

JAK2 Inhibition Augments the Anti-Proliferation Effects by AKT and MEK Inhibition in Triple-Negative Breast Cancer Cells

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
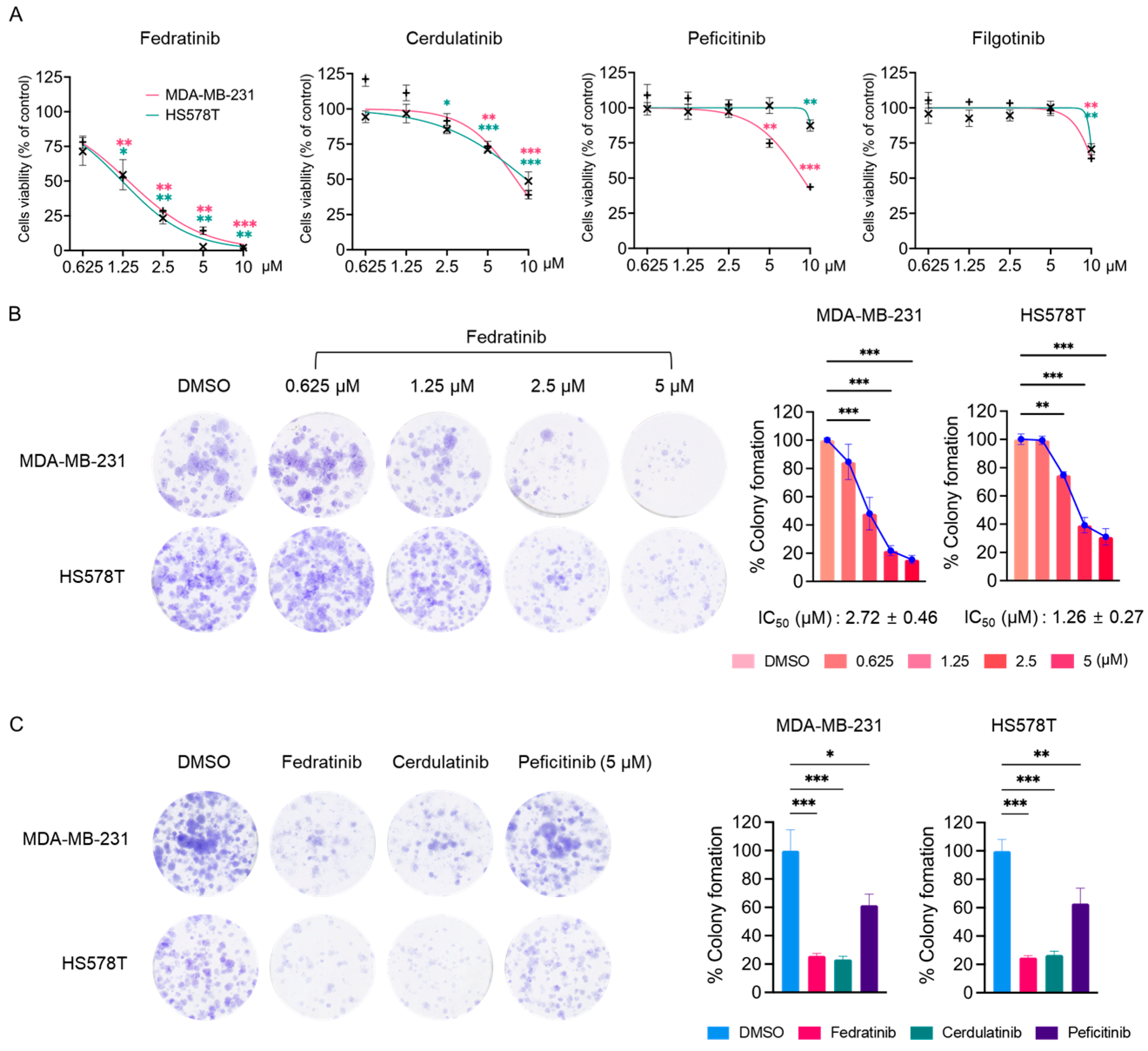
2.1. Fedratinib Inhibits the Proliferation of TNBC Cells
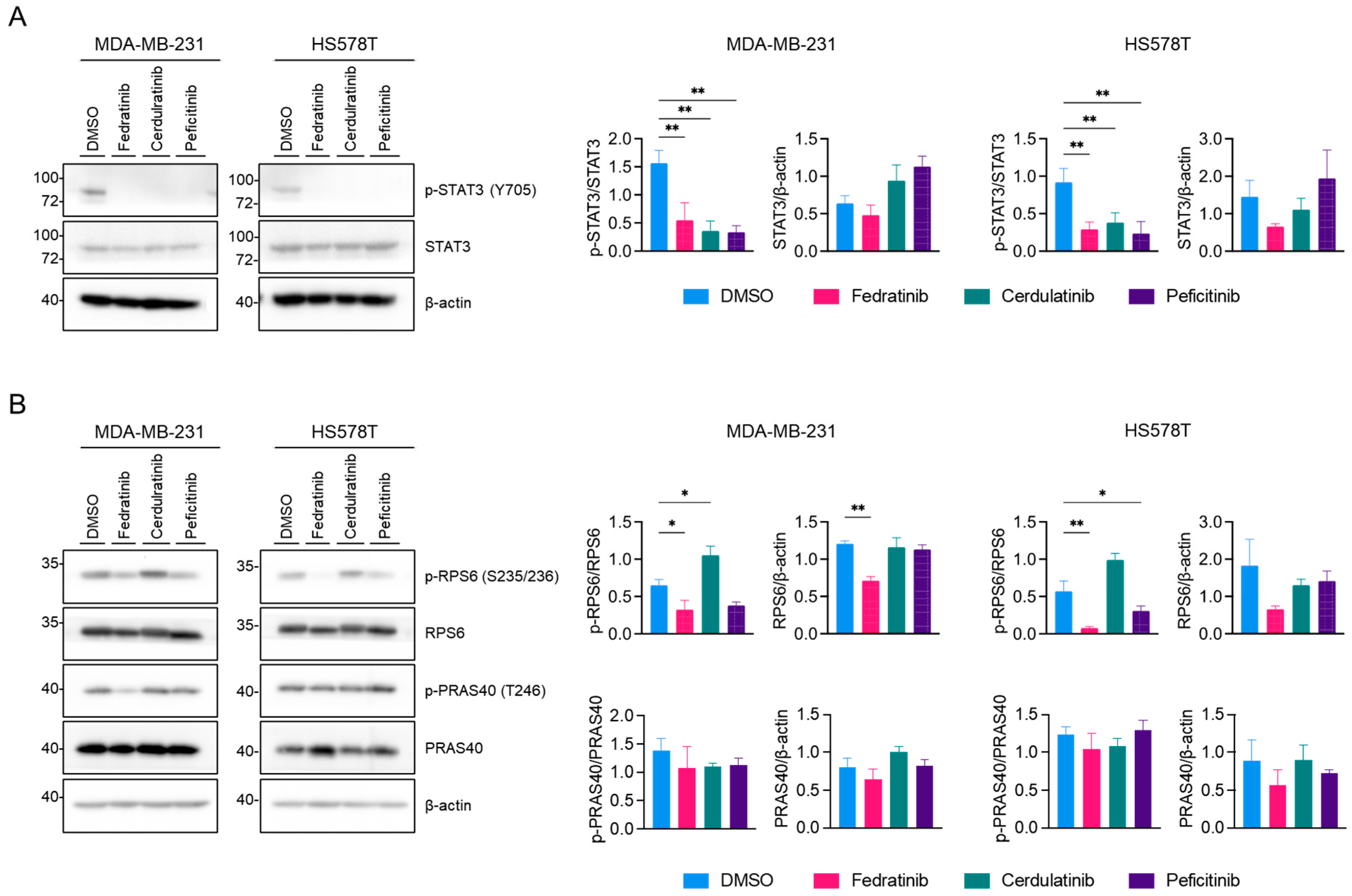
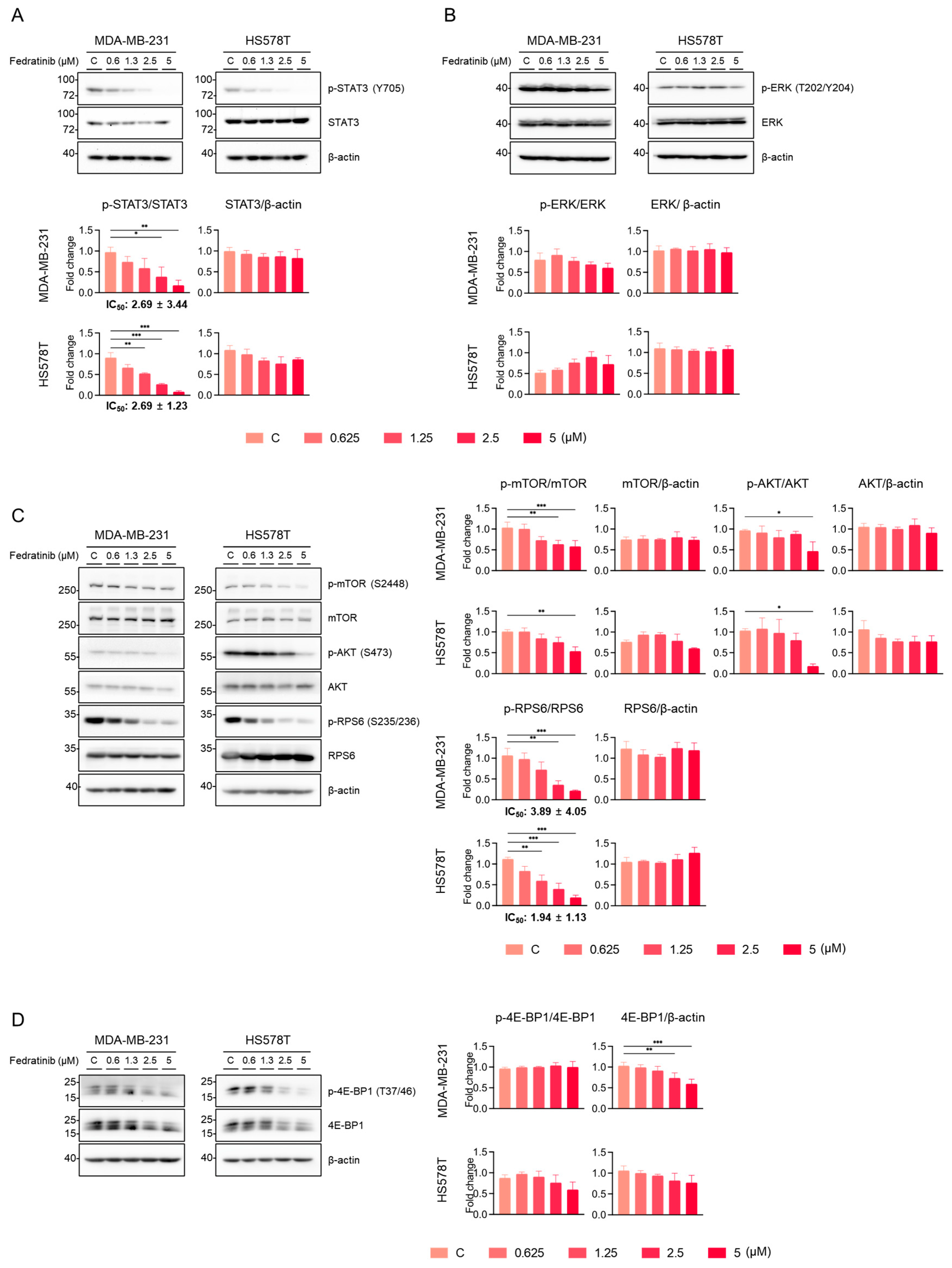
2.2. Fedratinib Inhibits the PI3K/AKT Pathway and Marginally Modulates the MEK/ERK Pathway in TNBC Cells
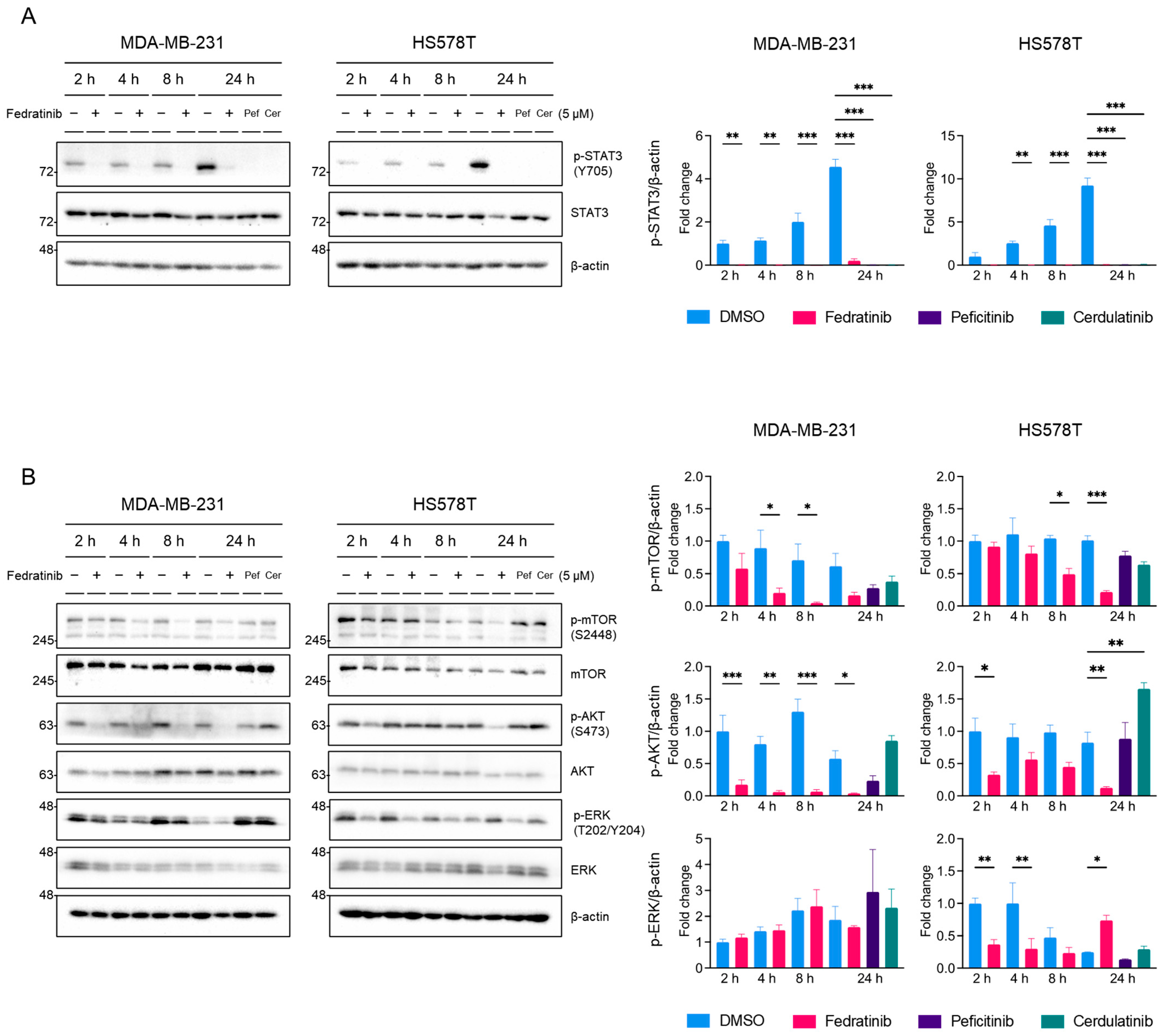
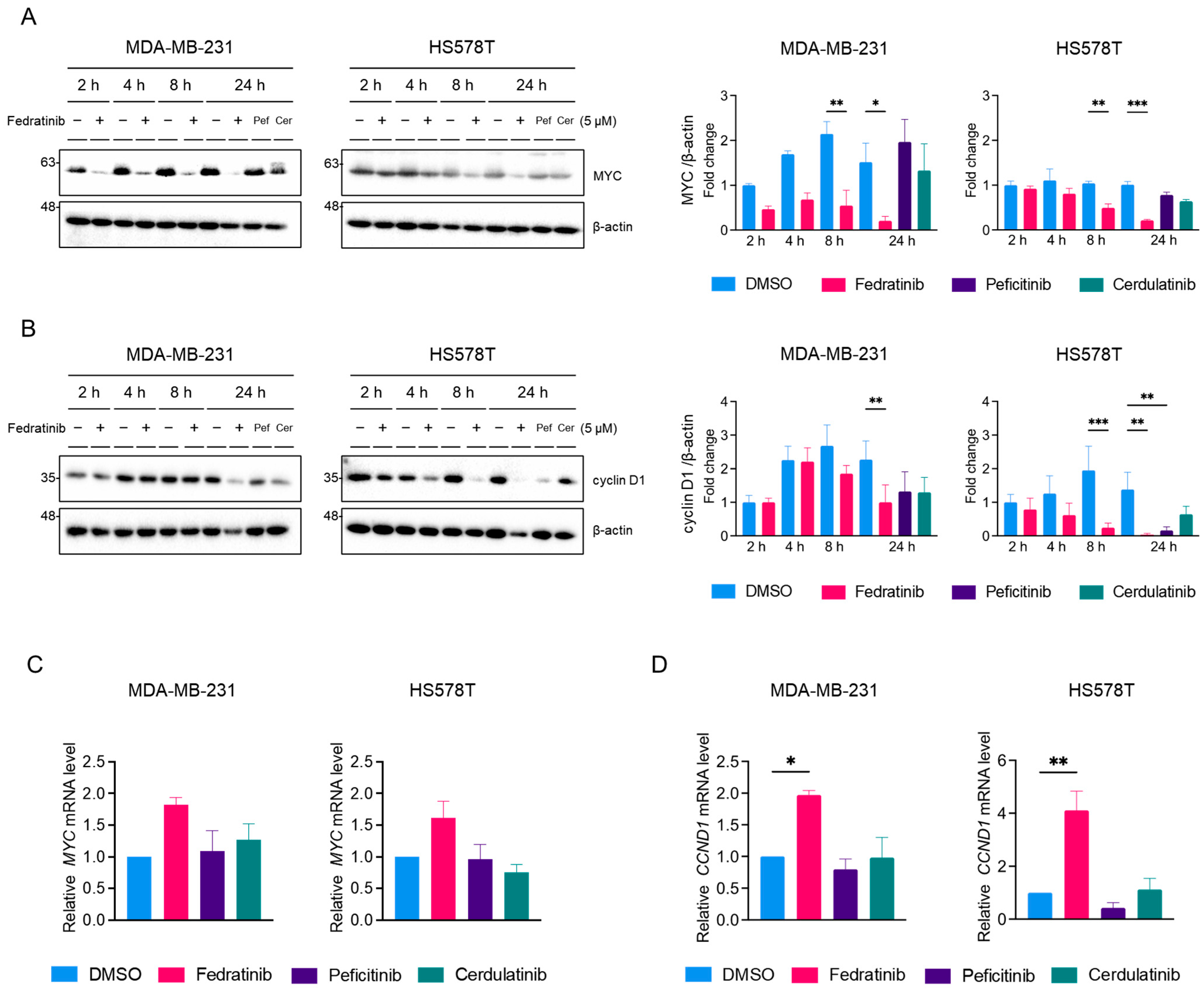
2.3. Fedratinib Inhibited the MEK/ERK Pathway in TNBC Cells in a Time- and Cell Line-Dependent Manners
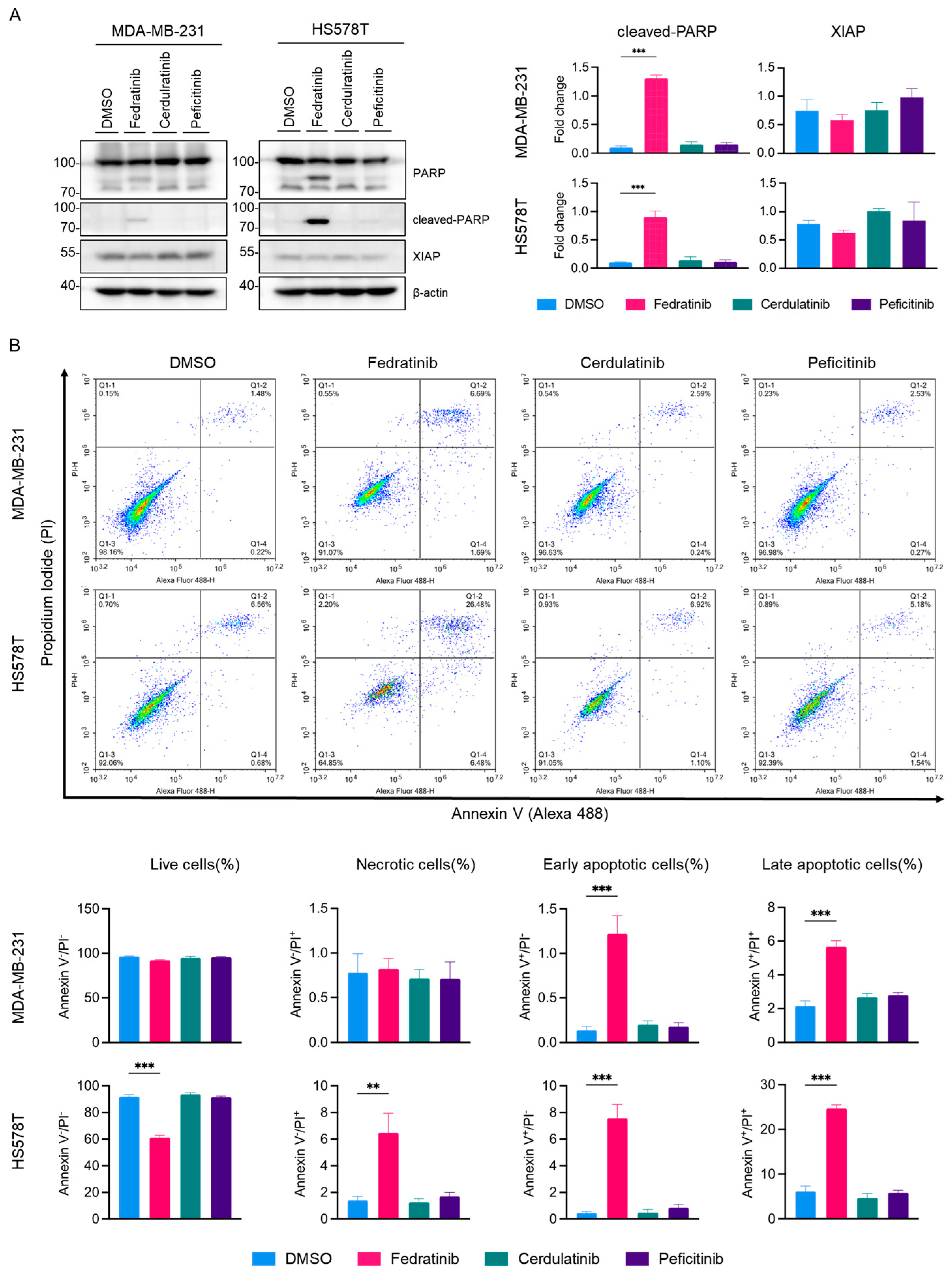
2.4. Fedratinib Induces Apoptotic Cell Death in TNBC Cells
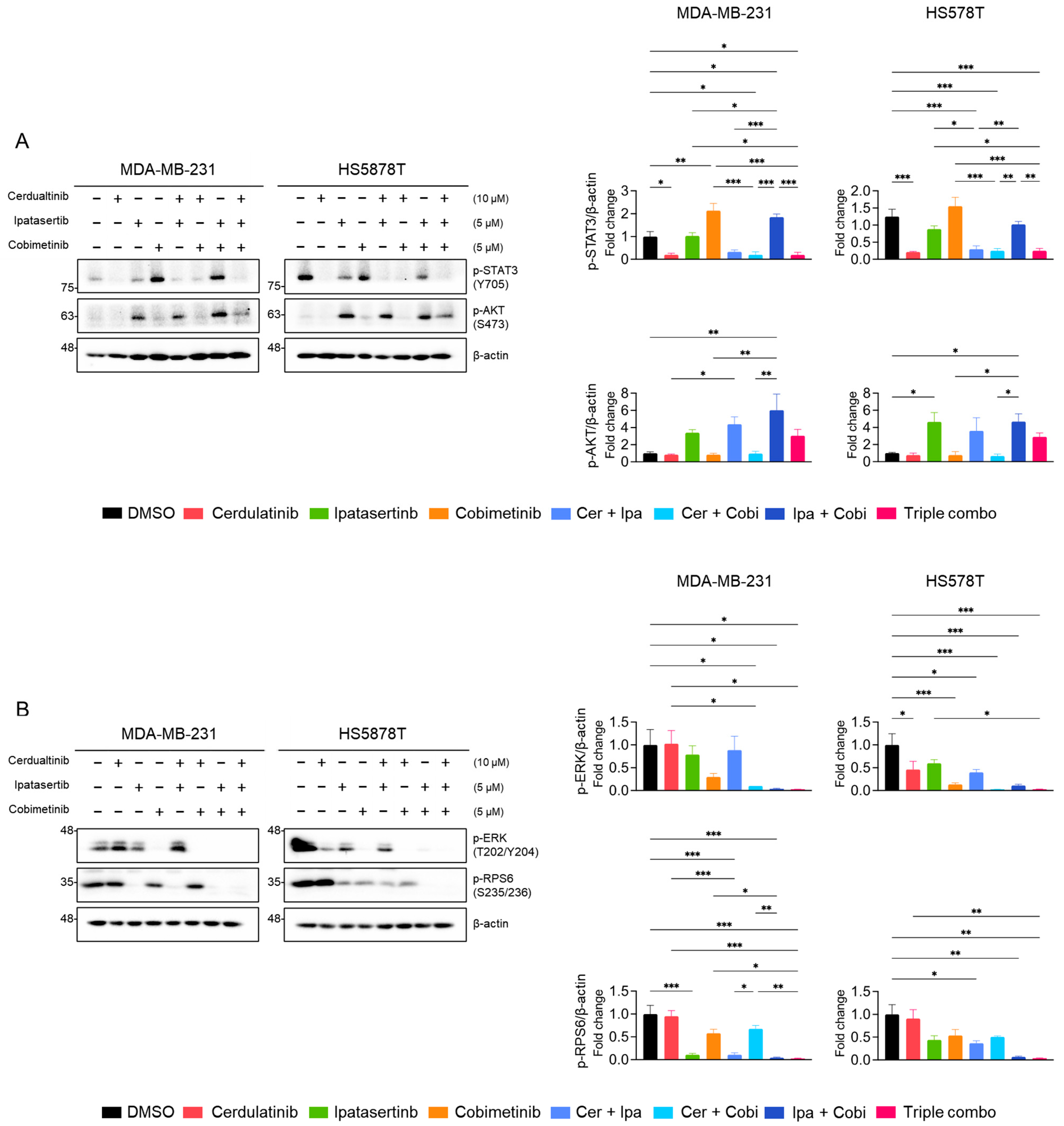
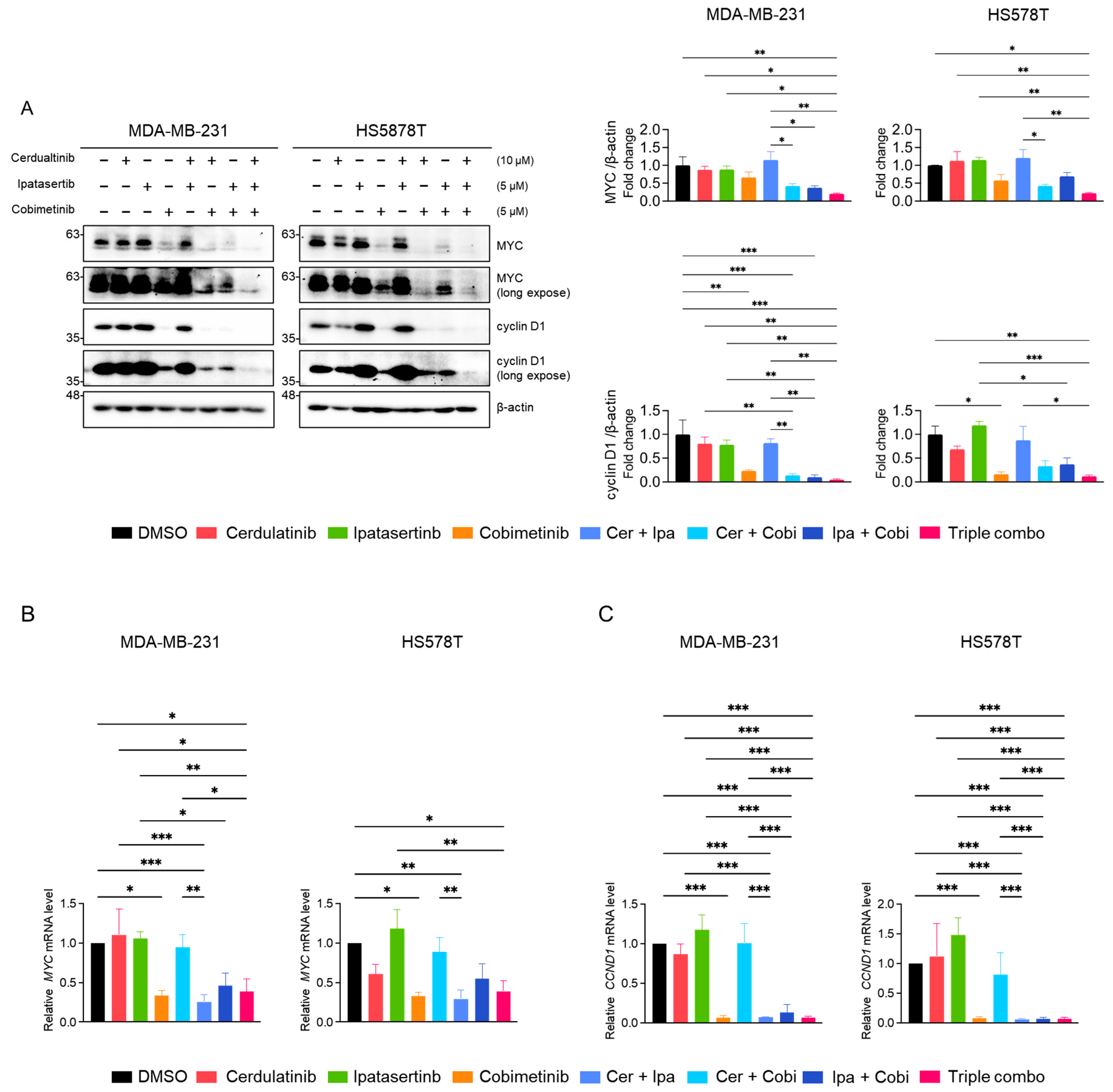
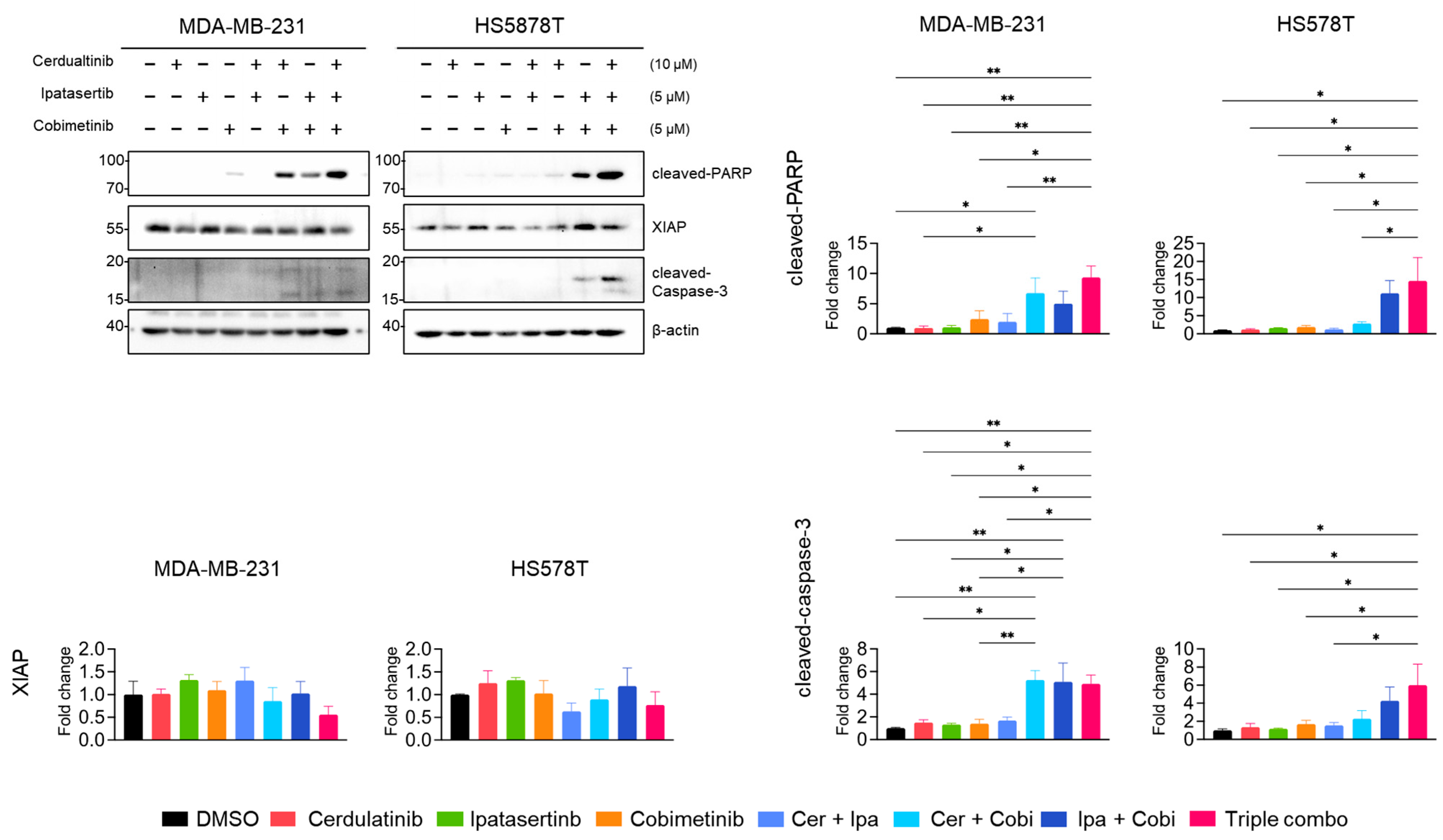
2.5. Inhibition of the JAK2/STAT3 Pathway Enhances the Effects of Blocking the PI3K/AKT and MEK/ERK Pathways in TNBC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. MTT Assay
4.3. Clonogenic Survival Assay
4.4. Western Blot Analysis
4.5. Fluorescence Cell Flow Cytometry
4.6. RNA Extraction, cDNA Synthesis, and Quantitative Real-Time PCR (RT-PCR)
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADC | antibody drug conjugate |
| AKTi | AKT inhibitor |
| ANOVA | analysis of variance |
| ATCC | American Type Culture Collection |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMSO | dimethyl sulfoxide |
| DNMT1 | DNA (cytosine-5)-methyltransferase 1 |
| DPBS | Dulbecco’s phosphate buffered saline |
| EGFR | epidermal growth factor receptor |
| ER | estrogen receptor |
| ERK | extracellular signal-regulated kinase |
| FBS | fetal bovine serum |
| FDA | Food and Drug Administration |
| FLT3 | FMS-like tyrosine kinase 3 |
| GvHD | graft-versus-host disease |
| HER2 | human epidermal growth factor 2 |
| IL6 | interleukin 6 |
| JAK | Janus kinase |
| JAK2i | JAK2 inhibitor |
| MEK | MAP kinase/ERK kinase |
| MEKi | MEK inhibitor |
| mTOR | mammalian target of rapamycin |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide |
| PARP1 | poly [adenosine diphosphate (ADP)-ribose] polymerase 1 |
| PD-L1 | programmed cell death ligand 1 |
| PDLIM4 | PDZ and LIM domain protein 4 |
| PKI | protein kinase inhibitor |
| PR | progesterone receptor |
| PVDF | polyvinylidene difluoride |
| RET | rearranged during transfection |
| RTK | receptor tyrosine kinase |
| STAT3 | signal transducer and activator of transcription 3 |
| SYK | spleen tyrosine kinase |
| TNBC | triple-negative breast cancer |
| TYK2 | tyrosine kinase 2 |
| VHL | von Hippel-Lindau disease tumor suppressor |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- You, K.S.; Yi, Y.W.; Cho, J.; Park, J.-S.; Seong, Y.-S. Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 589. [Google Scholar] [CrossRef]
- Diaz, L.K.; Cryns, V.L.; Symmans, W.F.; Sneige, N. Triple Negative Breast Carcinoma and the Basal Phenotype: From Expression Profiling to Clinical Practice. Adv. Anat. Pathol. 2007, 14, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.; Pambid, M.R.; Jayanthan, A.; Dorr, A.; Los, G.; Dunn, S.E. The Dawn of Targeted Therapies for Triple Negative Breast Cancer (TNBC): A Snapshot of Investigational Drugs in Phase I and II Trials. Expert Opin. Investig. Drugs 2020, 29, 1199–1208. [Google Scholar] [CrossRef]
- You, K.S.; Yi, Y.W.; Cho, J.; Seong, Y.-S. Dual Inhibition of AKT and MEK Pathways Potentiates the Anti-Cancer Effect of Gefitinib in Triple-Negative Breast Cancer Cells. Cancers 2021, 13, 1205. [Google Scholar] [CrossRef]
- YI, Y.W.; YOU, K.; BAE, E.J.; KWAK, S.-J.; SEONG, Y.-S.; BAE, I. Dual Inhibition of EGFR and MET Induces Synthetic Lethality in Triple-Negative Breast Cancer Cells through Downregulation of Ribosomal Protein S6. Int. J. Oncol. 2015, 47, 122–132. [Google Scholar] [CrossRef] [PubMed]
- You, K.S.; Yi, Y.W.; Kwak, S.-J.; Seong, Y.-S. Inhibition of RPTOR Overcomes Resistance to EGFR Inhibition in Triple-Negative Breast Cancer Cells. Int. J. Oncol. 2018, 52, 828–840. [Google Scholar] [CrossRef]
- DUONG, H.-Q.; YI, Y.W.; KANG, H.J.; HONG, Y.B.; TANG, W.; WANG, A.; SEONG, Y.-S.; BAE, I. Inhibition of NRF2 by PIK-75 Augments Sensitivity of Pancreatic Cancer Cells to Gemcitabine. Int. J. Oncol. 2013, 44, 959–969. [Google Scholar] [CrossRef]
- Eccles, S.A. The Epidermal Growth Factor Receptor/Erb-B/HER Family in Normal and Malignant Breast Biology. Int. J. Dev. Biol. 2011, 55, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB Network: At Last, Cancer Therapy Meets Systems Biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Nakai, K.; Hung, M.-C.; Yamaguchi, H. A Perspective on Anti-EGFR Therapies Targeting Triple-Negative Breast Cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar]
- Arteaga, C.L.; Truica, C.I. Challenges in the Development of Anti-Epidermal Growth Factor Receptor Therapies in Breast Cancer. Semin. Oncol. 2004, 31, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; He, J.; Yuan, Z.; Wang, S.; Peng, R.; Shi, Y.; Teng, X.; Qin, T. EGFR Expression Correlates with Decreased Disease-Free Survival in Triple-Negative Breast Cancer: A Retrospective Analysis Based on a Tissue Microarray. Med. Oncol. 2012, 29, 401–405. [Google Scholar] [CrossRef]
- Mandapati, A.; Lukong, K.E. Triple Negative Breast Cancer: Approved Treatment Options and Their Mechanisms of Action. J. Cancer Res. Clin. Oncol. 2023, 149, 3701–3719. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.; Darnell, J.E. The Role of STATs in Transcriptional Control and Their Impact on Cellular Function. Oncogene 2000, 19, 2468–2473. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E.; Silvennoinen, O.; O’Shea, J.J. The Janus Kinases (Jaks). Genome Biol. 2004, 5, 253. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in Cancer Inflammation and Immunity: A Leading Role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Mankan, A.K.; Greten, F.R. Inhibiting Signal Transducer and Activator of Transcription 3: Rationality and Rationale Design of Inhibitors. Expert Opin. Investig. Drugs 2011, 20, 1263–1275. [Google Scholar] [CrossRef]
- Debnath, B.; Xu, S.; Neamati, N. Small Molecule Inhibitors of Signal Transducer and Activator of Transcription 3 (Stat3) Protein. J. Med. Chem. 2012, 55, 6645–6668. [Google Scholar] [CrossRef]
- Horiguchi, A.; Oya, M.; Shimada, T.; Uchida, A.; Marumo, K.; Murai, M. Activation of Signal Transducer and Activator of Transcription 3 in Renal Cell Carcinoma: A Study of Incidence and Its Association With Pathological Features and Clinical Outcome. J. Urol. 2002, 168, 762–765. [Google Scholar] [CrossRef]
- Takemoto, S.; Ushijima, K.; Kawano, K.; Yamaguchi, T.; Terada, A.; Fujiyoshi, N.; Nishio, S.; Tsuda, N.; Ijichi, M.; Kakuma, T.; et al. Expression of Activated Signal Transducer and Activator of Transcription-3 Predicts Poor Prognosis in Cervical Squamous-Cell Carcinoma. Br. J. Cancer 2009, 101, 967–972. [Google Scholar] [CrossRef]
- Anderson, L.A. Which JAK Inhibitors Are Approved in the U.S? Available online: https://www.drugs.com/medical-answers/jak-inhibitors-approved-3575793/ (accessed on 20 June 2024).
- Shawky, A.M.; Almalki, F.A.; Abdalla, A.N.; Abdelazeem, A.H.; Gouda, A.M. A Comprehensive Overview of Globally Approved JAK Inhibitors. Pharmaceutics 2022, 14, 1001. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Fei, X.; Chen, L.; Yao, L.; Lei, X. Potential Therapeutic Targets of the JAK2/STAT3 Signaling Pathway in Triple-Negative Breast Cancer. Front. Oncol. 2024, 14, 1381251. [Google Scholar] [CrossRef]
- Flanagan, M.E.; Blumenkopf, T.A.; Brissette, W.H.; Brown, M.F.; Casavant, J.M.; Shang-Poa, C.; Doty, J.L.; Elliott, E.A.; Fisher, M.B.; Hines, M.; et al. Discovery of CP-690,550: A Potent and Selective Janus Kinase (JAK) Inhibitor for the Treatment of Autoimmune Diseases and Organ Transplant Rejection. J. Med. Chem. 2010, 53, 8468–8484. [Google Scholar] [CrossRef]
- Kang, H.J.; Yi, Y.W.; Hou, S.-J.; Kim, H.J.; Kong, Y.; Bae, I.; Brown, M.L. Disruption of STAT3-DNMT1 Interaction by SH-I-14 Induces Re-Expression of Tumor Suppressor Genes and Inhibits Growth of Triple-Negative Breast Tumor. Oncotarget 2017, 8, 83457–83468. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Hong, W.; Kang, H.J.; Kim, H.J.; Zhao, W.; Wang, A.; Seong, Y.; Bae, I. Inhibition of the PI3K/AKT Pathway Potentiates Cytotoxicity of EGFR Kinase Inhibitors in Triple-negative Breast Cancer Cells. J. Cell Mol. Med. 2013, 17, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Hwang, J.S.; Wang, A.; Bae, I. Inhibition of Constitutively Activated Phosphoinositide 3-kinase/AKT Pathway Enhances Antitumor Activity of Chemotherapeutic Agents in Breast Cancer Susceptibility Gene 1-defective Breast Cancer Cells. Mol. Carcinog. 2013, 52, 667–675. [Google Scholar] [CrossRef]
- Yi, Y.W.; Kang, H.J.; Bae, E.J.; Oh, S.; Seong, Y.-S.; Bae, I. β-TrCP1 Degradation Is a Novel Action Mechanism of PI3K/MTOR Inhibitors in Triple-Negative Breast Cancer Cells. Exp. Mol. Medicine 2015, 47, e143. [Google Scholar] [CrossRef]
- Yi, Y.W.; Park, J.-S.; Kwak, S.-J.; Seong, Y.-S. Co-Treatment with BEZ235 Enhances Sensitivity of BRCA1-Negative Breast Cancer Cells to Olaparib. Anticancer. Res. 2015, 35, 3829–3838. [Google Scholar]
- Yi, Y.W.; You, K.S.; Han, S.; Ha, I.J.; Park, J.-S.; Lee, S.-G.; Seong, Y.-S. Inhibition of IκB Kinase Is a Potential Therapeutic Strategy to Circumvent Resistance to Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer Cells. Cancers 2022, 14, 5215. [Google Scholar] [CrossRef]
- Wernig, G.; Kharas, M.G.; Okabe, R.; Moore, S.A.; Leeman, D.S.; Cullen, D.E.; Gozo, M.; McDowell, E.P.; Levine, R.L.; Doukas, J.; et al. Efficacy of TG101348, a Selective JAK2 Inhibitor, in Treatment of a Murine Model of JAK2V617F-Induced Polycythemia Vera. Cancer Cell 2008, 13, 311–320. [Google Scholar] [CrossRef]
- Coffey, G.; Betz, A.; DeGuzman, F.; Pak, Y.; Inagaki, M.; Baker, D.C.; Hollenbach, S.J.; Pandey, A.; Sinha, U. The Novel Kinase Inhibitor PRT062070 (Cerdulatinib) Demonstrates Efficacy in Models of Autoimmunity and B-Cell Cancer. J. Pharmacol. Exp. Ther. 2014, 351, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Rompaey, L.V.; Galien, R.; van der Aar, E.M.; Clement-Lacroix, P.; Nelles, L.; Smets, B.; Lepescheux, L.; Christophe, T.; Conrath, K.; Vandeghinste, N.; et al. Preclinical Characterization of GLPG0634, a Selective Inhibitor of JAK1, for the Treatment of Inflammatory Diseases. J. Immunol. 2013, 191, 3568–3577. [Google Scholar] [CrossRef]
- Ito, M.; Yamazaki, S.; Yamagami, K.; Kuno, M.; Morita, Y.; Okuma, K.; Nakamura, K.; Chida, N.; Inami, M.; Inoue, T.; et al. A Novel JAK Inhibitor, Peficitinib, Demonstrates Potent Efficacy in a Rat Adjuvant-Induced Arthritis Model. J. Pharmacol. Sci. 2017, 133, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, H.; Amano, Y.; Moritomo, A.; Shirakami, S.; Nakajima, Y.; Nakai, K.; Nomura, N.; Ito, M.; Higashi, Y.; Inoue, T. Discovery and Structural Characterization of Peficitinib (ASP015K) as a Novel and Potent JAK Inhibitor. Bioorg. Med. Chem. 2018, 26, 4971–4983. [Google Scholar] [CrossRef]
- Lin, L.; Hutzen, B.; Zuo, M.; Ball, S.; Deangelis, S.; Foust, E.; Pandit, B.; Ihnat, M.A.; Shenoy, S.S.; Kulp, S.; et al. Novel STAT3 Phosphorylation Inhibitors Exhibit Potent Growth-Suppressive Activity in Pancreatic and Breast Cancer Cells. Cancer Res. 2010, 70, 2445–2454. [Google Scholar] [CrossRef]
- Yi, Y.W.; You, K.S.; Park, J.-S.; Lee, S.-G.; Seong, Y.-S. Ribosomal Protein S6: A Potential Therapeutic Target against Cancer? Int. J. Mol. Sci. 2022, 23, 48. [Google Scholar] [CrossRef] [PubMed]
- Kovacina, K.S.; Park, G.Y.; Bae, S.S.; Guzzetta, A.W.; Schaefer, E.; Birnbaum, M.J.; Roth, R.A. Identification of a Proline-Rich Akt Substrate as a 14-3-3 Binding Partner*. J. Biol. Chem. 2003, 278, 10189–10194. [Google Scholar] [CrossRef] [PubMed]
- Subbannayya, T.; Leal-Rojas, P.; Zhavoronkov, A.; Ozerov, I.V.; Korzinkin, M.; Babu, N.; Radhakrishnan, A.; Chavan, S.; Raja, R.; Pinto, S.M.; et al. PIM1 Kinase Promotes Gallbladder Cancer Cell Proliferation via Inhibition of Proline-Rich Akt Substrate of 40 KDa (PRAS40). J. Cell Commun. Signal. 2019, 13, 163–177. [Google Scholar] [CrossRef]
- Brasó-Maristany, F.; Filosto, S.; Catchpole, S.; Marlow, R.; Quist, J.; Francesch-Domenech, E.; Plumb, D.A.; Zakka, L.; Gazinska, P.; Liccardi, G.; et al. PIM1 Kinase Regulates Cell Death, Tumor Growth and Chemotherapy Response in Triple-Negative Breast Cancer. Nat. Med. 2016, 22, 1303–1313. [Google Scholar] [CrossRef]
- Horiuchi, D.; Camarda, R.; Zhou, A.Y.; Yau, C.; Momcilovic, O.; Balakrishnan, S.; Corella, A.N.; Eyob, H.; Kessenbrock, K.; Lawson, D.A.; et al. PIM1 Kinase Inhibition as a Targeted Therapy against Triple-Negative Breast Tumors with Elevated MYC Expression. Nat. Med. 2016, 22, 1321–1329. [Google Scholar] [CrossRef]
- Kunder, R.; Velyunskiy, M.; Dunne, S.F.; Cho, B.-K.; Kanojia, D.; Begg, L.; Orriols, A.M.; Fleming-Trujillo, E.; Vadlamani, P.; Vialichka, A.; et al. Synergistic PIM Kinase and Proteasome Inhibition as a Therapeutic Strategy for MYC-Overexpressing Triple-Negative Breast Cancer. Cell Chem. Biol. 2022, 29, 358–372.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tang, G. PIM-1 Kinase: A Potential Biomarker of Triple-Negative Breast Cancer. OncoTargets Ther. 2019, 12, 6267–6273. [Google Scholar] [CrossRef] [PubMed]
- Shaul, Y.D.; Seger, R. The MEK/ERK Cascade: From Signaling Specificity to Diverse Functions. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2007, 1773, 1213–1226. [Google Scholar] [CrossRef]
- Navé, B.T.; Ouwens, D.M.; Withers, D.J.; Alessi, D.R.; Shepherd, P.R. Mammalian Target of Rapamycin Is a Direct Target for Protein Kinase B: Identification of a Convergence Point for Opposing Effects of Insulin and Amino-Acid Deficiency on Protein Translation. Biochem. J. 1999, 344, 427–431. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- She, Q.-B.; Halilovic, E.; Ye, Q.; Zhen, W.; Shirasawa, S.; Sasazuki, T.; Solit, D.B.; Rosen, N. 4E-BP1 Is a Key Effector of the Oncogenic Activation of the AKT and ERK Signaling Pathways That Integrates Their Function in Tumors. Cancer Cell 2010, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Ayele, T.M.; Muche, Z.T.; Teklemariam, A.B.; Kassie, A.B.; Abebe, E.C. Role of JAK2/STAT3 Signaling Pathway in the Tumorigenesis, Chemotherapy Resistance, and Treatment of Solid Tumors: A Systemic Review. J. Inflamm. Res. 2022, 15, 1349–1364. [Google Scholar] [CrossRef]
- Gera, J.F.; Mellinghoff, I.K.; Shi, Y.; Rettig, M.B.; Tran, C.; Hsu, J.; Sawyers, C.L.; Lichtenstein, A.K. AKT Activity Determines Sensitivity to Mammalian Target of Rapamycin (MTOR) Inhibitors by Regulating Cyclin D1 and c-Myc Expression*. J. Biol. Chem. 2004, 279, 2737–2746. [Google Scholar] [CrossRef]
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Signal Transduction Mediated by the Ras/Raf/MEK/ERK Pathway from Cytokine Receptors to Transcription Factors: Potential Targeting for Therapeutic Intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef]
- Benary, M.; Bohn, S.; Lüthen, M.; Nolis, I.K.; Blüthgen, N.; Loewer, A. Disentangling Pro-Mitotic Signaling during Cell Cycle Progression Using Time-Resolved Single-Cell Imaging. Cell Rep. 2020, 31, 107514. [Google Scholar] [CrossRef]
- Hoeflich, K.P.; Merchant, M.; Orr, C.; Chan, J.; Otter, D.D.; Berry, L.; Kasman, I.; Koeppen, H.; Rice, K.; Yang, N.-Y.; et al. Intermittent Administration of MEK Inhibitor GDC-0973 plus PI3K Inhibitor GDC-0941 Triggers Robust Apoptosis and Tumor Growth Inhibition. Cancer Res. 2012, 72, 210–219. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Kakefuda, R.; Tajima, N.; Sowa, Y.; Sakai, T. Antitumor Activities of JTP-74057 (GSK1120212), a Novel MEK1/2 Inhibitor, on Colorectal Cancer Cell Lines in Vitro and in Vivo. Int. J. Oncol. 2011, 39, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Blake, J.F.; Xu, R.; Bencsik, J.R.; Xiao, D.; Kallan, N.C.; Schlachter, S.; Mitchell, I.S.; Spencer, K.L.; Banka, A.L.; Wallace, E.M.; et al. Discovery and Preclinical Pharmacology of a Selective ATP-Competitive Akt Inhibitor (GDC-0068) for the Treatment of Human Tumors. J. Med. Chem. 2012, 55, 8110–8127. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a Potent and Selective Phosphatidylinositol-3 Kinase Alpha Inhibitor Selected for Clinical Evaluation. Bioorg Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef]
- Han, E.K.-H.; Leverson, J.D.; McGonigal, T.; Shah, O.J.; Woods, K.W.; Hunter, T.; Giranda, V.L.; Luo, Y. Akt Inhibitor A-443654 Induces Rapid Akt Ser-473 Phosphorylation Independent of MTORC1 Inhibition. Oncogene 2007, 26, 5655–5661. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination Therapy in Combating Cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Yap, T.A.; Omlin, A.; Bono, J.S. de Development of Therapeutic Combinations Targeting Major Cancer Signaling Pathways. J. Clin. Oncol. 2013, 31, 1592–1605. [Google Scholar] [CrossRef]
- Qin, S.-Y.; Cheng, Y.-J.; Lei, Q.; Zhang, A.-Q.; Zhang, X.-Z. Combinational Strategy for High-Performance Cancer Chemotherapy. Biomaterials 2018, 171, 178–197. [Google Scholar] [CrossRef]
- Yao, H.; He, G.; Yan, S.; Chen, C.; Song, L.; Rosol, T.J.; Deng, X. Triple-Negative Breast Cancer: Is There a Treatment on the Horizon? Oncotarget 2016, 8, 1913–1924. [Google Scholar] [CrossRef]
- Nedeljković, M.; Damjanović, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer—How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef]
- Chalakur-Ramireddy, N.K.R.; Pakala, S.B. Combined Drug Therapeutic Strategies for the Effective Treatment of Triple Negative Breast Cancer. Biosci. Rep. 2018, 38, BSR20171357. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W. Therapeutic Implications of the Drug Resistance Conferred by Extracellular Vesicles Derived from Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2023, 24, 3704. [Google Scholar] [CrossRef] [PubMed]
- Britton, K.M.; Eyre, R.; Harvey, I.J.; Stemke-Hale, K.; Browell, D.; Lennard, T.W.J.; Meeson, A.P. Breast Cancer, Side Population Cells and ABCG2 Expression. Cancer Lett. 2012, 323, 97–105. [Google Scholar] [CrossRef]
- Roda, E.; Luca, F.D.; Locatelli, C.A.; Ratto, D.; Iorio, C.D.; Savino, E.; Bottone, M.G.; Rossi, P. From a Medicinal Mushroom Blend a Direct Anticancer Effect on Triple-Negative Breast Cancer: A Preclinical Study on Lung Metastases. Molecules 2020, 25, 5400. [Google Scholar] [CrossRef]
- Roda, E.; Luca, F.D.; Iorio, C.D.; Ratto, D.; Siciliani, S.; Ferrari, B.; Cobelli, F.; Borsci, G.; Priori, E.C.; Chinosi, S.; et al. Novel Medicinal Mushroom Blend as a Promising Supplement in Integrative Oncology: A Multi-Tiered Study Using 4T1 Triple-Negative Mouse Breast Cancer Model. Int. J. Mol. Sci. 2020, 21, 3479. [Google Scholar] [CrossRef]
- Luca, F.D.; Roda, E.; Rossi, P.; Bottone, M.G. Medicinal Mushrooms in Metastatic Breast Cancer: What Is Their Therapeutic Potential as Adjuvant in Clinical Settings? Curr. Issues Mol. Biol. 2024, 46, 7577–7591. [Google Scholar] [CrossRef]
- Talpaz, M.; Kiladjian, J.-J. Fedratinib, a Newly Approved Treatment for Patients with Myeloproliferative Neoplasm-Associated Myelofibrosis. Leukemia 2021, 35, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Rah, B.; Rather, R.A.; Bhat, G.R.; Baba, A.B.; Mushtaq, I.; Farooq, M.; Yousuf, T.; Dar, S.B.; Parveen, S.; Hassan, R.; et al. JAK/STAT Signaling: Molecular Targets, Therapeutic Opportunities, and Limitations of Targeted Inhibitions in Solid Malignancies. Front. Pharmacol. 2022, 13, 821344. [Google Scholar] [CrossRef]
- Ni, Y.; Low, J.T.; Silke, J.; O’Reilly, L.A. Digesting the Role of JAK-STAT and Cytokine Signaling in Oral and Gastric Cancers. Front. Immunol. 2022, 13, 835997. [Google Scholar] [CrossRef]
- Tang, J.J.H.; Thng, D.K.H.; Lim, J.J.; Toh, T.B. JAK/STAT Signaling in Hepatocellular Carcinoma. Hepatic Oncol. 2020, 7, HEP18. [Google Scholar] [CrossRef]
- Park, H.; Lee, S.; Lee, J.; Moon, H.; Ro, S.W. Exploring the JAK/STAT Signaling Pathway in Hepatocellular Carcinoma: Unraveling Signaling Complexity and Therapeutic Implications. Int. J. Mol. Sci. 2023, 24, 13764. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.Y.; Johnson, F.M. Defining the Role of the JAK-STAT Pathway in Head and Neck and Thoracic Malignancies: Implications for Future Therapeutic Approaches. Drug Resist. Updates 2010, 13, 67–78. [Google Scholar] [CrossRef]
- Tabassum, S.; Abbasi, R.; Ahmad, N.; Farooqi, A.A. Breast Cancer Metastasis and Drug Resistance, Challenges and Progress. Adv. Exp. Med. Biol. 2019, 1152, 271–281. [Google Scholar] [CrossRef]
- Standing, D.; Feess, E.; Kodiyalam, S.; Kuehn, M.; Hamel, Z.; Johnson, J.; Thomas, S.M.; Anant, S. The Role of STATs in Ovarian Cancer: Exploring Their Potential for Therapy. Cancers 2023, 15, 2485. [Google Scholar] [CrossRef]
- Barrett, M.T.; Anderson, K.S.; Lenkiewicz, E.; Andreozzi, M.; Cunliffe, H.E.; Klassen, C.L.; Dueck, A.C.; McCullough, A.E.; Reddy, S.K.; Ramanathan, R.K.; et al. Genomic Amplification of 9p24.1 Targeting JAK2, PD-L1, and PD-L2 Is Enriched in High-Risk Triple Negative Breast Cancer. Oncotarget 2015, 6, 26483–26493. [Google Scholar] [CrossRef] [PubMed]
- Marotta, L.L.C.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 Signaling Pathway Is Required for Growth of CD44+CD24– Stem Cell–like Breast Cancer Cells in Human Tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Britschgi, A.; Andraos, R.; Brinkhaus, H.; Klebba, I.; Romanet, V.; Müller, U.; Murakami, M.; Radimerski, T.; Bentires-Alj, M. JAK2/STAT5 Inhibition Circumvents Resistance to PI3K/MTOR Blockade: A Rationale for Cotargeting These Pathways in Metastatic Breast Cancer. Cancer Cell 2012, 22, 796–811. [Google Scholar] [CrossRef]
- Weng, Y.-S.; Tseng, H.-Y.; Chen, Y.-A.; Shen, P.-C.; Haq, A.T.A.; Chen, L.-M.; Tung, Y.-C.; Hsu, H.-L. MCT-1/MiR-34a/IL-6/IL-6R Signaling Axis Promotes EMT Progression, Cancer Stemness and M2 Macrophage Polarization in Triple-Negative Breast Cancer. Mol. Cancer 2019, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Stover, D.G.; Alcazar, C.R.G.D.; Brock, J.; Guo, H.; Overmoyer, B.; Balko, J.; Xu, Q.; Bardia, A.; Tolaney, S.M.; Gelman, R.; et al. Phase II Study of Ruxolitinib, a Selective JAK1/2 Inhibitor, in Patients with Metastatic Triple-Negative Breast Cancer. npj Breast Cancer 2018, 4, 10. [Google Scholar] [CrossRef]
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial–Mesenchymal Transition. Cells 2020, 9, 217. [Google Scholar] [CrossRef]
- Balko, J.M.; Schwarz, L.J.; Luo, N.; Estrada, M.V.; Giltnane, J.M.; Dávila-González, D.; Wang, K.; Sánchez, V.; Dean, P.T.; Combs, S.E.; et al. Triple-Negative Breast Cancers with Amplification of JAK2 at the 9p24 Locus Demonstrate JAK2-Specific Dependence. Sci. Transl. Med. 2016, 8, 334ra53. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Song, L.; Xu, Y.; Wu, Y.; Dai, C.; Wang, X.; Sun, X.; Hou, Y.; Li, W.; Zhan, X.; et al. Loss of Wwox Drives Metastasis in Triple-Negative Breast Cancer by JAK2/STAT3 Axis. Nat. Commun. 2018, 9, 3486. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, Z.; Xu, J.; Lai, M.; Liu, L.; Zuo, M.; Dang, L. Fat Mass and Obesity-Associated Protein (FTO) Mediates Signal Transducer and Activator of Transcription 3 (STAT3)-Drived Resistance of Breast Cancer to Doxorubicin. Bioengineered 2021, 12, 1874–1889. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.-S.; Wang, S.; Deng, A.; Liu, B.; Edgerton, S.M.; Lind, S.E.; Wahdan-Alaswad, R.; Thor, A.D. Metformin Targets Stat3 to Inhibit Cell Growth and Induce Apoptosis in Triple-Negative Breast Cancers. Cell Cycle 2012, 11, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Regua, A.T.; Bindal, S.; Najjar, M.K.; Zhuang, C.; Khan, M.; Arrigo, A.B.J.; Gonzalez, A.O.; Zhang, X.R.; Zhu, J.-J.; Watabe, K.; et al. Dual Inhibition of the TrkA and JAK2 Pathways Using Entrectinib and Pacritinib Suppresses the Growth and Metastasis of HER2-Positive and Triple-Negative Breast Cancers. Cancer Lett. 2024, 597, 217023. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, J.; Du, Y.; Qin, X.; Miao, R.; Nan, J.; Chen, X.; Sun, J.; Zhao, R.; Zhang, X.; et al. Loss of ZIP Facilitates JAK2-STAT3 Activation in Tamoxifen-Resistant Breast Cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 15047–15054. [Google Scholar] [CrossRef]
- Han, J.; Yun, J.; Quan, M.; Kang, W.; Jung, J.-G.; Heo, W.; Li, S.; Lee, K.J.; Son, H.-Y.; Kim, J.H.; et al. JAK2 Regulates Paclitaxel Resistance in Triple Negative Breast Cancers. J. Mol. Med. 2021, 99, 1783–1795. [Google Scholar] [CrossRef]
- Changelian, P.S.; Flanagan, M.E.; Ball, D.J.; Kent, C.R.; Magnuson, K.S.; Martin, W.H.; Rizzuti, B.J.; Sawyer, P.S.; Perry, B.D.; Brissette, W.H.; et al. Prevention of Organ Allograft Rejection by a Specific Janus Kinase 3 Inhibitor. Science 2003, 302, 875–878. [Google Scholar] [CrossRef]
- Hou, S.; Yi, Y.W.; Kang, H.J.; Zhang, L.; Kim, H.J.; Kong, Y.; Liu, Y.; Wang, K.; Kong, H.-S.; Grindrod, S.; et al. Novel Carbazole Inhibits Phospho-STAT3 through Induction of Protein–Tyrosine Phosphatase PTPN6. J. Med. Chem. 2014, 57, 6342–6353. [Google Scholar] [CrossRef]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 Sequence-Specific DNA Binding and Transcription by P300/CREB-Binding Protein-Mediated Acetylation*. J. Biol. Chem. 2005, 280, 11528–11534. [Google Scholar] [CrossRef]
- Nadiminty, N.; Lou, W.; Lee, S.O.; Lin, X.; Trump, D.L.; Gao, A.C. Stat3 Activation of NF-ΚB P100 Processing Involves CBP/P300-Mediated Acetylation. Proc. Natl. Acad. Sci. USA 2006, 103, 7264–7269. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, M.; Unal, H.; Willard, B.; Yang, J.; Karnik, S.S.; Stark, G.R. Critical Role for Lysine 685 in Gene Expression Mediated by Transcription Factor Unphosphorylated STAT3*. J. Biol. Chem. 2014, 289, 30763–30771. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Kim, H.J.; Lee, H.-J.; Lee, K.; Hong, D.; Lim, H.; Cho, K.; Jung, N.; Yi, Y.W. Application of a Non-Hazardous Vital Dye for Cell Counting with Automated Cell Counters. Anal. Biochem. 2016, 492, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Kong, Y.; Brown, M.M.; Bae, I. Targeting Mutant P53 by a SIRT1 Activator YK-3-237 Inhibits the Proliferation of Triple-Negative Breast Cancer Cells. Oncotarget 2013, 4, 984–994. [Google Scholar] [CrossRef]
- Duong, H.-Q.; You, K.; Oh, S.; Kwak, S.-J.; Seong, Y.-S. Silencing of NRF2 Reduces the Expression of ALDH1A1 and ALDH3A1 and Sensitizes to 5-FU in Pancreatic Cancer Cells. Antioxidants 2017, 6, 52. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PKI (Ref) | Known Targets (IC50 Values in nM) | IC50 in HS578T (μM) | IC50 in MDA-MB-231 (μM) | |
|---|---|---|---|---|
| JAK Family | Other PKs | |||
| Fedratinib [31] | JAK2 (3), JAK1 (105 *), JAK3 (1002 *) | FLT3 (15), RET (48) | 1.23 ± 0.19 | 1.38 ± 0.06 |
| Cerdulatinib [32] | JAK1 (12), JAK2 (6), JAK3 (8), TYK2 (0.5) | SYK (32) | 9.66 ± 1.15 | 8.07 ± 0.62 |
| Filgotinib [33] | JAK1 (10), JAK2 (28), JAK3 (810), TYK2 (116) | >10 | >10 | |
| Peficitinib [34,35] | JAK1 (3.9), JAK2 (5.0), JAK3 (0.71), TYK2 (4.8) | >10 | 8.75 ± 0.58 | |
| Name | Cat # | M.W. (kDa) | Source | Company |
|---|---|---|---|---|
| AKT | 9272 | 60 | rabbit | Cell Signaling Technology (Denver, MA, USA) |
| p-AKT (S473) | 9271 | 60 | rabbit | |
| ERK1/2 | 9102 | 42, 44 | rabbit | |
| p-ERK1/2 (T202/Y204) | 9101 | 42, 44 | rabbit | |
| mTOR (L27D4) | 4517 | 289 | mouse | |
| p-mTOR (S2448) | 5536 | 289 | rabbit | |
| RPS6 | 2217 | 32 | rabbit | |
| p-RPS6 (S235/236) | 4856 | 32 | rabbit | |
| STAT3 | 9139 | 78, 86 | mouse | |
| p-STAT3 (Y705) | 9145 | 79, 86 | rabbit | |
| 4E-BP1 | 9452 | 15, 20 | rabbit | |
| p-4E-BP1 (T37/46) | 2855 | 15, 20 | rabbit | |
| PRAS40 | 2691 | 40 | rabbit | |
| p-PRAS40 (T246) | 13175 | 40 | rabbit | |
| XIAP | 2042 | 53 | rabbit | |
| PARP | 9542 | 89, 116 | rabbit | |
| MYC | 5605 | 62 | rabbit | |
| Cyclin D1 | 55506 | 36 | Rabbit | |
| Cleaved PARP | 5625 | 89 | rabbit | |
| Cleaved caspase-3 | 9664 | 17, 19 | rabbit | |
| β-actin | SC-47778 | 43 | mouse | Santa Cruz Biotechnology (Dallas, TX, USA) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, K.S.; Kim, T.-S.; Back, S.M.; Park, J.-S.; Liu, K.; Seong, Y.-S.; Kim, D.J.; Yi, Y.W. JAK2 Inhibition Augments the Anti-Proliferation Effects by AKT and MEK Inhibition in Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2025, 26, 6139. https://doi.org/10.3390/ijms26136139
You KS, Kim T-S, Back SM, Park J-S, Liu K, Seong Y-S, Kim DJ, Yi YW. JAK2 Inhibition Augments the Anti-Proliferation Effects by AKT and MEK Inhibition in Triple-Negative Breast Cancer Cells. International Journal of Molecular Sciences. 2025; 26(13):6139. https://doi.org/10.3390/ijms26136139
Chicago/Turabian StyleYou, Kyu Sic, Tae-Sung Kim, Su Min Back, Jeong-Soo Park, Kangdong Liu, Yeon-Sun Seong, Dong Joon Kim, and Yong Weon Yi. 2025. "JAK2 Inhibition Augments the Anti-Proliferation Effects by AKT and MEK Inhibition in Triple-Negative Breast Cancer Cells" International Journal of Molecular Sciences 26, no. 13: 6139. https://doi.org/10.3390/ijms26136139
APA StyleYou, K. S., Kim, T.-S., Back, S. M., Park, J.-S., Liu, K., Seong, Y.-S., Kim, D. J., & Yi, Y. W. (2025). JAK2 Inhibition Augments the Anti-Proliferation Effects by AKT and MEK Inhibition in Triple-Negative Breast Cancer Cells. International Journal of Molecular Sciences, 26(13), 6139. https://doi.org/10.3390/ijms26136139