
Gut Microbiota-Bile Acid Crosstalk Contributes to Meat Quality and Carcass Traits of Tan and Dorper Sheep

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
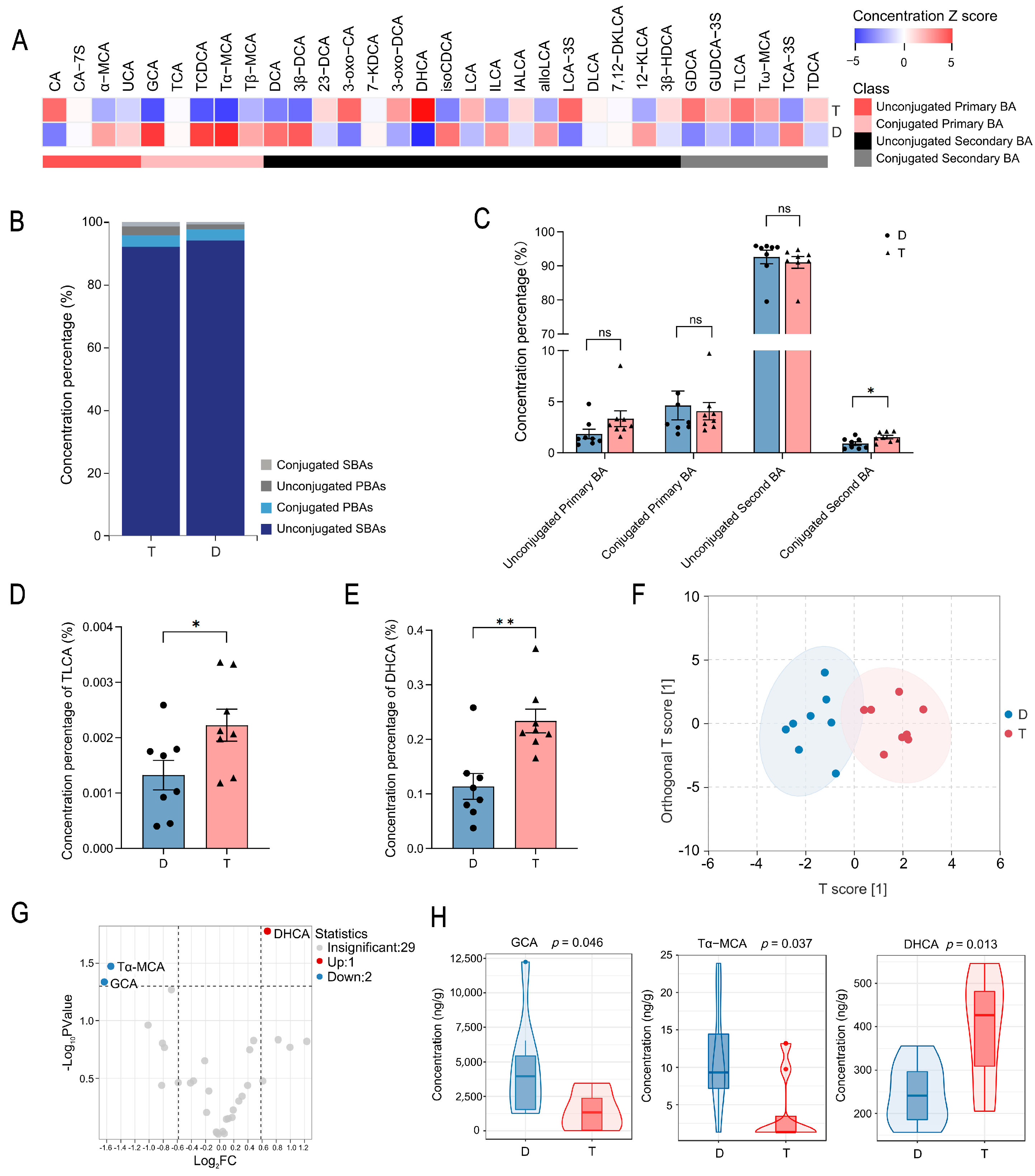
2.1. Comparative Analysis of Colonic Bile Acid Metabolic Profiles in Tan and Dorper Sheep
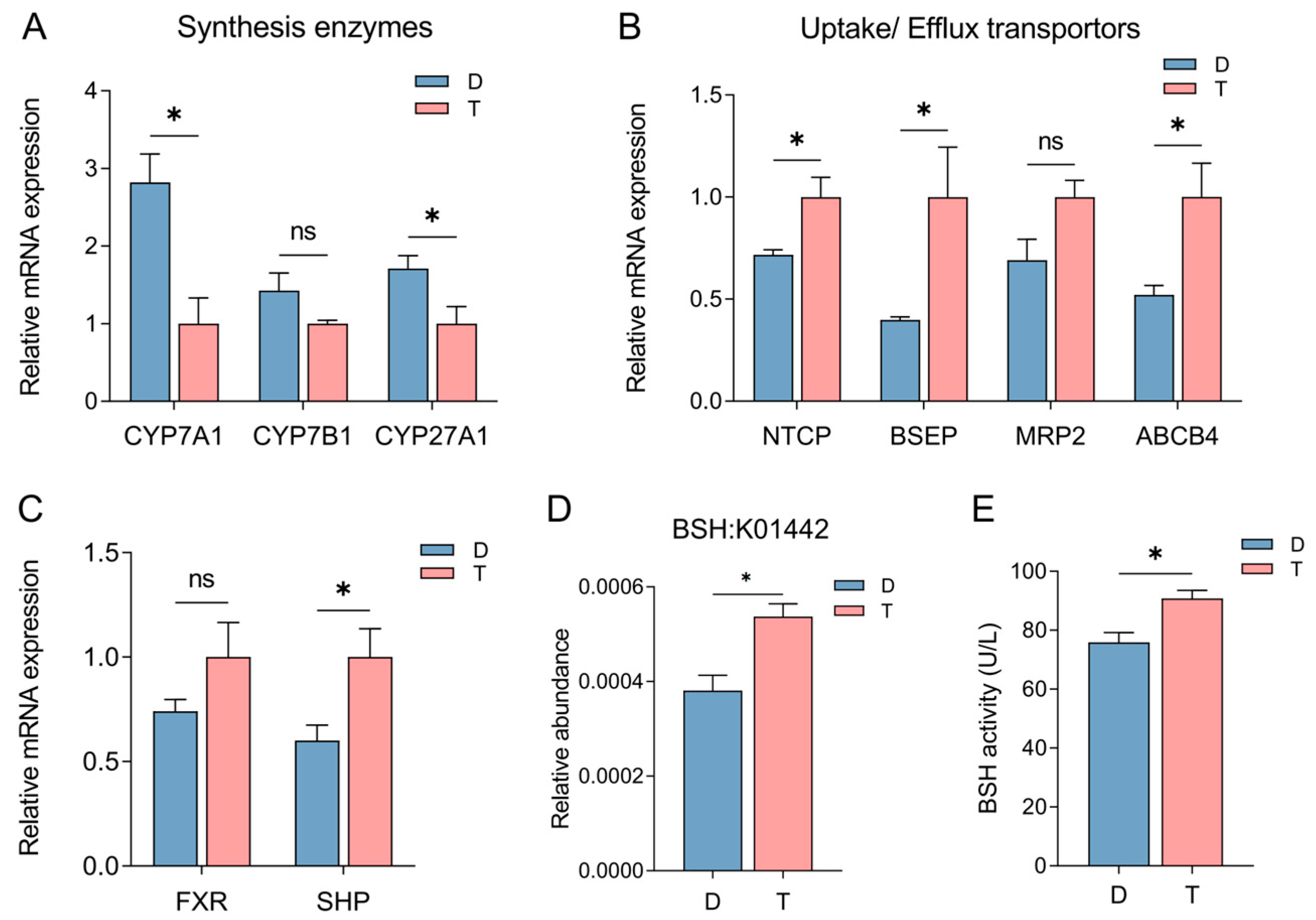
2.2. Influence of Host Genetics and Gut Microbiota on Bile Acid Synthesis, Transport, and Metabolism in Tan and Dorper Sheep
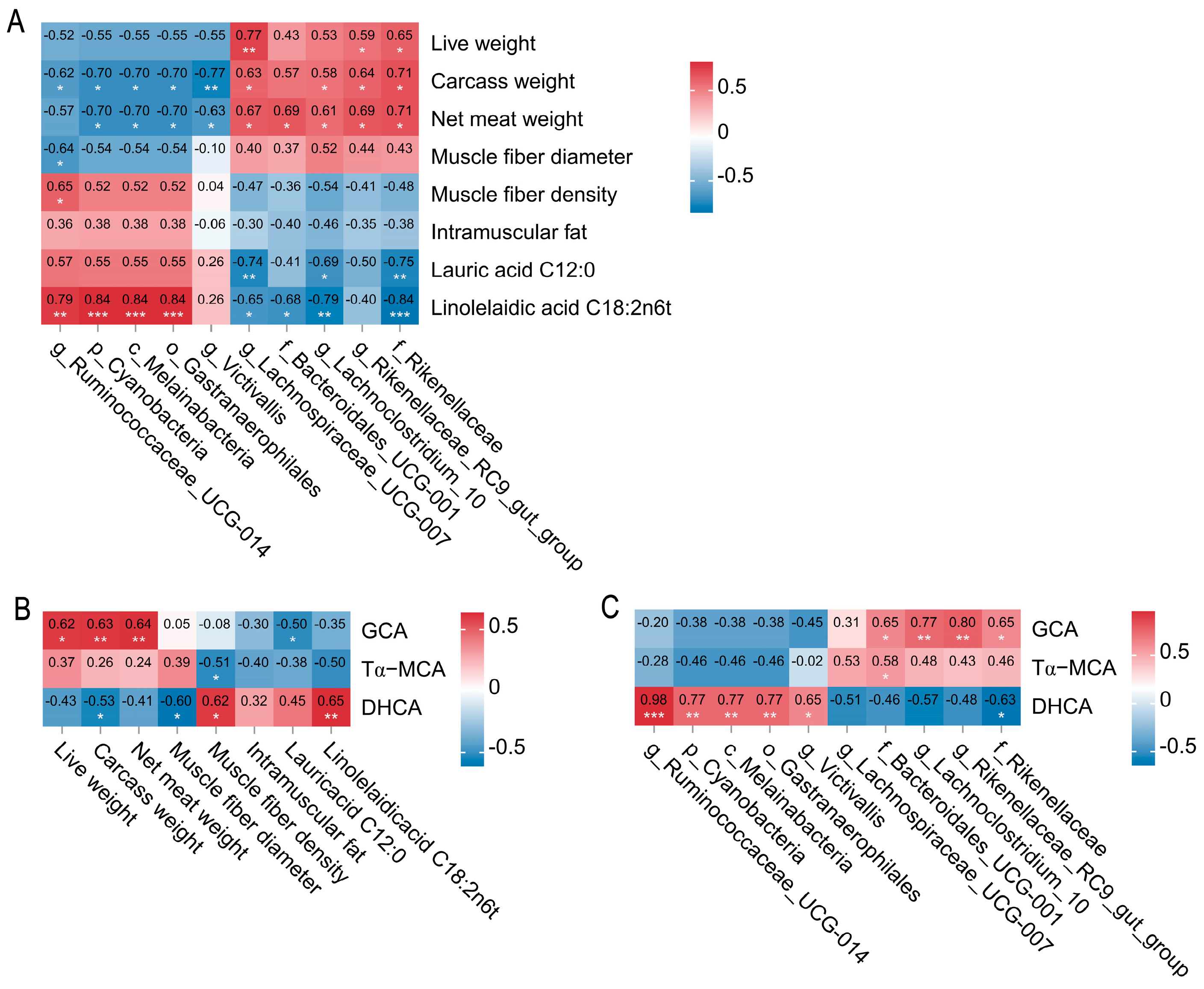
2.3. Spearman Correlation Networks Between Colonic Microbes, Bile Acid Metabolites, Carcass Traits, and Meat Quality in Tan and Dorper Sheep
2.4. Regulatory Role of Bile Acid Metabolites on Host Skeletal Muscle Transcript Expression
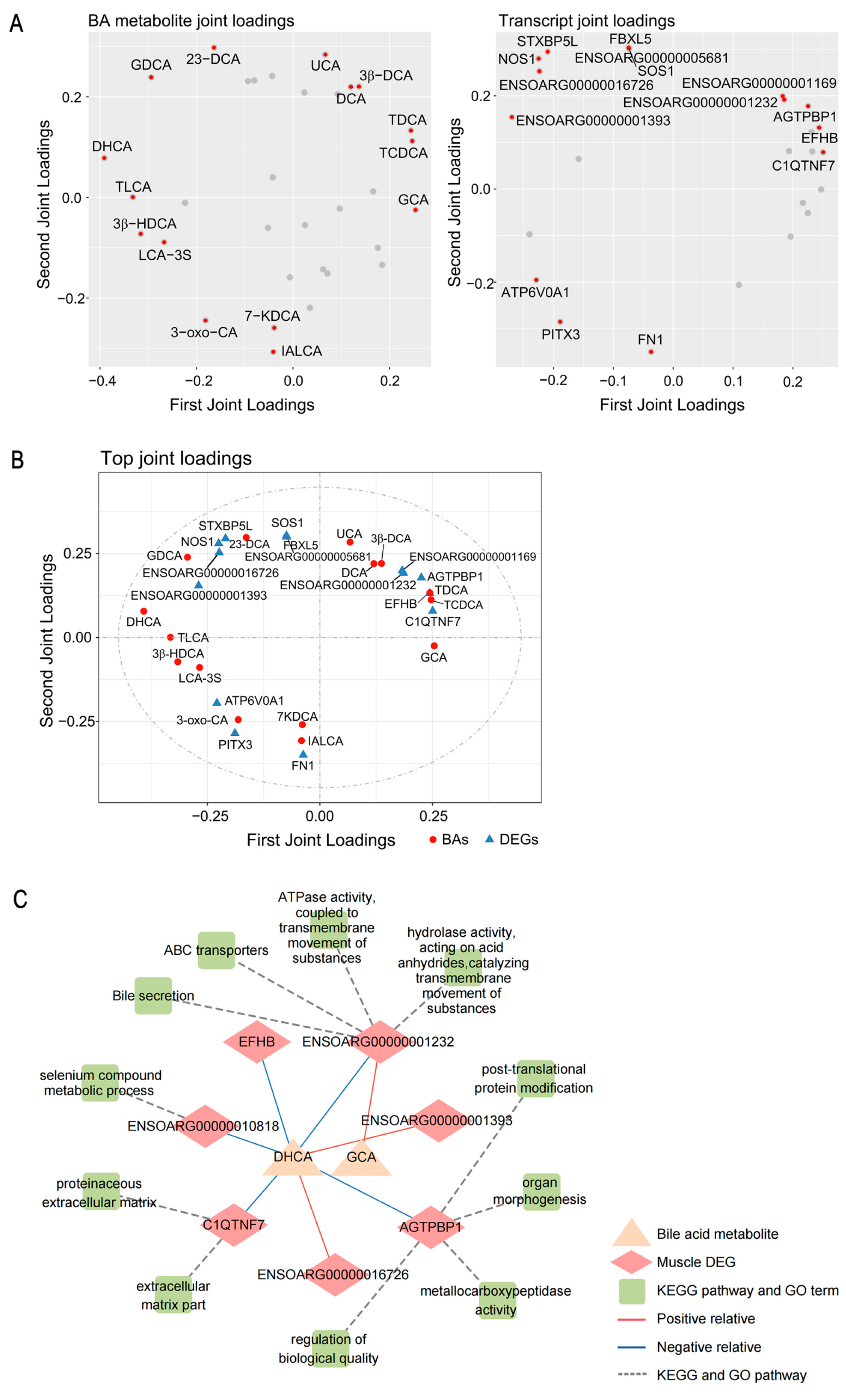
2.5. Multi-Omics Integration Analysis Reveals the Effect of Microbiota–Bile Acid–Gene Crosstalk on Carcass and Meat-Quality Traits of Sheep
3. Discussion
4. Materials and Methods
4.1. Animals and Sample Collection
4.2. Measurement of Meat-Quality Parameters
4.3. Targeted Metabolomics Analysis of Colonic Bile Acids
4.3.1. Sample Preparation and Extraction
4.3.2. High-Performance Liquid Chromatography Conditions
4.3.3. ESI-MS/MS
4.3.4. Qualitative and Quantification
4.3.5. Compositional Profiling of Colonic Bile Acids
4.4. Colon Bile Salt Hydrolases Activity Analysis
4.5. RNA Extraction and Real-Time Quantitative PCR
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BA | bile acid |
| BSH | bile salt hydrolase |
| DHCA | deoxycholic acid |
| GCA | glycochenodeoxycholic acid |
| CYP7A1 | cholesterol 7α-hydroxylase |
| CYP27A1 | cholesterol 27-hydroxylase |
| NCTP | sodium taurocholate cotransporting polypeptide |
| BSEP | bile salt export pump |
| ABCB4 | ATP binding cassette subfamily B member 4 |
| FXR | farnesoid X receptor |
| IMF | intramuscular fat |
References
- Ma, Y.; Yang, X.; Hua, G.; Deng, X.; Xia, T.; Li, X.; Feng, D.; Deng, X. Contribution of gut microbiomes and their metabolomes to the performance of Dorper and Tan sheep. Front. Microbiol. 2022, 13, 1047744. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Kang, X.; Liu, Y.; Liu, X.; Chan, S.; Wang, Y.; Li, Z.; Ling, Y.; Feng, D.; Li, M.; et al. Integrated analysis of the whole transcriptome of skeletal muscle reveals the ceRNA regulatory network related to the formation of muscle fibers in Tan sheep. Front. Genet. 2022, 13, 991606. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef]
- Xie, C.; Huang, W.; Young, R.L.; Jones, K.L.; Horowitz, M.; Rayner, C.K.; Wu, T. Role of Bile Acids in the Regulation of Food Intake, and Their Dysregulation in Metabolic Disease. Nutrients 2021, 13, 1104. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.R.; Want, E.J.; Geier, F.M.; Spagou, K.; Wilson, I.D.; Sidaway, J.E.; Nicholson, J.K.; Holmes, E. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4523–4530. [Google Scholar] [CrossRef] [PubMed]
- Pathak, P.; Xie, C.; Nichols, R.G.; Ferrell, J.M.; Boehme, S.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J.; Chiang, J.Y.L. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatol. Baltim. Md. 2018, 68, 1574–1588. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef]
- Irie, M.; Kouda, M.; Matono, H. Effect of ursodeoxycholic acid supplementation on growth, carcass characteristics, and meat quality of Wagyu heifers (Japanese Black cattle). J. Anim. Sci. 2011, 89, 4221–4226. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, Z.; Yu, Z.; Li, H.; Luo, H.; Wang, B. Transcriptome and targeted metabolome analysis provide insights into bile acids’ new roles and mechanisms on fat deposition and meat quality in lamb. Food Res. Int. Ott. Ont. 2022, 162, 111941. [Google Scholar] [CrossRef]
- Yu, X.; Xue, M.; Liu, Y.; Zhou, Z.; Jiang, Y.; Sun, T.; Liang, H. Effect of nicotinamide riboside on lipid metabolism and gut microflora-bile acid axis in alcohol-exposed mice. Food Sci. Nutr. 2021, 9, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Joyce, S.A.; MacSharry, J.; Casey, P.G.; Kinsella, M.; Murphy, E.F.; Shanahan, F.; Hill, C.; Gahan, C.G. Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc. Natl. Acad. Sci. USA 2014, 111, 7421–7426. [Google Scholar] [CrossRef] [PubMed]
- Clifford, B.L.; Sedgeman, L.R.; Williams, K.J.; Morand, P.; Cheng, A.; Jarrett, K.E.; Chan, A.P.; Brearley-Sholto, M.C.; Wahlström, A.; Ashby, J.W.; et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 2021, 33, 1671–1684.e1674. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Sleator, R.D.; Gahan, C.G.; Hill, C. Contribution of three bile-associated loci, bsh, pva, and btlB, to gastrointestinal persistence and bile tolerance of Listeria monocytogenes. Infect. Immun. 2005, 73, 894–904. [Google Scholar] [CrossRef]
- Liang, L.; Yi, Y.; Lv, Y.; Qian, J.; Lei, X.; Zhang, G. A Comprehensive Genome Survey Provides Novel Insights into Bile Salt Hydrolase (BSH) in Lactobacillaceae. Molecules 2018, 23, 1157. [Google Scholar] [CrossRef]
- Dong, Z.; Lee, B.H. Bile salt hydrolases: Structure and function, substrate preference, and inhibitor development. Protein Sci. A Publ. Protein Soc. 2018, 27, 1742–1754. [Google Scholar] [CrossRef]
- Qiu, Y.; Yu, J.; Li, Y.; Yang, F.; Yu, H.; Xue, M.; Zhang, F.; Jiang, X.; Ji, X.; Bao, Z. Depletion of gut microbiota induces skeletal muscle atrophy by FXR-FGF15/19 signalling. Ann. Med. 2021, 53, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Yu, J.; Ji, X.; Yu, H.; Xue, M.; Zhang, F.; Li, Y.; Bao, Z. Ileal FXR-FGF15/19 signaling activation improves skeletal muscle loss in aged mice. Mech. Ageing Dev. 2022, 202, 111630. [Google Scholar] [CrossRef]
- Sasaki, T.; Kuboyama, A.; Mita, M.; Murata, S.; Shimizu, M.; Inoue, J.; Mori, K.; Sato, R. The exercise-inducible bile acid receptor Tgr5 improves skeletal muscle function in mice. J. Biol. Chem. 2018, 293, 10322–10332. [Google Scholar] [CrossRef]
- Li, Z.; Cui, R.; Wang, Y.B.; Luo, Y.B.; Xue, P.X.; Tang, Q.G.; Fang, M.Y. Specific gastrointestinal microbiota profiles in Chinese Tan sheep are associated with lauric acid content in muscle. BMC Microbiol. 2023, 23, 331. [Google Scholar] [CrossRef]
- MahmoudianDehkordi, S.; Arnold, M.; Nho, K.; Ahmad, S.; Jia, W.; Xie, G.; Louie, G.; Kueider-Paisley, A.; Moseley, M.A.; Thompson, J.W.; et al. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease-An emerging role for gut microbiome. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2019, 15, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.A.; Theriot, C.M. Diversification of host bile acids by members of the gut microbiota. Gut Microbes 2020, 11, 158–171. [Google Scholar] [CrossRef]
- Houghton, M.J.; Kerimi, A.; Mouly, V.; Tumova, S.; Williamson, G. Gut microbiome catabolites as novel modulators of muscle cell glucose metabolism. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 1887–1898. [Google Scholar] [CrossRef]
- Frampton, J.; Murphy, K.G.; Frost, G.; Chambers, E.S. Short-chain fatty acids as potential regulators of skeletal muscle metabolism and function. Nat. Metab. 2020, 2, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Franco, P.; Porru, E.; Fiori, J.; Gioiello, A.; Cerra, B.; Roda, G.; Caliceti, C.; Simoni, P.; Roda, A. Identification and quantification of oxo-bile acids in human faeces with liquid chromatography-mass spectrometry: A potent tool for human gut acidic sterolbiome studies. J. Chromatogr. A 2019, 1585, 70–81. [Google Scholar] [CrossRef]
- Just, S.; Mondot, S.; Ecker, J.; Wegner, K.; Rath, E.; Gau, L.; Streidl, T.; Hery-Arnaud, G.; Schmidt, S.; Lesker, T.R.; et al. The gut microbiota drives the impact of bile acids and fat source in diet on mouse metabolism. Microbiome 2018, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Macierzanka, A.; Torcello-Gómez, A.; Jungnickel, C.; Maldonado-Valderrama, J. Bile salts in digestion and transport of lipids. Adv. Colloid Interface Sci. 2019, 274, 102045. [Google Scholar] [CrossRef]
- Ahmad, T.R.; Haeusler, R.A. Bile acids in glucose metabolism and insulin signalling—Mechanisms and research needs. Nat. Rev. Endocrinol. 2019, 15, 701–712. [Google Scholar] [CrossRef]
- Ren, T.; Pang, L.; Dai, W.; Wu, S.; Kong, J. Regulatory mechanisms of the bile salt export pump (BSEP/ABCB11) and its role in related diseases. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101641. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Meier, P.J. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J. Clin. Investig. 1994, 93, 1326–1331. [Google Scholar] [CrossRef]
- Oude Elferink, R.P.; Paulusma, C.C. Function and pathophysiological importance of ABCB4 (MDR3 P-glycoprotein). Pflug. Arch. Eur. J. Physiol. 2007, 453, 601–610. [Google Scholar] [CrossRef]
- Kerr, T.A.; Saeki, S.; Schneider, M.; Schaefer, K.; Berdy, S.; Redder, T.; Shan, B.; Russell, D.W.; Schwarz, M. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev. Cell 2002, 2, 713–720. [Google Scholar] [CrossRef]
- Fang, S. Bile Acid Receptor Farnesoid X Receptor: A Novel Therapeutic Target for Metabolic Diseases. J. Lipid Atheroscler. 2017, 6, 1. [Google Scholar] [CrossRef]
- Benoit, B.; Meugnier, E.; Castelli, M.; Chanon, S.; Vieille-Marchiset, A.; Durand, C.; Bendridi, N.; Pesenti, S.; Monternier, P.A.; Durieux, A.C.; et al. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat. Med. 2017, 23, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.P.; Hudson, L.L. Cloning and characterization of a conjugated bile acid hydrolase gene from Clostridium perfringens. Appl. Environ. Microbiol. 1995, 61, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Déjean, G.; Tudela, H.; Bruno, L.; Kissi, D.; Rawadi, G.; Claus, S.P. Identifying a Novel Bile Salt Hydrolase from the Keystone Gut Bacterium Christensenella minuta. Microorganisms 2021, 9, 1252. [Google Scholar] [CrossRef]
- Zhang, W.Y.; Wu, R.N.; Sun, Z.H.; Sun, T.S.; Meng, H.; Zhang, H.P. Molecular cloning and characterization of bile salt hydrolase inLactobacillus casei Zhang. Ann. Microbiol. 2009, 59, 721–726. [Google Scholar] [CrossRef]
- Menssen, A.; Häupl, T.; Sittinger, M.; Delorme, B.; Charbord, P.; Ringe, J. Differential gene expression profiling of human bone marrow-derived mesenchymal stem cells during adipogenic development. BMC Genom. 2011, 12, 461. [Google Scholar] [CrossRef]
- Li, B.; Qiao, L.; An, L.; Wang, W.; Liu, J.; Ren, Y.; Pan, Y.; Jing, J.; Liu, W. Transcriptome analysis of adipose tissues from two fat-tailed sheep breeds reveals key genes involved in fat deposition. BMC Genom. 2018, 19, 338. [Google Scholar] [CrossRef]
- Yasukochi, Y.; Sakuma, J.; Takeuchi, I.; Kato, K.; Oguri, M.; Fujimaki, T.; Horibe, H.; Yamada, Y. Identification of three genetic variants as novel susceptibility loci for body mass index in a Japanese population. Physiol. Genom. 2018, 50, 179–189. [Google Scholar] [CrossRef]
- Chen, Y.T.; Chiou, S.Y.; Hsu, A.H.; Lin, Y.C.; Lin, J.S. Lactobacillus rhamnosus Strain LRH05 Intervention Ameliorated Body Weight Gain and Adipose Inflammation via Modulating the Gut Microbiota in High-Fat Diet-Induced Obese Mice. Mol. Nutr. Food Res. 2022, 66, e2100348. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, D.; Ke, W.; Liang, D.; Hu, X.; Chen, F. Resveratrol-induced gut microbiota reduces obesity in high-fat diet-fed mice. Int. J. Obes. 2020, 44, 213–225. [Google Scholar] [CrossRef]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Dong, J.; Sun, Z.; Wang, T.; Guan, Y.; Sun, Y.; Qin, G.; Zhang, X.; Zhen, Y. Effects of antimicrobial peptide and tributyrin on fecal microflora and blood indices of female calves. Food Sci. Nutr. 2023, 11, 5248–5257. [Google Scholar] [CrossRef] [PubMed]
- GB/T 9695.7-2008; Meat and Meat Products-Determination of Total Fat Content. National Health and Family Planning Commission of the People’s Republic of China: Beijing, China, 2008.
- GB 5009.168-2016; Determination of Fatty Acids in Foods. National Health and Family Planning Commission of the People’s Republic of China: Beijing, China, 2016.
- Perwaiz, S.; Tuchweber, B.; Mignault, D.; Gilat, T.; Yousef, I.M. Determination of bile acids in biological fluids by liquid chromatography-electrospray tandem mass spectrometry. J. Lipid Res. 2001, 42, 114–119. [Google Scholar] [CrossRef]
- Yin, Q.; Yu, J.; Li, J.; Zhang, T.; Wang, T.; Zhu, Y.; Zhang, J.; Yao, J. Enhancing milk quality and modulating rectal microbiota of dairy goats in starch-rich diet: The role of bile acid supplementation. J. Anim. Sci. Biotechnol. 2024, 15, 7. [Google Scholar] [CrossRef]
- Zhai, Z.; Niu, K.M.; Liu, Y.; Lin, C.; Wu, X. The Gut Microbiota-Bile Acids-TGR5 Axis Mediates Eucommia ulmoides Leaf Extract Alleviation of Injury to Colonic Epithelium Integrity. Front. Microbiol. 2021, 12, 727681. [Google Scholar] [CrossRef]
- Huang, J.; Bathena, S.P.; Csanaky, I.L.; Alnouti, Y. Simultaneous characterization of bile acids and their sulfate metabolites in mouse liver, plasma, bile, and urine using LC-MS/MS. J. Pharm. Biomed. Anal. 2011, 55, 1111–1119. [Google Scholar] [CrossRef]
- Bouhaddani, S.E.; Houwing-Duistermaat, J.; Salo, P.; Perola, M.; Jongbloed, G.; Uh, H.W. Evaluation of O2PLS in Omics data integration. BMC Bioinform. 2016, 17 (Suppl. S2), 11. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Cui, R.; Li, Z.; Xue, M.; Chan, S.; Xue, P.; Yang, X.; Zhang, L.; Lv, F.; Fang, M. Gut Microbiota-Bile Acid Crosstalk Contributes to Meat Quality and Carcass Traits of Tan and Dorper Sheep. Int. J. Mol. Sci. 2025, 26, 6224. https://doi.org/10.3390/ijms26136224
Yang L, Cui R, Li Z, Xue M, Chan S, Xue P, Yang X, Zhang L, Lv F, Fang M. Gut Microbiota-Bile Acid Crosstalk Contributes to Meat Quality and Carcass Traits of Tan and Dorper Sheep. International Journal of Molecular Sciences. 2025; 26(13):6224. https://doi.org/10.3390/ijms26136224
Chicago/Turabian StyleYang, Lixian, Ran Cui, Zhen Li, Mingming Xue, Shuheng Chan, Pengxiang Xue, Xiaoyang Yang, Longmiao Zhang, Fenghua Lv, and Meiying Fang. 2025. "Gut Microbiota-Bile Acid Crosstalk Contributes to Meat Quality and Carcass Traits of Tan and Dorper Sheep" International Journal of Molecular Sciences 26, no. 13: 6224. https://doi.org/10.3390/ijms26136224
APA StyleYang, L., Cui, R., Li, Z., Xue, M., Chan, S., Xue, P., Yang, X., Zhang, L., Lv, F., & Fang, M. (2025). Gut Microbiota-Bile Acid Crosstalk Contributes to Meat Quality and Carcass Traits of Tan and Dorper Sheep. International Journal of Molecular Sciences, 26(13), 6224. https://doi.org/10.3390/ijms26136224