, A.; Salih, F.; Abualainin, W.; Abdelrazek, M.; Szabados, L.; Al-Bozom, I.; Omar, N.E. Leptomeningeal metastatic L858R EGFR-mutant lung cancer: Prompt response to osimertinib in the absence of T790M-mutation and effective subsequent pulsed erlotinib. OncoTargets Ther. 2022, 15, 659–667. [Google Scholar] [CrossRef]
- Martin-Fernandez, M.L.; Clarke, D.T.; Roberts, S.K.; Zanetti-Domingues, L.C.; Gervasio, F.L. Structure and dynamics of the EGF receptor as revealed by experiments and simulations and its relevance to non-small cell lung cancer. Cells 2019, 8, 316. [Google Scholar] [CrossRef]
- Yang, J.C.; Ahn, M.J.; Kim, D.W.; Ramalingam, S.S.; Sequist, L.V.; Su, W.C.; Kim, S.W.; Kim, J.H. Osimertinib in pretreated T790M-positive advanced non–small-cell lung cancer: AURA study phase II extension component. J. Clin. Oncol. 2017, 35, 1288–1296. [Google Scholar] [CrossRef]
- Sutto, L.; Gervasio, F.L. Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 10616–10621. [Google Scholar] [CrossRef]
- Astl, L.; Tse, A.; Verkhivker, G.M. Interrogating regulatory mechanisms in signaling proteins by allosteric inhibitors and activators: A dynamic view through the lens of residue interaction networks. In Protein Allostery in Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2019; pp. 187–223. [Google Scholar]
- Maity, A.; Majumdar, S.; Priya, P.; De, P.; Saha, S.; Dastidar, S.G. Adaptability in protein structures: Structural dynamics and implications in ligand design. J. Biomol. Struct. Dyn. 2015, 33, 298–321. [Google Scholar] [CrossRef]
- Yang, M.; Bonati, L.; Polino, D.; Parrinello, M. Using metadynamics to build neural network potentials for reactive events: The case of urea decomposition in water. Catal. Today 2022, 387, 143–149. [Google Scholar] [CrossRef]
- Singh, H.; Raja, A.; Shekhar, N.; Chauhan, A.; Prakash, A.; Avti, P.; Medhi, B. Computational attributes of protein kinase-C gamma C2-domain & virtual screening for small molecules: Elucidation from meta-dynamics simulations & free-energy calculations. J. Biomol. Struct. Dyn. 2023, 41, 4981–4992. [Google Scholar]
- Kipf, T.; Fetaya, E.; Wang, K.C.; Welling, M.; Zemel, R. Neural relational inference for interacting systems. In Proceedings of the 35th International Conference on Machine Learning, Stockholm, Sweden, 10–15 July 2018; pp. 2688–2697. [Google Scholar]
- Webb, E.; Day, B.; Andres-Terre, H.; Lió, P. Factorised neural relational inference for multi-interaction systems. arXiv 2019, arXiv:1905.08721. [Google Scholar]
- Papakyriakou, A.; Vourloumis, D.; Tzortzatou-Stathopoulou, F.; Karpusas, M. Conformational dynamics of the EGFR kinase domain reveals structural features involved in activation. Proteins Struct. Funct. Bioinform. 2009, 76, 375–386. [Google Scholar] [CrossRef]
- Tamirat, M.Z.; Kurppa, K.J.; Elenius, K.; Johnson, M.S. Deciphering the structural effects of activating EGFR somatic mutations with molecular dynamics simulation. J. Vis. Exp. 2020, 159, 61125. [Google Scholar]
- Schrodinger, L.L. The PyMOL molecular graphics system. Version 2015, 1, 8. [Google Scholar]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef]
- Hashemi, S.; Seradj, H.; Zare Gheshlaghi, S.; Behrouz, M.; Shahraki, A.; Emami, L. Computational studies of Salvurmin A and Salvurmin B, two ursane triterpenoids of Salvia Urmiensis as anti-cancer agents: Molecular docking, molecular dynamic simulation, and DFT and ADMET analysis. Mol. Phys. 2024, 122, e2253334. [Google Scholar] [CrossRef]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef]
- Aliev, A.E.; Kulke, M.; Khaneja, H.S.; Chudasama, V.; Sheppard, T.D.; Lanigan, R.M. Motional timescale predictions by molecular dynamics simulations: Case study using proline and hydroxyproline sidechain dynamics. Proteins Struct. Funct. Bioinform. 2014, 82, 195–215. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Pires, D.E.; de Melo-Minardi, R.C.; Santos, M.A.D.; da Silveira, C.H.; Santoro, M.M.; Meira, W. An iterative workflow for mining the human intestinal metaproteome. BMC Genom. 2011, 12, 6. [Google Scholar]
- Sang, P.; Yang, Q.; Du, X.; Yang, N.; Yang, L.Q.; Ji, X.L.; Fu, Y.X.; Meng, Z.H.; Liu, S.Q. Effect of the solvent temperatures on dynamics of serine protease proteinase K. Int. J. Mol. Sci. 2016, 17, 254. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Husic, B.E.; Pande, V.S. Markov state models: From an art to a science. J. Am. Chem. Soc. 2018, 140, 2386–2396. [Google Scholar] [CrossRef]
- Noé, F.; Clementi, C. Kinetic distance and kinetic maps from molecular dynamics simulation. J. Chem. Theory Comput. 2015, 11, 5002–5011. [Google Scholar] [CrossRef]
- Pérez-Hernández, G.; Paul, F.; Giorgino, T.; De Fabritiis, G.; Noé, F. Identification of slow molecular order parameters for Markov model construction. J. Chem. Phys. 2013, 139, 015102. [Google Scholar] [CrossRef] [PubMed]
- Prinz, J.H.; Wu, H.; Sarich, M.; Keller, B.; Senne, M. Markov models of molecular kinetics: Generation and validation. J. Chem. Phys. 2011, 134, 174105. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.R.; Pande, V.S.; Noé, F. An Introduction to Markov State Models and Their Application to Long Timescale Molecular Simulation; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Scherer, M.K.; Trendelkamp-Schroer, B.; Paul, F.; Pérez-Hernández, G.; Hoffmann, M.; Plattner, N.; Wehmeyer, C.; Prinz, J.-H.; Noé, F. PyEMMA 2: A software package for estimation, validation, and analysis of Markov models. J. Chem. Theory Comput. 2015, 11, 5525–5542. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, J.; Li, G. Neural relational inference with efficient message passing mechanisms. Proc. AAAI Conf. Artif. Intell. 2021, 35, 7055–7063. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, J.; Han, W.; Xu, D. Neural relational inference to learn allosteric long-range interactions in proteins from molecular dynamics simulations. bioRxiv 2021. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Javaid, A. Understanding Dijkstra’s Algorithm. 2013. Available online: https://ssrn.com/abstract=2340905 (accessed on 20 September 2024).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, H.; Zhao, D.-R.; Liu, M.-T.; Yang, L.-Q.; Sang, P. Conformational Remodeling and Allosteric Regulation Underlying EGFR Mutant-Induced Activation: A Multi-Scale Analysis Using MD, MSMs, and NRI. Int. J. Mol. Sci. 2025, 26, 6226. https://doi.org/10.3390/ijms26136226
Duan H, Zhao D-R, Liu M-T, Yang L-Q, Sang P. Conformational Remodeling and Allosteric Regulation Underlying EGFR Mutant-Induced Activation: A Multi-Scale Analysis Using MD, MSMs, and NRI. International Journal of Molecular Sciences. 2025; 26(13):6226. https://doi.org/10.3390/ijms26136226
Chicago/Turabian StyleDuan, Hui, De-Rui Zhao, Meng-Ting Liu, Li-Quan Yang, and Peng Sang. 2025. "Conformational Remodeling and Allosteric Regulation Underlying EGFR Mutant-Induced Activation: A Multi-Scale Analysis Using MD, MSMs, and NRI" International Journal of Molecular Sciences 26, no. 13: 6226. https://doi.org/10.3390/ijms26136226
APA StyleDuan, H., Zhao, D.-R., Liu, M.-T., Yang, L.-Q., & Sang, P. (2025). Conformational Remodeling and Allosteric Regulation Underlying EGFR Mutant-Induced Activation: A Multi-Scale Analysis Using MD, MSMs, and NRI. International Journal of Molecular Sciences, 26(13), 6226. https://doi.org/10.3390/ijms26136226