
Modeling the Bone Marrow Niche in Multiple Myeloma: From 2D Cultures to 3D Systems


Abstract
1. Introduction
1.1. General Considerations for Multiple Myeloma
1.2. Bioprinting: An Innovation in the Field of Regenerative Medicine
1.3. Multiple Myeloma Biology: In Vivo Models
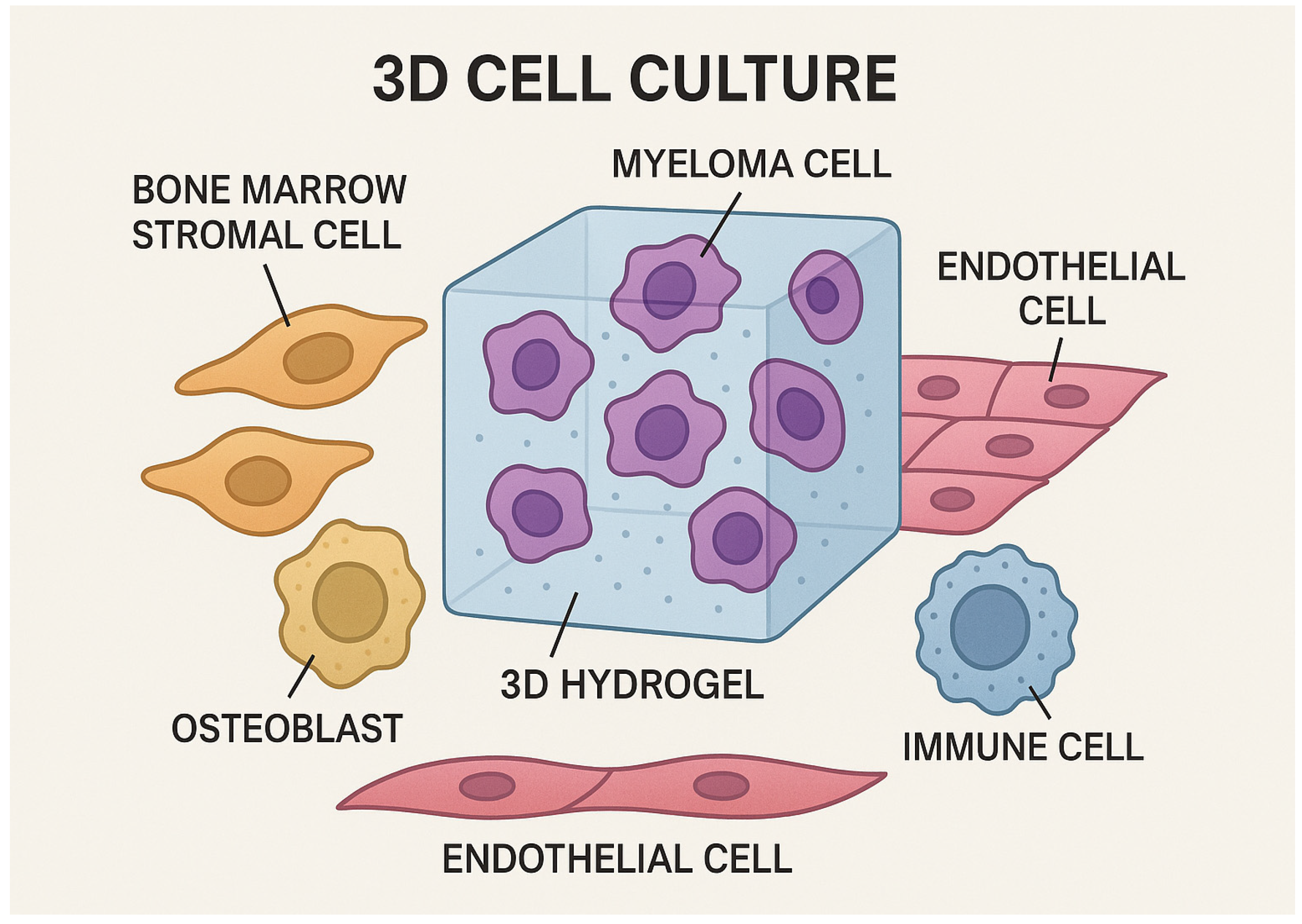
2. Three-Dimensional Cultures: Innovation in the Field of Research
General Considerations for 3D Cultures
3. Three-Dimensional Cultures in Multiple Myeloma
Modeling Multiple Myeloma in 3D: Insights into Molecular Mechanisms
4. Multiple Myeloma Therapy: Applications of 3D Cultures
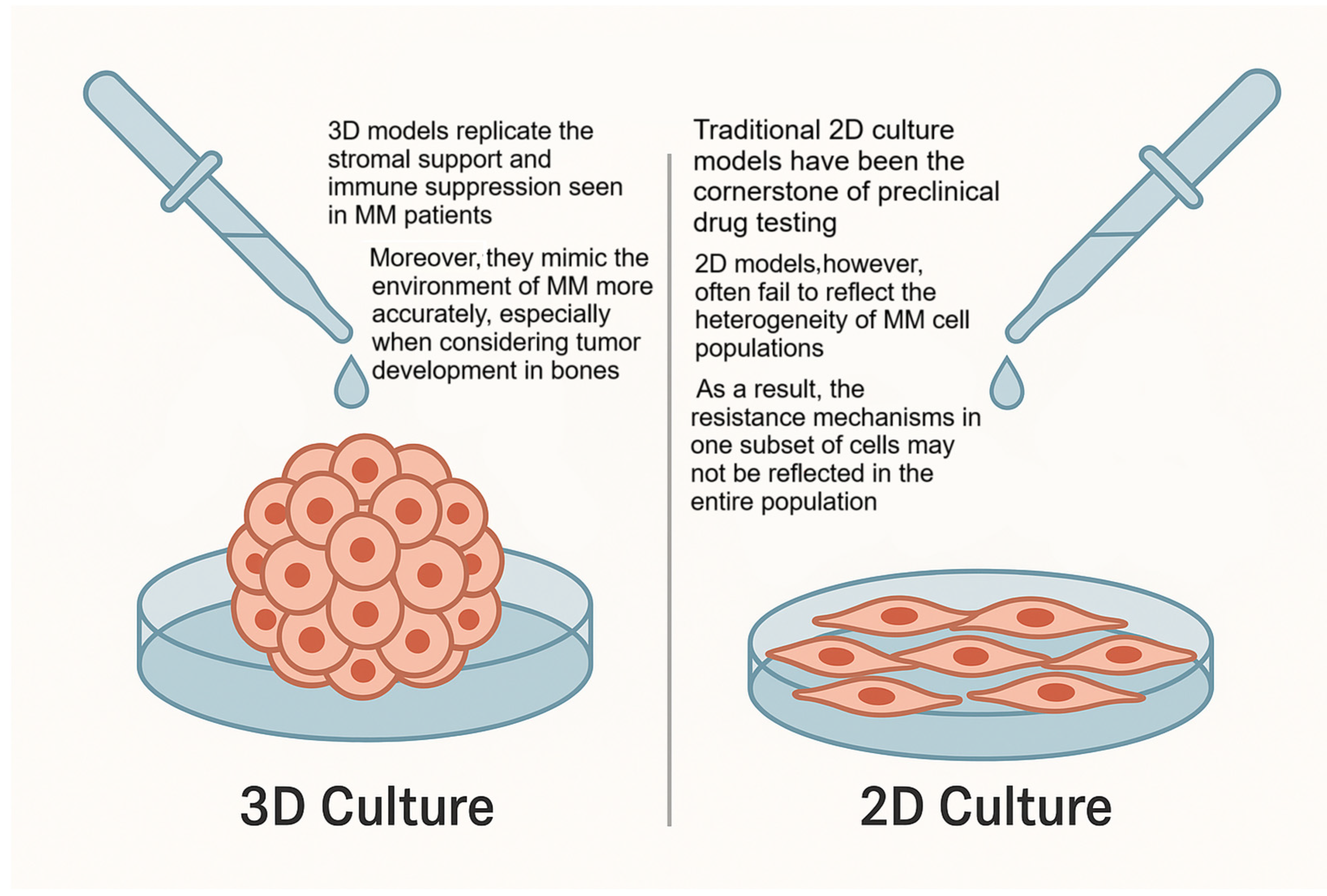
4.1. Three-Dimensional Models and Drug Resistance in Multiple Myeloma
4.2. Controversies and Limitations in the Application of 3D Models
5. A Concise Overview on Organoids and Their Potential Role in Personalized Medicine: A Future Perspective
6. CAR-T: A New Perspective
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| WHO | World Health Organization. |
| MM | Multiple myeloma. |
| PCs | Plasma cells. |
| FDM | Fused deposition modeling. |
| SLA | Stereolithography. |
| DIW | Direct ink writing. |
| LGDW | Laser-guided direct writing. |
| PCL | Polycaprolactone. |
| PLA | Poly(lactic acid). |
| MGUS | Monoclonal gammopathy of undetermined significance. |
| TNF-α | Tumor necrosis factor alpha. |
| IL-1 | Interleukin 1. |
| TEM | Effector memory T. |
| TEMRA | Terminally differentiated effector memory RA. |
| MDSCs | Myeloid-derived suppressor cells. |
| PFS | Progression free survival. |
| OS | Overall survival. |
| T-regs | Regulatory T cells. |
| TGF-β | Transforming growth factor beta. |
| SDF-1 | Stromal cells derived factor 1. |
| IMiDs | Immunomodulatory drugs. |
| CAR-T | Chimeric antigen receptor T-cell. |
| TME | Tumor microenvironment. |
| ASCs | Adult stem cells. |
| ESCs | Embryonic stem cells. |
| iPSCs | Induced pluripotent stem cells. |
| CLL | Chronic lymphocytic leukemia. |
| BM | Bone marrow. |
| RCCS™ | Rotary cell culture system. |
| HS-5 | Human bone marrow stromal cell. |
| HARV | High aspect ratio vessels. |
| ECM | Extracellular matrix. |
| WNT | Wingless/integrated. |
| APC | Antigen-presenting cell. |
| GSK-3β | Glycogen synthase kinase-3 beta. |
| CK1 | Casein kinase 1. |
| FZD | Frizzled. |
| LRP5/6 | Lung resistance-related protein. |
| PCP | Planar cell polarity. |
| ROR2 | Receptor tyrosine kinase-like orphan receptor 2. |
| PI3K | Phosphoinositide 3-kinase. |
| AKT | Protein kinase B. |
| mTOR | Mammalian target of rapamycin. |
| EMD | Extramedullary. |
| STAT3 | Signal transducer and activator of transcription 3. |
| ERK 3 | Extracellular signal-regulated kinase. |
| MAPK | Mitogen-activated protein kinase. |
| NFKb | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| EGFR | Epidermal growth factor receptor. |
| SAHA | Suberoylanilide hydroxamic acid. |
| HDAC | Histone deacetylase inhibitor. |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand. |
| MSCs | Mesenchymal stromal cells. |
| BM-MSCs | Bone marrow mesenchymal stromal cells. |
| PTX | Paclitaxel. |
| CAM-DR | Cell adhesion-mediated drug resistance. |
| VEGF | Vascular endothelial growth factor. |
| RAF | Rapidly accelerated fibrosarcoma. |
| MEK | Mitogen-activated protein kinase. |
| CRISPR-Cas9 | Clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9. |
| DMEM | Dulbecco’s Modified Eagle Medium. |
| EGF | Epidermal growth factor. |
| FGF10 | Fibroblast growth factor 10. |
| HEPES | 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid. |
| HGF | Hepatocyte growth factor. |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Jomehpour, M.; Khosravi, M.; Janfada, M.; Abroun, S.; Vahdat, S. Establishment of A Three-Dimensional Culture Condition for The U266 Cell Line Based on Peripheral Blood Plasma-Derived Fibrin Gels. Cell J. 2023, 25, 229–237. [Google Scholar] [CrossRef] [PubMed]
- De La Puente, P.; Muz, B.; Gilson, R.C.; Azab, F.; Luderer, M.; King, J.; Achilefu, S.; Vij, R.; Azab, A.K. 3D Tissue-Engineered Bone Marrow as a Novel Model to Study Pathophysiology and Drug Resistance in Multiple Myeloma. Biomaterials 2015, 73, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Ignatz-Hoover, J.J.; Driscoll, J.J. Therapeutics to Harness the Immune Microenvironment in Multiple Myeloma. Cancer Drug Resist. 2022, 5, 647–661. [Google Scholar] [CrossRef]
- Kirshner, J.; Thulien, K.J.; Martin, L.D.; Debes Marun, C.; Reiman, T.; Belch, A.R.; Pilarski, L.M. A Unique Three-Dimensional Model for Evaluating the Impact of Therapy on Multiple Myeloma. Blood 2008, 112, 2935–2945. [Google Scholar] [CrossRef]
- Arhoma, A.; Chantry, A.D.; Haywood-Small, S.L.; Cross, N.A. SAHA-Induced TRAIL-Sensitisation of Multiple Myeloma Cells Is Enhanced in 3D Cell Culture. Exp. Cell Res. 2017, 360, 226–235. [Google Scholar] [CrossRef]
- Anderson, K.C.; Carrasco, R.D. Pathogenesis of Myeloma. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 249–274. [Google Scholar] [CrossRef]
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispenzieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical Course and Prognosis of Smoldering (Asymptomatic) Multiple Myeloma. N. Engl. J. Med. 2007, 356, 2582–2590. [Google Scholar] [CrossRef]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2018, 378, 241–249. [Google Scholar] [CrossRef]
- Bustoros, M.; Sklavenitis-Pistofidis, R.; Park, J.; Redd, R.; Zhitomirsky, B.; Dunford, A.J.; Salem, K.; Tai, Y.-T.; Anand, S.; Mouhieddine, T.H.; et al. Genomic Profiling of Smoldering Multiple Myeloma Identifies Patients at a High Risk of Disease Progression. J. Clin. Oncol. 2020, 38, 2380–2389. [Google Scholar] [CrossRef]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The Genetic Architecture of Multiple Myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Otero, P.; Paiva, B.; San-Miguel, J.F. Roadmap to Cure Multiple Myeloma. Cancer Treat. Rev. 2021, 100, 102284. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Molina, L.; Espert, L.; Mechti, N. Hyaluronan, a Major Non-protein Glycosaminoglycan Component of the Extracellular Matrix in Human Bone Marrow, Mediates Dexamethasone Resistance in Multiple Myeloma. Br. J. Haematol. 2003, 121, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Cooke, R.E.; Koldej, R.; Ritchie, D. Immunotherapeutics in Multiple Myeloma: How Can Translational Mouse Models Help? J. Oncol. 2019, 2019, 2186494. [Google Scholar] [CrossRef]
- Kantaros, A.; Ganetsos, T. From Static to Dynamic: Smart Materials Pioneering Additive Manufacturing in Regenerative Medicine. Int. J. Mol. Sci. 2023, 24, 15748. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kantaros, A. 3D Printing in Regenerative Medicine: Technologies and Resources Utilized. Int. J. Mol. Sci. 2022, 23, 14621. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yaneva, A.; Shopova, D.; Bakova, D.; Mihaylova, A.; Kasnakova, P.; Hristozova, M.; Semerdjieva, M. The Progress in Bioprinting and Its Potential Impact on Health-Related Quality of Life. Bioengineering 2023, 10, 910. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vanderkerken, K.; Van Camp, B.; De Greef, C.; Vande Broek, I.; Asosingh, K.; Van Riet, I. Homing of the Myeloma Cell Clone. Acta Oncol. 2000, 39, 771–776. [Google Scholar] [CrossRef]
- Vanderkerken, K.; Asosingh, K.; Croucher, P.; Van Camp, B. Multiple Myeloma Biology: Lessons from the 5TMM Models. Immunol. Rev. 2003, 194, 196–206. [Google Scholar] [CrossRef]
- Guillerey, C.; Ferrari De Andrade, L.; Vuckovic, S.; Miles, K.; Ngiow, S.F.; Yong, M.C.R.; Teng, M.W.L.; Colonna, M.; Ritchie, D.S.; Chesi, M.; et al. Immunosurveillance and Therapy of Multiple Myeloma Are CD226 Dependent. J. Clin. Investig. 2015, 125, 2077–2089. [Google Scholar] [CrossRef]
- Coder, B.; Su, D.-M. Thymic Involution beyond T-Cell Insufficiency. Oncotarget 2015, 6, 21777–21778. [Google Scholar] [CrossRef] [PubMed]
- Den Braber, I.; Mugwagwa, T.; Vrisekoop, N.; Westera, L.; Mögling, R.; Bregje de Boer, A.; Willems, N.; Schrijver, E.H.R.; Spierenburg, G.; Gaiser, K.; et al. Maintenance of Peripheral Naive T Cells Is Sustained by Thymus Output in Mice but Not Humans. Immunity 2012, 36, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Görgün, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-Promoting Immune-Suppressive Myeloid-Derived Suppressor Cells in the Multiple Myeloma Microenvironment in Humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [PubMed]
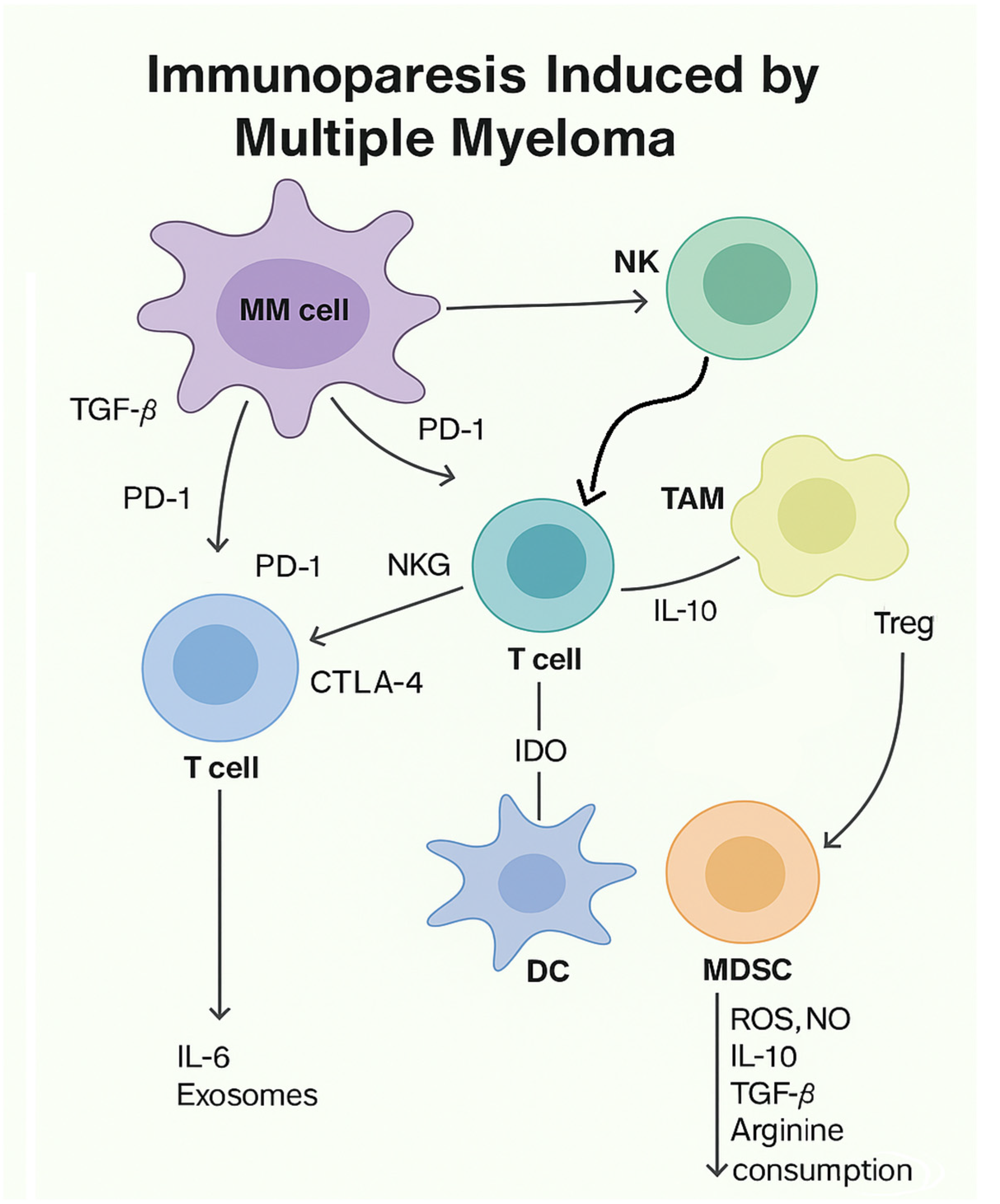
- Sørrig, R.; Klausen, T.W.; Salomo, M.; Vangsted, A.J.; Frølund, U.C.; Andersen, K.T.; Klostergaard, A.; Helleberg, C.; Pedersen, R.S.; Pedersen, P.T.; et al. Immunoparesis in Newly Diagnosed Multiple Myeloma Patients: Effects on Overall Survival and Progression Free Survival in the Danish Population. PLoS ONE 2017, 12, e0188988. [Google Scholar] [CrossRef]
- Pratt, G.; Goodyear, O.; Moss, P. Immunodeficiency and Immunotherapy in Multiple Myeloma. Br. J. Haematol. 2007, 138, 563–579. [Google Scholar] [CrossRef]
- Minnema, M.C.; Van der Veer, M.S.; Aarts, T.; Lokhorst, H.M. The role of T-cell dysfunction in the pathogenesis and treatment of multiple myeloma. Leukemia 2010, 24, 452–462. [Google Scholar]
- Dosani, T.; Carlsten, M.; Maric, I.; Landgren, O. Therapeutic strategies targeting immune dysfunction in multiple myeloma. Br. J. Haematol. 2015, 169, 385–401. [Google Scholar]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2019, 9, 3176. [Google Scholar] [CrossRef]
- Winkler, W.; Farré Díaz, C.; Blanc, E.; Napieczynska, H.; Langner, P.; Werner, M.; Walter, B.; Wollert-Wulf, B.; Yasuda, T.; Heuser, A.; et al. Mouse Models of Human Multiple Myeloma Subgroups. Proc. Natl. Acad. Sci. USA 2023, 120, e2219439120. [Google Scholar] [CrossRef]
- Cordeiro, S.; Oliveira, B.B.; Valente, R.; Ferreira, D.; Luz, A.; Baptista, P.V.; Fernandes, A.R. Breaking the Mold: 3D Cell Cultures Reshaping the Future of Cancer Research. Front. Cell Dev. Biol. 2024, 12, 1507388. [Google Scholar] [CrossRef]
- Abuwatfa, W.H.; Pitt, W.G.; Husseini, G.A. Scaffold-Based 3D Cell Culture Models in Cancer Research. J. Biomed. Sci. 2024, 31, 7. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; An, B.S.; Kim, M.J.; Jeoung, Y.J.; Byun, J.-H.; Lee, J.H.; Oh, S.H. Signaling Molecule-Immobilized Porous Particles with a Leaf-Stacked Structure as a Bioactive Filler System. ACS Biomater. Sci. Eng. 2020, 6, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.; Choudhury, D.; Fang, Y.; Hunziker, W. Towards Manufacturing of Human Organoids. Biotechnol. Adv. 2020, 39, 107460. [Google Scholar] [CrossRef] [PubMed]
- 1Belloni, D.; Ferrarini, M.; Ferrero, E.; Guzzeloni, V.; Barbaglio, F.; Ghia, P.; Scielzo, C. Protocol for Generation of 3D Bone Marrow Surrogate Microenvironments in a Rotary Cell Culture System. STAR Protoc. 2022, 3, 101601. [Google Scholar] [CrossRef]
- Baker, B.M.; Chen, C.S. Deconstructing the Third Dimension—How 3D Culture Microenvironments Alter Cellular Cues. J. Cell Sci. 2012, 125 Pt 13, 3015–3024. [Google Scholar] [CrossRef]
- Kenny, P.A.; Lee, G.Y.; Myers, C.A.; Neve, R.M.; Semeiks, J.R.; Spellman, P.T.; Lorenz, K.; Lee, E.H.; Barcellos-Hoff, M.H.; Petersen, O.W.; et al. The Morphologies of Breast Cancer Cell Lines in Three-dimensional Assays Correlate with Their Profiles of Gene Expression. Mol. Oncol. 2007, 1, 84–96. [Google Scholar] [CrossRef]
- Zlei, M.; Egert, S.; Bayer, C.; Wider, D.; Schüler, J.; Engelhardt, M. Assessment of the culture requirements for optimal in vitro growth and survival of multiple myeloma cells. Leuk. Res. 2006, 30, 955–964. [Google Scholar] [CrossRef]
- Misund, K.; Baranowska, K.A.; Holien, T.; Rampa, C.; Klein, D.C.G.; Børset, M.; Waage, A.; Sundan, A. A Method for Measurement of Drug Sensitivity of Myeloma Cells Co-Cultured with Bone Marrow Stromal Cells. SLAS Discov. Adv. Sci. Drug Discov. 2013, 18, 637–646. [Google Scholar] [CrossRef]
- Ndubuisi, C.; Dang, T.; Staskiewicz, A.; Wang, X. Development of Cell Culture Models for Multiple Myeloma Drug Discovery: Comparison of Traditional 2D, Transwell 2D and 3D-Bioprinted Models. Ph.D. Thesis, 2024. [Google Scholar]
- Giliberto, G.; Botta, C.; Tagliaferri, P.; Tassone, P. Ex vivo drug sensitivity screening in multiple myeloma identifies drug combinations that act synergistically. Br. J. Haematol. 2022, 198, 830–842. [Google Scholar] [CrossRef]
- Poincloux, R.; Collin, O.; Lizárraga, F.; Romao, M.; Debray, M.; Piel, M.; Chavrier, P. Contractility of the Cell Rear Drives Invasion of Breast Tumor Cells in 3D Matrigel. Proc. Natl. Acad. Sci. USA 2011, 108, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Gargotti, M.; Lopez-Gonzalez, U.; Byrne, H.J.; Casey, A. Comparative Studies of Cellular Viability Levels on 2D and 3D in Vitro Culture Matrices. Cytotechnology 2018, 70, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Rabionet, M.; Yeste, M.; Puig, T.; Ciurana, J. Electrospinning PCL Scaffolds Manufacture for Three-Dimensional Breast Cancer Cell Culture. Polymers 2017, 9, 328. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, M.; Piccinini, F.; Arienti, C.; Zamagni, A.; Santi, S.; Polico, R.; Bevilacqua, A.; Tesei, A. 3D Tumor Spheroid Models for in Vitro Therapeutic Screening: A Systematic Approach to Enhance the Biological Relevance of Data Obtained. Sci. Rep. 2016, 6, 19103. [Google Scholar] [CrossRef]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; et al. Comparison of 2D- and 3D-Culture Models as Drug-Testing Platforms in Breast Cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef]
- He, J.; Liang, X.; Luo, F.; Chen, X.; Xu, X.; Wang, F.; Zhang, Z. P53 Is Involved in a Three-Dimensional Architecture-Mediated Decrease in Chemosensitivity in Colon Cancer. J. Cancer 2016, 7, 900–909. [Google Scholar] [CrossRef]
- Hou, J.; Chen, Y.; Meng, X.; Shi, C.; Li, C.; Chen, Y.; Sun, H. Compressive force regulates ephrinB2 and EphB4 in osteoblasts and osteoclasts contributing to alveolar bone resorption during experimental tooth movement. Korean J. Orthod. 2014, 44, 320–329. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, G.; Xu, F. Engineering Biomaterials and Approaches for Mechanical Stretching of Cells in Three Dimensions. Front. Bioeng. Biotechnol. 2020, 8, 589590. [Google Scholar] [CrossRef]
- Kahlert, C.; Kalluri, R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J. Mol. Med. 2013, 91, 431–437. [Google Scholar] [CrossRef]
- Leverett, L.B.; Hellums, J.D.; Alfrey, C.P.; Lynch, E.C. Red Blood Cell Damage by Shear Stress. Biophys. J. 1972, 12, 257–273. [Google Scholar] [CrossRef]
- Minsky, B.D.; Chlapowski, F.J. Morphometric analysis of the translocation of lumenal membrane between cytoplasm and cell surface of transitional epithelial cells during the expansion-contraction cycles of mammalian urinary bladder. J. Cell Biol. 1978, 77, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Ilic, J.; Koelbl, C.; Simon, F.; Wußmann, M.; Ebert, R.; Trivanovic, D.; Herrmann, M. Liquid Overlay and Collagen-Based Three-Dimensional Models for In Vitro Investigation of Multiple Myeloma. Tissue Eng. Part C Methods 2024, 30, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Hinsenkamp, A.; Kun, K.; Gajnut, F.; Majer, A.; Lacza, Z.; Hornyák, I. Cell Attachment Capacity and Compounds of Fibrin Membranes Isolated from Fresh Frozen Plasma and Cryoprecipitate. Membranes 2021, 11, 783. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Tripathi, K.; Pathak, C.; Vernon, B.L. Plasma-Based Fast-Gelling Biohybrid Gels for Biomedical Applications. Sci. Rep. 2019, 9, 10881. [Google Scholar] [CrossRef]
- Martini, S.; Drzeniek, N.M.; Stark, R.; Kollert, M.R.; Du, W.; Reinke, S.; Ort, M.; Hardt, S.; Kotko, I.; Kath, J.; et al. Long-Term in Vitro Maintenance of Plasma Cells in a Hydrogel-Enclosed Human Bone Marrow Microphysiological 3D Model System. Biofabrication 2024, 16, 045005. [Google Scholar] [CrossRef]
- Xu, K.; Narayanan, K.; Lee, F.; Bae, K.H.; Gao, S.; Kurisawa, M. Enzyme-Mediated Hyaluronic Acid–Tyramine Hydrogels for the Propagation of Human Embryonic Stem Cells in 3D. Acta Biomater. 2015, 24, 159–171. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Almowaled, M.; Li, J.; Venner, C.; Sandhu, I.; Peters, A.; Lavasanifar, A.; Lai, R. Three-Dimensional Reconstructed Bone Marrow Matrix Culture Improves the Viability of Primary Myeloma Cells In-Vitro via a STAT3-Dependent Mechanism. Curr. Issues Mol. Biol. 2021, 43, 313–323. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/Beta-Catenin Pathway in Cancer: Update on Effectors and Inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Frenquelli, M.; Caridi, N.; Antonini, E.; Storti, F.; Viganò, V.; Gaviraghi, M.; Occhionorelli, M.; Bianchessi, S.; Bongiovanni, L.; Spinelli, A.; et al. The WNT Receptor ROR2 Drives the Interaction of Multiple Myeloma Cells with the Microenvironment through AKT Activation. Leukemia 2020, 34, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Boulogeorgou, K.; Papaioannou, M.; Chatzileontiadou, S.; Georgiou, E.; Fola, A.; Tzorakoleftheraki, S.-E.; Hatjiharissi, E.; Koletsa, T. Unveiling Extramedullary Myeloma Immune Microenvironment: A Systematic Review. Cancers 2025, 17, 1081. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Molavi, O.; Alshareef, A.; Haque, M.; Wang, Q.; Chu, M.P.; Venner, C.P.; Sandhu, I.; Peters, A.C.; Lavasanifar, A.; et al. Constitutive Activation of STAT3 in Myeloma Cells Cultured in a Three-Dimensional, Reconstructed Bone Marrow Model. Cancers 2018, 10, 206. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Janan, M.; Poohadsuan, J.; Rodboon, N.; Samart, P.; Rungarunlert, S.; Issaragrisil, S. A High-Throughput, Three-Dimensional Multiple Myeloma Model Recapitulating Tumor-Stroma Interactions for CAR-Immune Cell-Mediated Cytotoxicity Assay. Immunotargets Ther. 2025, 14, 321–338. [Google Scholar] [CrossRef]
- Martínez-Lorenzo, M.J.; Anel, A.; Gamen, S.; Monle n, I.; Lasierra, P.; Larrad, L.; Piñeiro, A.; Alava, M.A.; Naval, J. Activated Human T Cells Release Bioactive Fas Ligand and APO2 Ligand in Microvesicles. J. Immunol. 1999, 163, 1274–1281. [Google Scholar] [CrossRef]
- Janssen, E.M.; Droin, N.M.; Lemmens, E.E.; Pinkoski, M.J.; Bensinger, S.J.; Ehst, B.D.; Griffith, T.S.; Green, D.R.; Schoenberger, S.P. CD4+ T-Cell Help Controls CD8+ T-Cell Memory via TRAIL-Mediated Activation-Induced Cell Death. Nature 2005, 434, 88–93. [Google Scholar] [CrossRef]
- Rimondi, E.; Secchiero, P.; Quaroni, A.; Zerbinati, C.; Capitani, S.; Zauli, G. Involvement of TRAIL/TRAIL-receptors in Human Intestinal Cell Differentiation. J. Cell. Physiol. 2006, 206, 647–654. [Google Scholar] [CrossRef]
- Mirandola, P. Activated Human NK and CD8+ T Cells Express Both TNF-Related Apoptosis-Inducing Ligand (TRAIL) and TRAIL Receptors but Are Resistant to TRAIL-Mediated Cytotoxicity. Blood 2004, 104, 2418–2424. [Google Scholar] [CrossRef]
- Özören, N.; El-Deiry, W.S. Defining Characteristics of Types I and II Apoptotic Cells in Response to TRAIL. Neoplasia 2002, 4, 551–557. [Google Scholar] [CrossRef]
- Jablonska, E.; Jablonski, J.; Marcinczyk, M.; Grabowska, Z.; Piotrowski, L. The Release of Soluble Forms of TRAIL and DR5 by Neutrophils of Oral Cavity Cancer Patients. Folia Histochem. Cytobiol. 2008, 46, 177–183. [Google Scholar] [CrossRef] [PubMed]
- İYaylım, T.; Ozkan, N.E.; Turan, S.; Korkmaz, G.; Yıldız, Y.; Cacina, C.; Toptas, B.; Arıkan, S. sTRAIL serum levels and TRAIL 1595 Genotypes: Associations with progress and prognosis of colorectal carcinoma. J. Cancer Ther. 2012, 3, 941–9471. [Google Scholar] [CrossRef]
- Mielczarek-Palacz, A.; Sikora, J.; Kondera-Anasz, Z. Assessment of Concentrations of sTRAIL Ligand and Its Receptors sTRAIL-R1 and sTRAIL-R2—Markers Monitoring the Course of the Extrinsic Pathway of Apoptosis Induction: Potential Application in Ovarian Cancer Diagnostics. Aoms 2017, 3, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, J.M.; Zhou, J.-Y.; Wu, G.S. The Role of TRAIL in Apoptosis and Immunosurveillance in Cancer. Cancers 2023, 15, 2752. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, J.E.; Estes, J.M.; Straughn, J.M., Jr.; Alvarez, R.D.; Buchsbaum, D.J. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) and Its Therapeutic Potential in Breast and Gynecologic Cancers. Gynecol. Oncol. 2007, 106, 614–621. [Google Scholar] [CrossRef]
- Palumbo, A.; Anderson, K. Multiple Myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef]
- Di Ianni, M.; Moretti, L.; Del Papa, B.; Gaozza, E.; Bell, A.S.; Falzetti, F.; Tabilio, A. A Microelectronic DNA Chip Detects the V617F JAK-2 Mutation in Myeloproliferative Disorders. Leukemia 2006, 20, 1895–1897. [Google Scholar] [CrossRef]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef]
- Scalzulli, E.; Grammatico, S.; Vozella, F.; Petrucci, M.T. Proteasome Inhibitors for the Treatment of Multiple Myeloma. Expert. Opin. Pharmacother. 2018, 19, 375–386. [Google Scholar] [CrossRef]
- Adams, J.; Palombella, V.J.; Sausville, E.A.; Johnson, J.; Destree, A.; Lazarus, D.D.; Maas, J.; Pien, C.S.; Prakash, S.; Elliott, P.J. Proteasome inhibitors: A novel class of potent and effective antitumor agents. Cancer Res. 1999, 59, 2615–2622. [Google Scholar]
- Schmitt, S.M.; Deshmukh, R.R.; Dou, Q.P. Proteasome Inhibitors and Lessons Learned from Their Mechanisms of Action and resistance in Human Cancer. In Resistance to Proteasome Inhibitors in Cancer: Molecular Mechanisms and Strategies to Overcome Resistance; Springer International Publishing: Cham, Switzerland, 2014; pp. 1–46. [Google Scholar]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-Drug Resistance in Cancer Chemotherapeutics: Mechanisms and Lab Approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-C.; Lin, S.-F. Mechanisms of Drug Resistance in Relapse and Refractory Multiple Myeloma. BioMed Res. Int. 2015, 2015, 341430. [Google Scholar] [CrossRef] [PubMed]
- Cusack, J. Rationale for the Treatment of Solid Tumors with the Proteasome Inhibitor Bortezomib. Cancer Treat. Rev. 2003, 29, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Fabre, C.; Mimura, N.; Bobb, K.; Kong, S.-Y.; Gorgun, G.; Cirstea, D.; Hu, Y.; Minami, J.; Ohguchi, H.; Zhang, J.; et al. Correction: Dual Inhibition of Canonical and Noncanonical NF-κB Pathways Demonstrates Significant Antitumor Activities in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 2938. [Google Scholar] [CrossRef]
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Kim, J.; Werndli, J.E.; Raschko, M.; Leith, C.P.; Kahl, B.S.; Kim, K.; Miyamoto, S. Bortezomib-Resistant Nuclear Factor-κB Activity in Multiple Myeloma Cells. Mol. Cancer Res. 2008, 6, 1356–1364. [Google Scholar] [CrossRef]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-Mediated Drug Resistance: A Major Contributor to Minimal Residual Disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef]
- Yetisgin, A.A.; Cetinel, S.; Zuvin, M.; Kosar, A.; Kutlu, O. Therapeutic Nanoparticles and Their Targeted Delivery Applications. Molecules 2020, 25, 2193. [Google Scholar] [CrossRef]
- Iannazzo, D.; Ettari, R.; Giofrè, S.; Eid, A.H.; Bitto, A. Recent Advances in Nanotherapeutics for Multiple Myeloma. Cancers 2020, 12, 3144. [Google Scholar] [CrossRef]
- De La Puente, P.; Luderer, M.J.; Federico, C.; Jin, A.; Gilson, R.C.; Egbulefu, C.; Alhallak, K.; Shah, S.; Muz, B.; Sun, J.; et al. Enhancing Proteasome-Inhibitory Activity and Specificity of Bortezomib by CD38 Targeted Nanoparticles in Multiple Myeloma. J. Control. Release 2018, 270, 158–176. [Google Scholar] [CrossRef]
- Nigro, A.; Frattaruolo, L.; Fava, M.; De Napoli, I.; Greco, M.; Comandè, A.; De Santo, M.; Pellegrino, M.; Ricci, E.; Giordano, F.; et al. Bortezomib-Loaded Mesoporous Silica Nanoparticles Selectively Alter Metabolism and Induce Death in Multiple Myeloma Cells. Cancers 2020, 12, 2709. [Google Scholar] [CrossRef]
- Habanjar, O.; Diab-Assaf, M.; Caldefie-Chezet, F.; Delort, L. 3D Cell Culture Systems: Tumor Application, Advantages, and Disadvantages. Int. J. Mol. Sci. 2021, 22, 12200. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kalla, J.; Pfneissl, J.; Mair, T.; Tran, L.; Egger, G. A systematic review on the culture methods and applications of 3D tumoroids for cancer research and personalized medicine. Cell Oncol. 2025, 48, 1–26. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rossi, G.; Manfrin, A.; Lutolf, M.P. Progress and Potential in Organoid Research. Nat. Rev. Genet. 2018, 19, 671–687. [Google Scholar] [CrossRef]
- Yoshida, G.J. Applications of Patient-Derived Tumor Xenograft Models and Tumor Organoids. J. Hematol. Oncol. 2020, 13, 4. [Google Scholar] [CrossRef]
- Wang, V.M.-Y.; Ferreira, R.M.M.; Almagro, J.; Evan, T.; Legrave, N.; Zaw Thin, M.; Frith, D.; Carvalho, J.; Barry, D.J.; Snijders, A.P.; et al. CD9 Identifies Pancreatic Cancer Stem Cells and Modulates Glutamine Metabolism to Fuel Tumour Growth. Nat. Cell Biol. 2019, 21, 1425–1435. [Google Scholar] [CrossRef]
- Wilson, H.V. A New Method by Which Sponges May Be Artificially Reared. Science 1907, 25, 912–915. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Xu, H.; Lyu, X.; Yi, M.; Zhao, W.; Song, Y.; Wu, K. Organoid Technology and Applications in Cancer Research. J. Hematol. Oncol. 2018, 11, 116. [Google Scholar] [CrossRef]
- Schutgens, F.; Clevers, H. Human Organoids: Tools for Understanding Biology and Treating Diseases. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 211–234. [Google Scholar] [CrossRef]
- Holokai, L.; Chakrabarti, J.; Lundy, J.; Croagh, D.; Adhikary, P.; Richards, S.S.; Woodson, C.; Steele, N.; Kuester, R.; Scott, A.; et al. Murine- and Human-Derived Autologous Organoid/Immune Cell Co-Cultures as Pre-Clinical Models of Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 3816. [Google Scholar] [CrossRef] [PubMed]
- Ning, R.-X.; Liu, C.-Y.; Wang, S.-Q.; Li, W.K.; Kong, X. Application status and optimization suggestions of tumor organoids and CAR-T cell co-culture models. Cancer Cell Int. 2024, 24, 98. [Google Scholar] [CrossRef] [PubMed]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell 2018, 22, 454–467. [Google Scholar] [CrossRef]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Dias, J.; Garcia, J.; Agliardi, G.; Roddie, C. CAR-T cell manufacturing landscape-Lessons from the past decade and considerations for early clinical development. Mol. Ther. Methods Clin. Dev. 2024, 32, 101250. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Feature | 2D Cell Culture Models | 3D Cell Culture Models |
|---|---|---|
| Architecture | Monolayer, flat surface | Multicellular aggregates or organoids in a 3D scaffold |
| Cell–cell/cell–matrix interaction | Limited and artificial | More physiologically relevant |
| Mimicking bone marrow microenvironment | Poor | Good simulation of bone marrow niche |
| Drug response predictivity | Low predictive value for clinical outcomes | Improved correlation with in vivo responses |
| Immune interaction modeling | Absent or highly reduced | Potential to integrate immune components |
| Key pathway activation (e.g., STAT3) | Often not activated | STAT3 and other signaling pathways are activated as in vivo |
| Complexity and cost | Low cost, easy to handle | Higher cost, but more informative |
| Suitability for high-throughput screening | High | Moderate to high (increasing with organ-on-chip and automation) |
| Aspect | Details |
|---|---|
| Limitations of 2D Culture |
|
| Advantages of 3D Culture |
|
| Static 3D Systems |
Scaffold-free:
|
| Dynamic 3D Systems |
|
| Bioreactors and Microcarriers |
|
| Organ-on-a-Chip |
|
| Challenges |
|
| Future Directions |
|
| 3D Model Structure | Materials/Scaffold | Biological Properties (Cells and Bioactivity) | Simulatable Dimensions |
|---|---|---|---|
| In situ vascularized bone via 3D bioprinting | GelMA hydrogel + BMSCs + endothelial cells in dual-extrusion printing with tubular channels | Promotes angiogenesis via endothelial sprouting; upregulates osteogenic genes; forms vascularized bone in vivo | Vascularization + bone regeneration |
| Dual-printed SLA + FDM vascularized bone | SLA-printed scaffold with PVA sacrificial channels + FDM PVA template | Mature co-culture of hMSCs and HUVECs; perfusable vessel formation; osteogenesis and angiogenesis coupling | Vascularization + bone differentiation |
| Cell-enhanced 3D-printed PCL/HAp scaffolds | 3D-printed PCL/HAp scaffolds seeded with MSCs and EPC or HUVECs | In vitro vascular network; capillary infiltration and anastomosis in vivo; enhanced bone repair | Vascularization + bone regeneration |
| Nanofiber scaffold with osteo/angiogenic and immunomod | Xonotlite nanofiber + silk fibroin/gelatin hydrogel | Enhances BMSC osteo- and angiogenesis; reprograms macrophages to anti-inflammatory M2—promotes osteoimmune microenvironment | Vascularization + bone regeneration + immunomodulation |
| Nanoparticle-containing porous scaffold | Agarose + nanocrystalline apatite + VEGF-loaded nanoparticles | Nanoparticles induce M2 macrophage polarization, promote IL-10 secretion, and support MSC osteogenesis and angiogenesis in vitro and in vivo | Vascularization + bone support + immunomodulation |
| 3D-printed GelMA-based hydrogel (multifunctional) | GelMA-CQD or GelMA-PPy-Fe in extrusion-based 3D printing | M2 macrophage polarization; anti-inflammatory; enhanced osteogenesis and angiogenesis; shows tumor-combatting + bone repair properties | Vascularization + bone regeneration + immunomodulation |
| 3D Model | Materials/Scaffold | Cell Sources | Bio-Functional Evaluation Metrics | Advantages/Limitations |
|---|---|---|---|---|
| Spheroids (scaffold-free) | No scaffold; use ultra-low attachment plates, hanging drop, spinner, magnetics | Cell lines, primary cells, co-cultures | Oxygen/nutrient gradient formation; necrotic core; drug response assays | Simple, scalable, easy, and cost-effective; however, there is size variability and mechanical/mechano-biological limitations |
| Organoids | ECM-based hydrogels (e.g., Matrigel), natural or synthetic hydrogels | Stem cells (iPSC/ESC), primary tissue | Structural organization, differentiation, gene/protein profiling, multi-cell type presence | Recapitulates organ complexity, heterogeneity; there is batch variability (Matrigel) and costly, time-intensive protocols |
| Hydrogel-based scaffolds | Natural (collagen, HA, alginate) or synthetic (PEG, nanocellulose) | Cell lines, primary cells, iPSC | Viability/growth, mechanical properties (rheology), adhesion, differentiation | ECM-like environment, tunable mechanics; natural gels are variable, synthetics may lack bioactivity |
| Organ-on-chip/microfluidics | PDMS, glass; integrated ECM like collagen, decellularized ECM | Primary cells, tumor, endothelial, immune | Fluid flow, barrier function, migration, toxicity response, multi-tissue interaction | Mimics physiological flow and dynamics, vascularization; complex, costly, low throughput |
| 3D bioprinting | Bio-inks: cell-laden hydrogels (PEG, collagen, gelatin), microgels | Cell lines, primary cells, stem cells | Structural precision, viability, function (e.g., albumin production), ADME gene benchmarking | Highly customizable and scalable; expensive equipment, bio-ink formulation challenges |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bottaro, A.; Nasso, M.E.; Stagno, F.; Fazio, M.; Allegra, A. Modeling the Bone Marrow Niche in Multiple Myeloma: From 2D Cultures to 3D Systems. Int. J. Mol. Sci. 2025, 26, 6229. https://doi.org/10.3390/ijms26136229
Bottaro A, Nasso ME, Stagno F, Fazio M, Allegra A. Modeling the Bone Marrow Niche in Multiple Myeloma: From 2D Cultures to 3D Systems. International Journal of Molecular Sciences. 2025; 26(13):6229. https://doi.org/10.3390/ijms26136229
Chicago/Turabian StyleBottaro, Adele, Maria Elisa Nasso, Fabio Stagno, Manlio Fazio, and Alessandro Allegra. 2025. "Modeling the Bone Marrow Niche in Multiple Myeloma: From 2D Cultures to 3D Systems" International Journal of Molecular Sciences 26, no. 13: 6229. https://doi.org/10.3390/ijms26136229
APA StyleBottaro, A., Nasso, M. E., Stagno, F., Fazio, M., & Allegra, A. (2025). Modeling the Bone Marrow Niche in Multiple Myeloma: From 2D Cultures to 3D Systems. International Journal of Molecular Sciences, 26(13), 6229. https://doi.org/10.3390/ijms26136229