1. Introduction
Radiotherapy is a cornerstone of cancer treatment, typically delivered in fractionated schemes in which the tumor receives daily doses over several weeks to achieve a therapeutic effect [
1]. While the primary goal is to eradicate malignant cells, healthy tissues surrounding the treatment field are inevitably exposed to varying levels of ionizing radiation, determined not only by their anatomical proximity to the targeted area but also by the inherent limitations of the radiation delivery method. Consequently, secondary organs are exposed to cumulative radiation doses that vary in magnitude and impact. While moderate-to-high doses are associated with adverse effects such as radiation-induced cardiotoxicity and pneumonitis, particularly in breast cancer patients [
2,
3,
4], lower doses, though not overtly toxic, can still induce biological changes over time [
4,
5,
6,
7,
8,
9].
Subtherapeutic doses, typically below 1 Gy, are encountered in several clinically relevant scenarios, including scatter radiation to adjacent healthy tissues during external beam radiotherapy and cumulative exposure from repeated diagnostic imaging procedures. Moreover, environmental or occupational exposure in medical or nuclear industries can also result in comparable low-dose radiation accumulation. These contexts highlight the clinical significance of SDIRs, particularly in patients undergoing repeated radiation treatments or those with tumors located near radiosensitive organs.
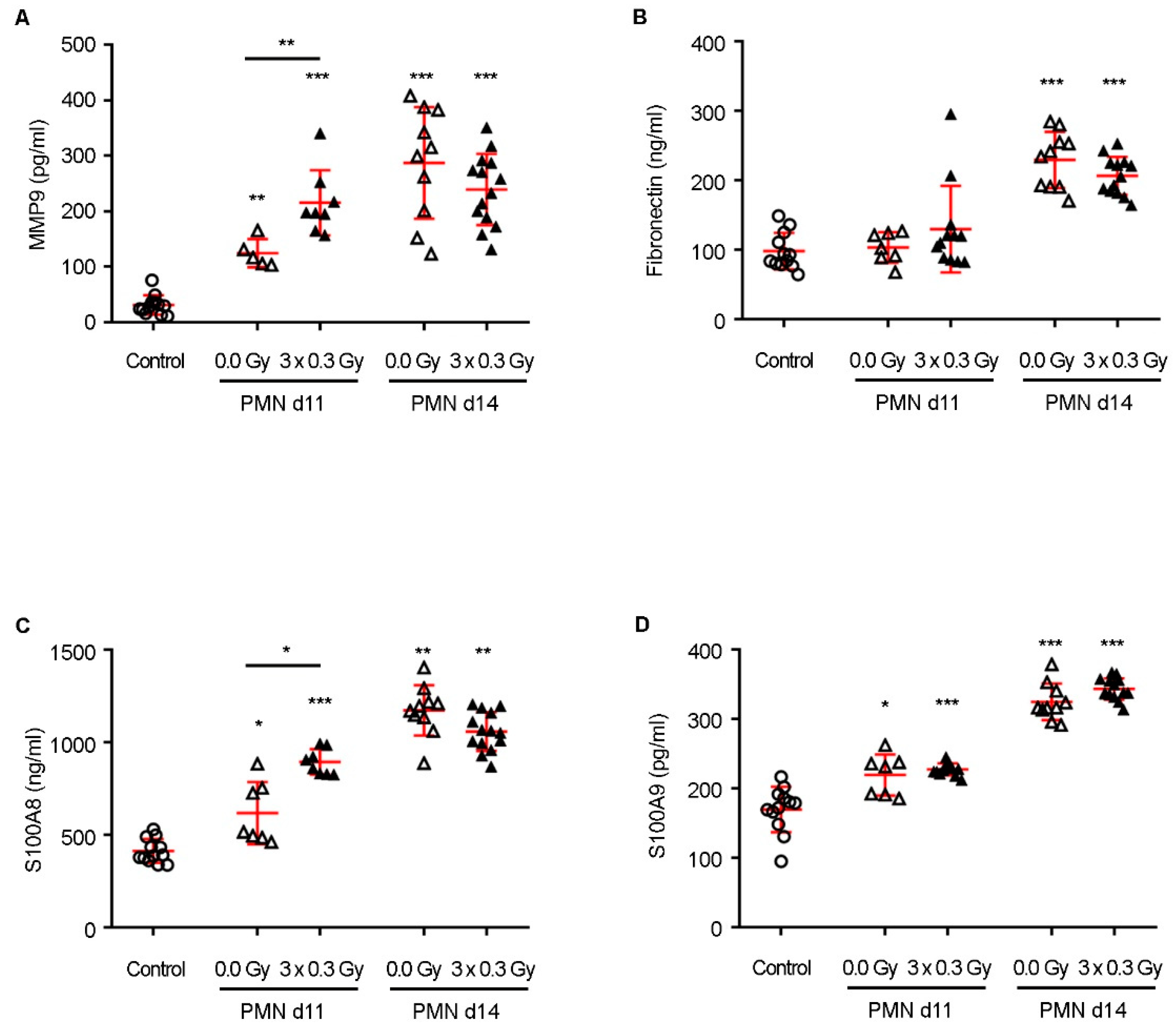
Recent studies indicate that, within the subtherapeutic range, doses below 0.8 Gy can phosphorylate VEGFR2, modulating signaling pathways and gene expression, ultimately leading to endothelial cell activation and increased microvascular density [
8,
9]. Interestingly, the SDIR has been shown to accelerate angiogenic sprouting during embryonic development and enhance inter-ray vessel density following caudal fin amputation and regeneration in adult zebrafish, further supporting its pro-angiogenic effects across different biological contexts [
6,
9]. These findings gain further relevance as low doses have also been shown to activate endothelial cells in human tissues [
5]. In patients with locally advanced rectal cancer undergoing neoadjuvant radiotherapy, paired biopsies revealed increased microvasculature density and the upregulation of angiogenic genes in endothelial cells exposed to low doses compared to internal calibrator tissues [
5]. These data strongly suggest that SDIR-induced vascular remodeling is not merely an experimental observation but a clinically relevant process. Moreover, following primary tumor induction, whole-body irradiation markedly increases metastases in the predicted target organ [
9,
10], suggesting that the SDIR plays a role in PMN modulation. The PMN is a dynamic microenvironment within distant organs that is primed to facilitate the colonization of circulating tumor cells before metastatic dissemination occurs [
11,
12,
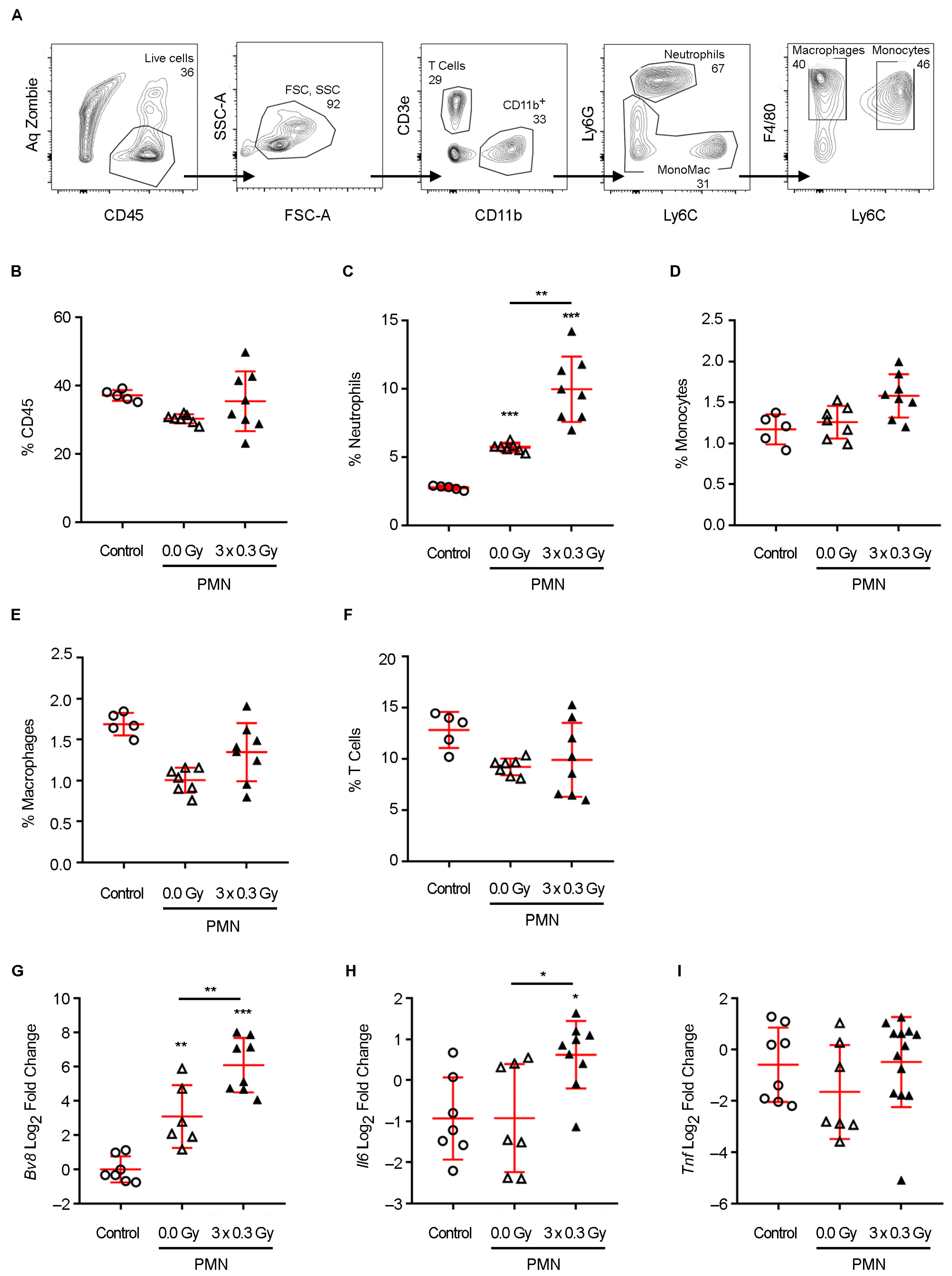
13]. Its formation is orchestrated by a complex interplay of molecular and cellular events, including stromal remodeling, endothelial activation, immune modulation, and ECM restructuring, collectively creating a permissive landscape for tumor cell seeding and outgrowth. A key hallmark of PMN formation is the recruitment of neutrophils, macrophages, and bone marrow-derived suppressor cells, guided by systemic signals that precondition distant organs for tumor cell arrival [
11,
13,
14,
15,
16,
17,
18,
19,
20]. Among these, Bv8 plays a pivotal role in neutrophil mobilization to pre-metastatic sites, fostering an inflammatory microenvironment that supports metastatic progression [
21,
22,
23]. Additionally, inflammatory mediators and endothelial activation induce the upregulation of proteins such as S100A8 and MMP9, which promote ECM remodeling and increase vascular permeability, further facilitating metastatic cell adhesion and extravasation [
17,
21,
22,
24]. IL-6, a pro-inflammatory cytokine elevated in response to SDIRs, reinforces immune suppression and enhances angiogenesis, amplifying the niche’s capacity to sustain disseminated tumor cells [
25,
26,
27]. Collectively, these alterations transform distant organs into a permissive “soil” for metastatic colonization, underscoring the intricate molecular landscape that precedes overt metastasis. Given the emerging evidence that the SDIR profoundly reshapes vascular and immune dynamics in distant organs, elucidating its impact on PMN reprogramming is essential. Whether this modulation fosters or restrains metastatic colonization remains an open question with critical implications for understanding the broader effects of radiation exposure on metastasis. Here, we investigate how the SDIR shapes the PMN, offering new insights into its role in metastatic initiation and progression. Within the SDIR range, the PMN was exposed to 0.3 Gy, previously described as both pro-angiogenic and anti-inflammatory, administered over three consecutive days. This fractionated schedule was designed to mimic clinical radiotherapy while targeting the PMN during a temporal window in which it remains uncolonized by tumor cells, thereby allowing us to evaluate the specific effects of radiation on niche reprogramming without interference from direct tumor cell interactions.
3. Discussion
Neutrophil recruitment plays a pivotal role in shaping the PMN, acting as a critical intermediary between systemic inflammatory signals and metastatic progression. Elevated Bv8 expression has been identified as a key driver of neutrophil mobilization to pre-metastatic organs, where these cells contribute to niche conditioning by secreting a repertoire of pro-metastatic mediators [
22,
34]. Among these, MMP9 facilitates ECM degradation, enhancing vascular permeability and promoting tumor cell extravasation, while S100A8 fosters a pro-inflammatory microenvironment that sustains neutrophil survival and function [
17,
22,
34]. Additionally, IL-6, an inflammatory cytokine elevated in response to radiation exposure, exerts pleiotropic effects by reinforcing immune suppression, stimulating angiogenesis, and sustaining a niche conducive to metastatic outgrowth [
25,
27]. The interplay between these factors establishes a permissive microenvironment that primes the lung for colonization by disseminated tumor cells [
29].
Our findings align with previous reports indicating that low-dose radiation can modulate immune responses and promote angiogenesis [
8,
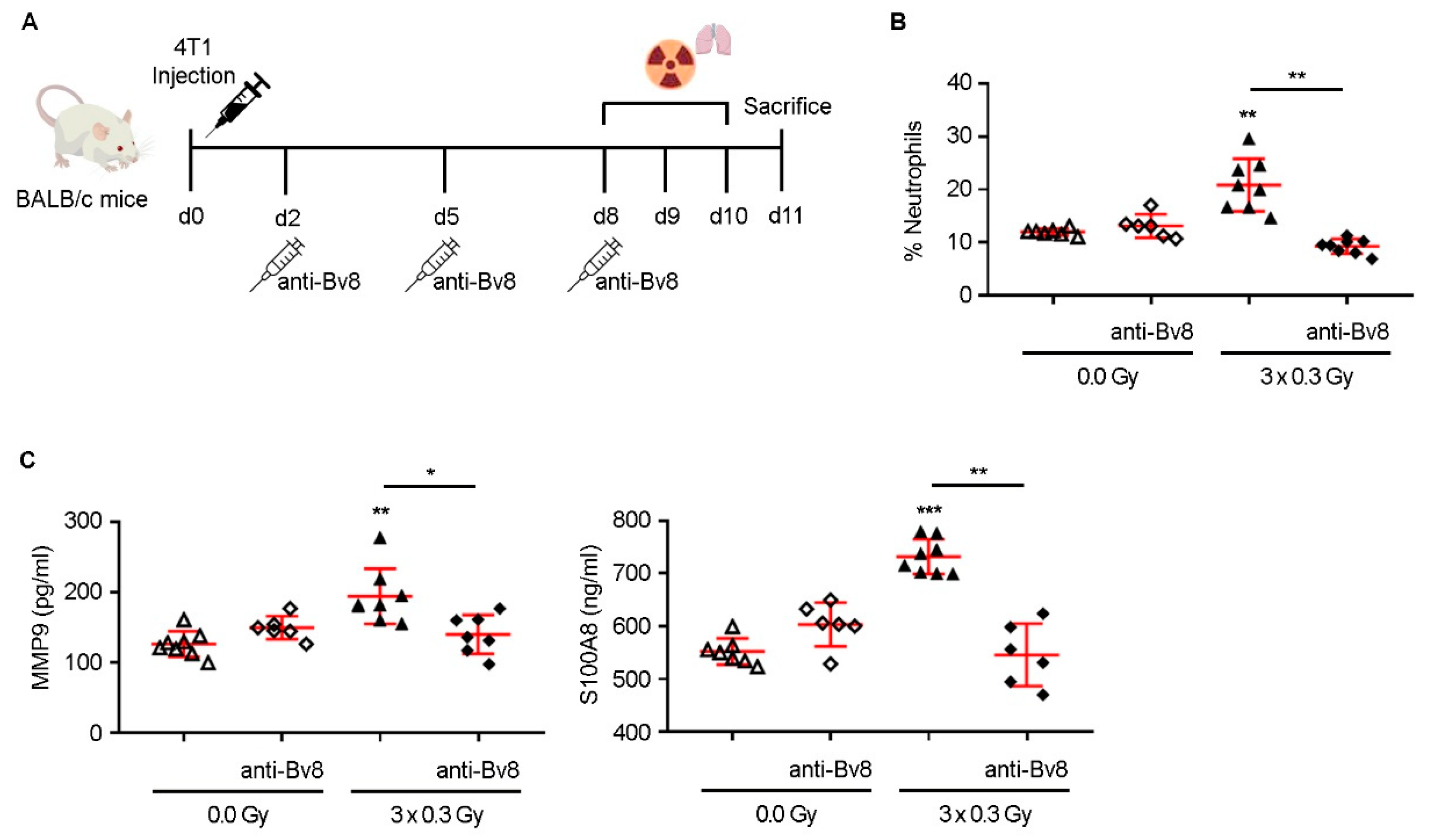
9]. However, our study expands upon these observations by identifying a mechanistic axis linking SDIR exposure to neutrophil-driven PMN remodeling and metabolic adaptations. Notably, SDIR-induced neutrophil accumulation was entirely abrogated by Bv8 blockade, which also prevented the associated upregulation of MMP9 and S100A8, confirming the pivotal role of Bv8 in mediating these effects.
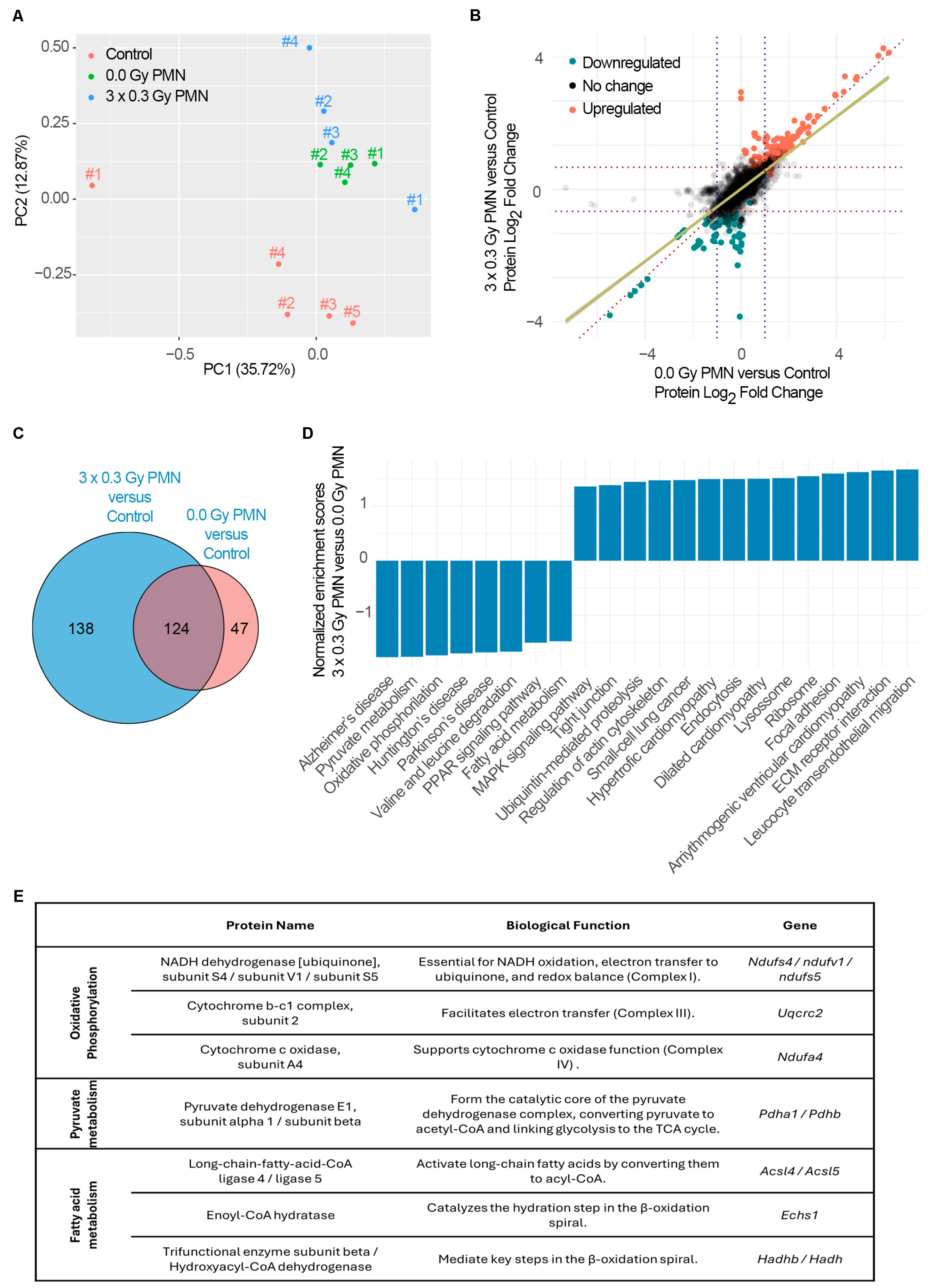
Importantly, while SDIRs significantly accelerated PMN formation at early time points, differences between irradiated and non-irradiated conditions for MMP9 and S100A8 were no longer observed by day 14, as their expression levels increased equally in both experimental groups. This suggests that SDIR functions primarily as an early catalyst of niche priming. This is further supported by our observation that the Bv8 blockade, despite effectively preventing SDIR-induced neutrophil recruitment, did not abrogate the increase in TMV observed at later time points. These findings indicate that SDIR-driven metastasis involves mechanisms extending beyond neutrophil accumulation, potentially encompassing stromal activation, metabolic reprogramming, and vascular remodeling. This prompted us to conduct a proteomic analysis, which revealed profound metabolic reprogramming within the PMN as early as day 11 following SDIR exposure.
In agreement with previous studies, the PMN undergoes significant metabolic reprogramming, which further facilitates tumor cell colonization and outgrowth [
29]. Increasing evidence suggests a shift from oxidative phosphorylation (OXPHOS) toward glycolysis and lactate production, a change that is essential for creating a permissive environment for metastasis [
11]. A key driver of this metabolic shift is the activation of inducible nitric oxide synthase (NOS2) in response to inflammatory signals [
11]. Nitric oxide inhibits key components of the mitochondrial electron transport chain, notably complex III (ubiquinol-cytochrome c reductase) and complex IV (cytochrome c oxidase), impairing mitochondrial respiration and further promoting a glycolytic phenotype [
11].
This metabolic reprogramming leads to lactate accumulation, resulting in microenvironment acidification and immune suppression, both of which are hallmarks of a metastasis-permissive niche [
30]. Our study contributes novel insights by demonstrating that SDIR exposure induces a significant downregulation of OXPHOS-related genes within the PMN. Proteome-wide GSEA revealed a significant depletion of the oxidative phosphorylation pathway (KEGG), with key components of the electron transport chain exhibiting reduced expression. This includes NDUFS4, NDUFV1, and NDUFS5, which are crucial for NADH oxidation and electron transfer in complex I, and UQCRC2, a core component of complex III involved in maintaining the proton gradient [
33]. Additionally, the downregulation of NDUFA4, which regulates cytochrome c oxidase activity in complex IV, suggests further inhibition of mitochondrial electron flow [
33]. Although we observed a reduction in ATP5PB and ATP5MG, key components of ATP synthase [
33], the downregulation of critical subunits in complexes I, III, and IV appears to be the primary driver of OXPHOS disruption. Given that ATP synthase function relies on the proton gradient established by upstream complexes, we propose that its downregulation is a downstream effect of impaired electron transport, rather than a direct cause of the metabolic reprogramming in the PMN following SDIR exposure. Furthermore, GSEA in this study pointed to a marked decrease in the expression of PDHA1 and PDHB, core subunits of the pyruvate dehydrogenase complex, specifically driven by SDIR exposure. These enzymes are essential for converting pyruvate to acetyl-CoA, a critical step for entry into the tricarboxylic acid (TCA) cycle, thereby further promoting the metabolic shift away from mitochondrial respiration [
32]. The accumulation of pyruvate in the cytoplasm is expected to drive its conversion to lactate, a process that would likely contribute to extracellular acidification, immune evasion, and stromal remodeling, all of which are key features that support metastatic progression [
30]. In addition to these metabolic alterations, a significant decrease in fatty acid metabolism pathways (KEGG) is observed in the SDIR-exposed PMN. The downregulation of ACSL4 and ACSL5, enzymes responsible for the activation of long-chain fatty acids for mitochondrial oxidation [
35], indicates a reduced capacity for fatty acid utilization. This alteration is further supported by a decrease in the expression of ECHS1 and HADHB, essential enzymes in the β-oxidation pathway, suggesting a reduction in fatty acid oxidation [
35]. These changes indicate a shift away from fatty acid metabolism, potentially driving increased reliance on glycolysis and facilitating lipid accumulation within the PMN. This shift contributes to ECM remodeling and immune modulation, fostering a microenvironment that is conducive to tumor progression [
30]. Our results also highlight the role of neutrophils in this altered metabolic landscape. Previous studies have established that neutrophils within the lung PMN accumulate neutral lipids, a process intricately regulated by lung-resident mesenchymal cells [
36]. This lipid sequestration is driven by the suppression of adipose triglyceride lipase (ATGL), a key enzyme in triglyceride hydrolysis, leading to intracellular lipid accumulation [
36]. Rather than serving as inert reservoirs, these lipid-laden neutrophils function as metabolic intermediaries, transferring stored lipids to metastatic tumor cells. This exchange fuels tumor cell bioenergetics, enhancing their survival, proliferation, and invasive potential. These findings underscore a broader metabolic crosstalk between immune and stromal components within the PMN, wherein neutrophils not only shape the inflammatory landscape but also actively reprogram the metabolic architecture of the metastatic niche [
36].
SDIR exposure drives profound metabolic reprogramming within the PMN, characterized by the suppression of both OXPHOS and fatty acid oxidation, alongside an increased dependence on glycolysis. The downregulation of key enzymes in fatty acid metabolism, coupled with lipid accumulation, further highlights the metabolic shift that supports tumor progression [
37]. Additionally, the accumulation of pyruvate and the consequent rise in lactate levels are expected to drive extracellular acidification, contributing to immune modulation [
38]. This rewiring promotes a pro-inflammatory phenotype in neutrophils and macrophages while fostering an immunosuppressive environment, a hallmark of metastatic niches [
29]. In parallel, the acidified environment is likely to activate fibroblasts and other stromal cells, inducing the secretion of matrix remodeling factors that facilitate tumor cell invasion [
29]. Metastatic progression is shaped not only by the intrinsic properties of circulating tumor cells but also by the permissiveness of the PMN. Our findings reveal, for the first time, that the SDIR profoundly reprograms the metabolic landscape of the lung PMN, establishing conditions that facilitate tumor colonization. This highlights the need to integrate organ-specific radiation exposure and dose ranges into predictive models of metastasis, particularly in tumors with defined organotropism. By identifying metabolic pathways and immune-stromal interactions that drive SDIR-induced PMN remodeling, this study uncovers actionable targets for intervention. While the current findings offer novel insights into the impact of SDIRs on pre-metastatic niche formation, certain considerations should be kept in mind when interpreting the data. The use of a single animal model, though well-characterized, may not capture the full spectrum of human physiological responses to low-dose radiation. Another important limitation of this study is the absence of long-term follow-up after SDIR exposure. While our data reveal early and intermediate effects on PMN remodeling, they do not capture the potential delayed consequences of SDIRs, which could manifest beyond the timeframe of our current experimental design. Nonetheless, as cancer survivorship improves and the cumulative impact of radiation exposure becomes more clinically relevant, future research should explore the temporal evolution of SDIR-induced changes over longer durations. Nonetheless, these results provide a mechanistic foundation that opens important avenues for future translational research. Validating these pathways in patient samples or organotypic models and integrating them into studies of clinical radiotherapy will be essential to assess their broader applicability. In this light, our study should be viewed as a step toward a more integrated understanding of radiation-induced metastatic risk, with future work needed to bridge the gap to clinical implementation. These findings pave the way for precision oncology strategies aimed at mitigating radiation-induced metastatic risk. These insights have direct implications for optimizing radiotherapy protocols, not only in terms of dose and field design but also with regard to therapeutic timing. For instance, thoracic radiation fields might be adjusted to minimize low-dose spillover to distant, organ-specific metastatic sites, particularly in cancers with well-characterized organotropism. Future strategies should leverage these findings to refine patient stratification, adjust radiation plans accordingly, and explore the use of metabolic or immunomodulatory agents to counteract SDIR-driven niche formation. Together, these approaches can help balance local tumor control with long-term metastatic risk, supporting a more holistic model of precision radiotherapy.
4. Materials and Methods
4.1. Cell Culture
The 4T1 breast carcinoma cells (American Type Culture Collection, RRID:CVCL_0125) were maintained in Dulbecco’s modified Eagle’s medium GlutaMAX (DMEM, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) with 10% (v/v) FBS (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and 1% (v/v) penicillin/streptomycin (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). Cells were used at passage 5 and confirmed mycoplasma-free.
4.2. Animal Procedures
Animals were housed in individually ventilated cages (IVCs), with a maximum of five mice per cage, using corn cob bedding. Housing conditions were maintained within standard parameters: temperature at 22 ± 2 °C, relative humidity at 55 ± 10%, and a 14/12 h light/dark cycle. Animals had ad libitum access to standard laboratory rodent chow and filtered water. Environmental enrichment was routinely provided and included nesting material and cardboard tunnels. Animal health and welfare were monitored daily by trained and competent personnel, and any signs of distress or illness were promptly addressed, ensuring compliance with institutional and European Union regulations on animal care and use. Eight-week-old female Balb/c mice (Charles River Laboratories, Barcelona, Spain, RRID:IMSR_JAX:001026) were anesthetized with intraperitoneal ketamine-medetomidine (75 mg/kg and 1 mg/kg BW). Adequate sedation was confirmed by the absence of the paw withdrawal reflex, and anesthesia was reversed with atipamezole (5 mg/kg BW). Mice were shaved in the abdominal region and anesthetized before 4T1 cell injection and radiotherapy planning. To induce tumors, 4T1 cells (5 × 104) were injected subcutaneously into the fourth right mammary gland in a 50 µL Matrigel/PBS (1:5) mixture. Mice received thoracic irradiation (0.3 Gy) on days 8–10 post-injection. At the experimental endpoint (days 11 or 14), lungs were collected for analysis by flow cytometry, ELISA, or qRT-PCR. For antibody treatment, Balb/c mice received intraperitoneal injections of anti-Bv8 monoclonal antibody (Genentech, Inc., South San Francisco, CA, USA) at 5 mg/kg, twice weekly for up to two weeks after 4T1 cell injection. Animals were randomly assigned to experimental groups using simple randomization, and all analyses were performed in a blinded manner to reduce bias. Sample size was determined by power analysis, assuming a significance level of 0.05 and a power of 0.9. Two animals were excluded from the final analysis on day 18 after tumor cell injection due to primary tumor weights below 0.05 g. Euthanasia was performed by cervical dislocation.
4.3. Irradiation Procedure
The setup for 3D reconstruction of the mice for treatment planning was as follows: the mice were positioned in an acrylic phantom to ensure proper thickness and enhance dose homogeneity during radiation therapy. A computed tomography (CT) scan was performed using a Siemens Somatom Sensation scanner, acquiring volumetric images reconstructed into 1 mm axial slices. These cross-sectional images were then transferred to the 3D computerized treatment planning system (TPS), Monaco (Elekta), for contouring the planning target volume (PTV), which encompassed the thoracic region of the mice. The radiotherapy treatment plan was designed within the same TPS, employing an isocentric dose distribution from two opposing fields (0° and 180°) with a 6 MV photon beam. The dose distribution was normalized to a reference point within the PTV. The dose calculation was performed using the clinical Monte Carlo algorithm, XVMC, for photons. A prescribed dose variation of 0.3 Gy was applied with a tolerance of ±10% within the PTV to ensure optimal dose coverage. Radiation treatment was administered at room temperature using a linear accelerator (Synergy, Elekta, Stockholm, Sweden) generating a 6 MV photon beam at a dose rate of 600 MU/min. To verify that the calculated dose matched the delivered dose to the PTV, regular quality control tests were conducted on the linear accelerator. These tests confirmed an uncertainty of less than 3% (k = 1) between the calculated and delivered doses. The absorbed dose to water for the radiation beam was determined following the IAEA TRS-398 code of practice. A total dose of 0.3 Gy was delivered to the thoracic region of the mice for three consecutive days. Control mice were sham irradiated (0.0 Gy) following the same procedures as the irradiated experimental groups. During the treatment protocol, the mice were anesthetized as previously described.
4.4. Protein Extraction and Digestion
Lung tissue proteins were extracted and solubilized with 100 mM Tris pH 8.5, 1% sodium deoxycholate, 10 mM tris(2-carboxyethyl)phosphine (TCEP), 40 mM of chloroacetamide and protease inhibitors for 10 min, at 95 °C and 100×
g. Protein extract was sonicated for 10 cycles, 30 s on/30 s off (Bioruptor Plus, Diagenode, Seraing, Belgium). After centrifugation at 16.560×
g, for 5 min, 100 μg of protein was processed for proteomic analysis following the solid-phase-enhanced sample-preparation (SP3) protocol [
39]. Enzymatic digestion was performed with Trypsin/LysC (2 μg), overnight at 37 °C and 100×
g. The resulting peptides were cleaned up and desalted with C18 micro-columns and further quantified.
4.5. NanoLC-MS/MS
Protein identification was performed by nanoLC-MS/MS. This equipment is composed of an Ultimate 3000 liquid chromatography system coupled to a Q-Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). In total, 500 ng of peptides were loaded onto a trapping cartridge (Acclaim PepMap C18 100 Å, 5 mm × 250 µm i.d., 160454, Thermo Fisher Scientific, Waltham, MA, USA) in a mobile phase of 2% ACN, 0.1% FA at 10 µL/min. After 3 min loading, the trap column was switched in-line to a 50 cm by 75 μm inner diameter EASY-Spray column (ES803, PepMap RSLC, C18, 2 μm, Thermo Fisher Scientific, Waltham, MA, USA) at 300 nL/min. Separation was generated by mixing A: 0.1% FA and B: 80% ACN, with the following gradient: 5 min (2.5% B to 10% B), 120 min (10% B to 30% B), 20 min (30% B to 50% B), 5 min (50% B to 99% B), and 10 min (hold 99% B). Subsequently, the column was equilibrated with 2.5% B for 17 min. Data acquisition was controlled by Tune 2.9 software and Xcalibur 4.0 (Thermo Fisher Scientific, Waltham, MA, USA, RRID:SCR_014593).
The mass spectrometer was operated in data-dependent (dd) positive acquisition mode alternating between a full scan (m/z 380–1580) and subsequent HCD MS/MS of the 10 most intense peaks from full scan (normalized collision energy of 27%). ESI spray voltage was 1.9 kV. Global settings: Lock masses best (m/z 445.12003), lock mass injection Full MS, chrom. Peak width (FWHM) of 15 s. Full scan settings: Resolution of 70 k (m/z 200), AGC target 3 × 106, and maximum injection time of 120 ms. dd settings: Minimum AGC target 8 × 103, intensity threshold of 7.3 × 104, charge exclusion: unassigned, 1, 8, >8, peptide match preferred, exclude isotopes on, and dynamic exclusion 45 s. MS2 settings: Microscans 1, resolution of 35 k (m/z 200), AGC target 2 × 105, maximum injection time of 110 ms, isolation window 2.0 m/z, isolation offset 0.0 m/z, and spectrum data type profile.
4.6. Protein Identification and Quantification
Protein identification and quantitative analysis were performed by Proteome Discoverer™ (Thermo Fisher Scientific, Waltham, MA, USA), MaxQuant Version 1.5.3.30 and VEMS considering the information from the UniProt protein database (RRID:SCR_002380) for Mus musculus taxonomic selection. Quantitative analysis was compared and merged to create a consensus quantitative report to minimize computational artifacts. Quantitation was performed on the peptide and protein level based on iBAQ ion counts.
4.7. Database-Dependent Search of MS Data
Mass accuracy was set to 5 ppm on the peptide level and 0.01 Da on the fragment ions. A maximum of four missed cleavages was used. Carbamidomethyl (C) was set as a fixed modification. Methionine oxidation and N-terminal protein acetylation were set as variable modifications. The MS/MS data was searched against all reviewed mouse proteins from UniProt with the concatenation of all the sequences in reverse, maintaining only lysine and arginine in place. The data was searched and quantified with both MaxQuant Version 1.5.3.30 and VEMS [
40]. For MaxQuant, the variable modifications such as methionine oxidation, N-terminal protein acetylation, tyrosine phosphorylation, threonine phosphorylation, and serine phosphorylation were included.
4.8. Quantitative Analysis of MS Data
Quantitative data from MaxQuant and VEMS were analyzed using the R statistical programming language. Protein iBAQ values from the two programs were preprocessed by removing common MS contaminants, followed by Log2(x + 1) transformation and quantile normalization. The distribution of quantitative values in each sample for each processing method was analyzed using boxplots in R. To identify differentially expressed proteins, R (v4.4.0) and the limma package (v3.60.4; RRID:SCR_010943) were used to compare each tumor-bearing experimental group with one another, as well as with the control group. Multiple testing correction was applied using the Benjamini-Hochberg method to control the false discovery rate (FDR). Proteins with an FDR < 0.1 were considered differentially expressed. No fold-change cutoff was applied.
4.9. Functional Enrichment Analysis
GSEA was performed to identify functionally enriched pathways among differentially expressed proteins. Enrichment analysis was carried out using KEGG pathway annotations (RRID:SCR_012773), considering the differential expression of proteins between experimental groups. p-values were estimated by comparing the empirical enrichment score (ES) of a gene set relative to a null distribution of ESs derived from permuting the gene set 1000 times and then adjusting for multiple hypothesis testing.
Proteins assigned to significantly enriched pathways were extracted and categorized based on functional annotations. The top enriched pathways were ranked according to statistical significance, and results were visualized as bar plots representing normalized enrichment scores. All data processing and statistical analysis were conducted using R statistical programming language.
4.10. Circulating Cytokine Quantification
Lungs were perfused with PBS and collected in tubes containing lysing matrix D (MP Biomedicals, Aurora, OH, USA) and 0.5 mL of PBS supplemented with an EDTA-free Protease Inhibitor Cocktail (Roche, Basel, Switzerland). Lung tissue was homogenized twice using a Mini-Beadbeater for 45 s. Homogenates were centrifuged at 16.060× g for 10 min, and the supernatant was collected. Matrix metallopeptidase 9 (MMP9), S100 calcium-binding protein A8 (S100A8), S100 calcium-binding protein A9 (S100A9), granulocyte-colony stimulating factor (GCSF), and fibronectin levels were quantified in the supernatant using ELISA kits (Abcam, Cambridge, UK), following the manufacturer’s instructions.
4.11. Tissue Digestion and Flow Cytometry
Pulmonary cells were isolated from well-perfused lungs. Lung tissue was finely chopped and digested with Type IV collagenase (0.5 mg/mL; Sigma-Aldrich, St. Louis, MO, USA) and DNase I (10 µg/mL; Sigma-Aldrich, St. Louis, MO, USA) for 30 min at 37 °C with agitation (100× g). The resulting cell suspension was filtered through a 70 µm nylon cell strainer (Falcon Corning Incorporated, Corning, NY, USA), and erythrocytes were lysed osmotically using RBC Lysis Buffer (BioLegend, San Diego, CA, USA) for 5 min in the dark. Cells were then counted.
For surface staining, cells were Fc-blocked with mouse anti-CD16/32 (Thermo Fisher Scientific, Waltham, MA, USA, Cat# 14-0161-82, RRID:AB_467133), followed by incubation with an antibody mix for 1 h at room temperature in the dark. The following monoclonal antibodies were used: anti-CD45 (BD Biosciences, San Jose, CA, USA, Cat# 559864, RRID:AB_398672), anti-CD3ε (Thermo Fisher Scientific, Waltham, MA, USA, Cat# 45-0031-82, RRID:AB_1107000), anti-CD11b (BD Biosciences, San Jose, CA, USA, Cat# 563168, RRID:AB_2716860), anti-F4/80 (Invitrogen, Carlsbad, CA, USA, Cat# 25-4801-82, RRID:AB_469653), and anti-Ly6C (Invitrogen, Carlsbad, CA, USA, Cat# 53-5932-82, RRID:AB_2574427). Zombie Aqua Dye (BioLegend, San Diego, CA, USA, Cat# 423101) as added to the final suspension for 30 min at room temperature to stain and exclude dead cells. Samples were acquired using a FACS Fortessa™ (BD Biosciences, San Jose, CA, USA), and data were analyzed with FlowJo™ v10 software (Tree Star, Inc., Ashland, OR, USA, RRID:SCR_008520).
4.12. RNA Extraction, cDNA Synthesis, and Quantitative Real-Time PCR
Total RNA was extracted using an RNeasy® Micro Kit (QIAGEN, Hilden, Germany) according to the instructions of the manufacturer, for animal tissue. For the synthesis and pre-amplification of cDNA, the RT2 Nano PreAMP cDNA synthesis kit (QIAGEN, Hilden, Germany) was used with two rounds of pre-amplification, using the same genes described below. The mRNA expression levels were assessed by qRT-PCR using a Power SYBR® Green system (Invitrogen, Carlsbad, CA, USA) on a 7500/7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The housekeeping gene was 18S. The initial denaturation step was 10 min at 95 °C, followed by 50 cycles of 15 s at 95 °C, and 1 min at 60 °C. The relative quantification was performed through the 2−ΔCt method, where ΔCt = Ct (sample) − Ct (reference gene). Data for each animal are represented as a Log2 of fold change (2−ΔCt), relative to the fold change average of the 0.0 Gy group. Gene-specific primers are detailed as follows (5′–3′):
Bv8 Fw: GAAGGGCGAAGAGAAGAAAGAG
Bv8 Rev: TCCGGGCCAAGCAAATAA
Il6 Fw: GTCTGTAGCTCATTCTGCTCTG
Il6 Rev: GAAGGCAACTGGATGGAAGT
Tnf Fw: CTACCTTGTTGCCTCCTCTTT
Tnf Rev: GAGCAGAGGTTCAGTGATGTAG
4.13. Stereology Analysis
Lungs were formalin-fixed, paraffin-embedded, and sectioned at a uniform interval (T) of 200 µm with a random start, yielding a total of eight sections per lung. All sections were stained with H&E. Volume estimates were obtained using the Cavalieri principle [
41]: V = T × a/p × ∑Pi, where T is the thickness of the slabs, Pi is the number of points hitting the structure of interest, and a/p is the area associated with each point. For point counting, step lengths in both the x- and y-directions were fixed, enabling the evaluation of 20% of the total area using a 10x objective. The meander sampling resulted in approximately 30 fields evaluated per section. Quantification was performed by overlaying a point grid onto 10× fields of view and counting the number of points hitting the lung and those hitting lung metastases. A 5 × 5 (25-point) grid was used. Each point has an associated area (a/p), which is multiplied by the number of points counted. The circled point in the upper left-hand corner was counted each time it hit the lung (A), with an associated area 25 times larger than the individual points counted when hitting metastases (B). The area fraction of lung metastases, representing the percentage of lung occupied by metastases, was calculated by dividing the number of points hitting metastases by the number hitting the lung. Digital slides were acquired using the Hamamatsu NanoZoomer slide scanner, and measurements were performed with Visiopharm stereology software (version 2019, Visiopharm A/S, Hørsholm, Denmark). Stereology was conducted at the Histopathology Unit at IGC.
4.14. Statistics
For multiple-group comparisons, one-way ANOVA with Bonferroni post hoc correction was used for normally distributed data. When variance was unequal, Welch’s ANOVA with Tamhane’s T2 post hoc test was applied. For non-normally distributed data, the Kruskal–Wallis test with Dunn’s multiple comparison adjustment was used.
For two-group comparisons, an independent two-tailed unpaired t-test was performed for normally distributed data, with Welch’s correction applied in cases of unequal variance. Assumptions of normality and homogeneity of variances were tested using the Shapiro-Wilk and Levene’s tests, respectively. Statistical analyses were performed using GraphPad Prism 8 (Graphpad Software, LLC Company Profile, San Diego, CA, USA, RRID:SCR_002798). All p-values are two-sided, with p < 0.05 considered statistically significant. Sample sizes (n) per experiment are provided in the corresponding figure legends.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}