
Naturally Occurring Angiotensin Peptides Enhance the SARS-CoV-2 Spike Protein Binding to Its Receptors

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Effects of Angiotensin II (1–8) on Spike Protein Binding to Host Cell Receptors
2.2. Effects of C-Terminal Deletion of Angiotensin II (1–8) on Spike–AXL Binding
2.3. Effects of N-Terminal Deletion of Angiotensin II (1–8) on Spike–AXL Binding
2.4. Effects of N-Terminal Deletion of Angiotensin (1–7) on Spike–AXL Binding
2.5. Effects of Angiotensin II (1–8) Mutants on Spike–AXL Binding
2.6. Effects of Angiotensin IV (3–8) on Spike Protein Binding to AXL, ACE2, and NRP1
3. Discussion
4. Materials and Methods
4.1. Chemicals
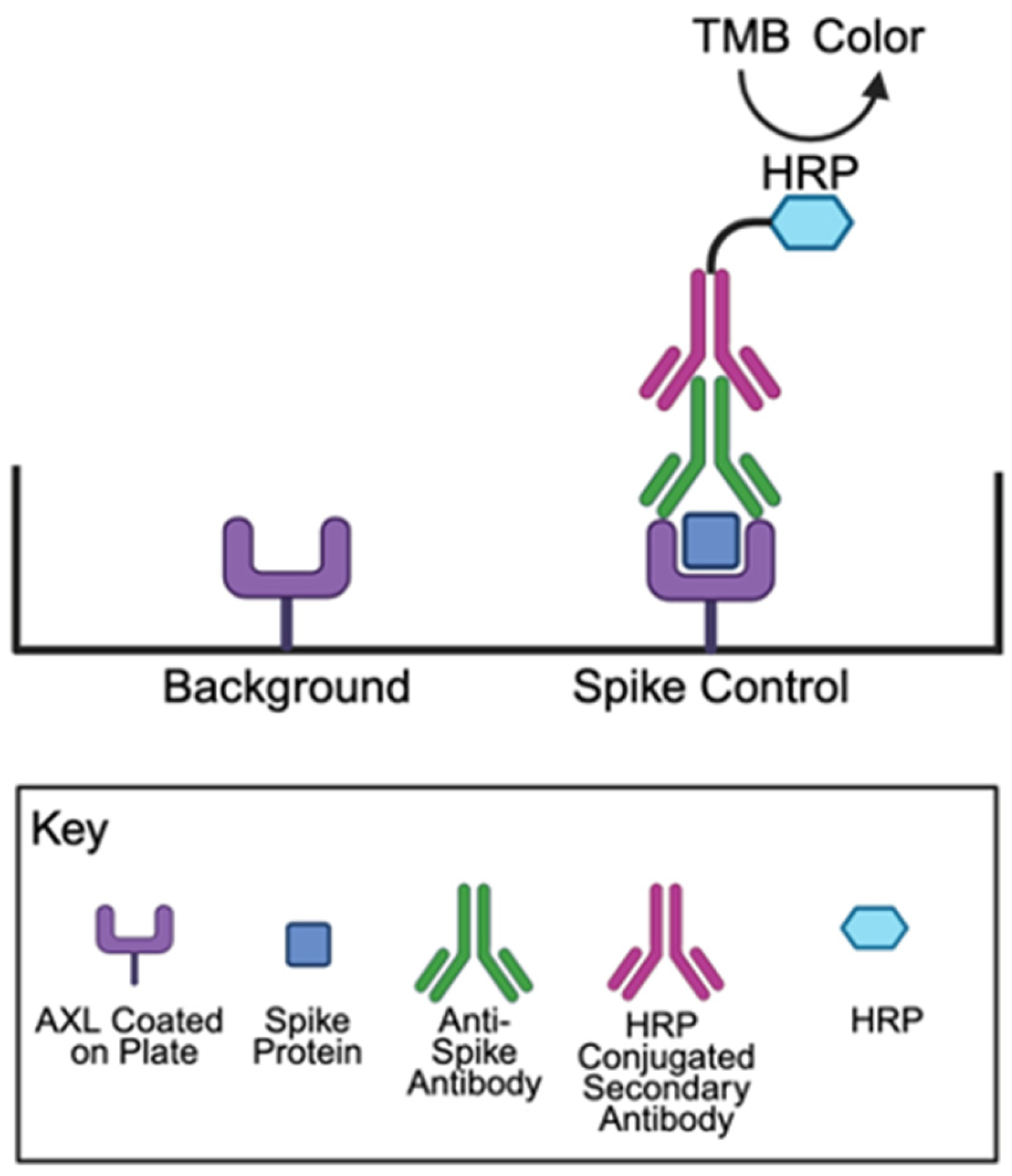
4.2. Spike Protein Binding Assays
4.3. Furin Activity Assay
4.4. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| COVID-19 | Coronavirus disease 2019 |
| ACE2 | Angiotensin-converting enzyme 2 |
| RAS | Renin–angiotensin system |
| ACE | Angiotensin-converting enzyme |
| AT1R | Type 1 angiotensin II receptor |
| GPCR | G protein-coupled receptor |
| ADH | Antidiuretic hormone |
| NO | Nitric oxide |
| AT2R | Type 2 angiotensin II receptor |
| HRP | Horseradish peroxidase |
| TMB | 3,3′,5,5′-tetramethylbenzidine |
| RFU | Relative fluorescence unit |
| SEM | Standard error of the mean |
References
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef]
- Yang, R.; Smolders, I.; Dupont, A.G. Blood pressure and renal hemodynamic effects of angiotensin fragments. Hypertens. Res. 2011, 34, 674–683. [Google Scholar] [CrossRef]
- Fountain, J.H.; Kaur, J.; Lappin, S.L. Physiology, Renin Angiotensin System; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Haulica, I.; Bild, W.; Serban, D.N. Angiotensin peptides and their pleiotropic actions. J. Renin Angiotensin Aldosterone Syst. 2005, 6, 121–131. [Google Scholar] [CrossRef]
- Beyerstedt, S.; Casaro, E.B.; Rangel, É.B. COVID-19: Angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 905–919. [Google Scholar] [CrossRef]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, J.A.; Tremblay, B.J.M.; Mansfield, M.J.; Woody, O.; Lobb, B.; Banerjee, A.; Chandiramohan, A.; Tiessen, N.; Cao, Q.; Dvorkin-Gheva, A.; et al. Gene expression and in situ protein profiling of candidate SARS-CoV-2 receptors in human airway epithelial cells and lung tissue. Eur. Respir. J. 2020, 56, 2001123. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Xu, W.; Zheng, T.; Wu, P.; Xie, S.; Bian, W.; Zhang, C.; et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021, 31, 126–140. [Google Scholar] [CrossRef]
- Goyette, M.A.; Côté, J.F. AXL receptor tyrosine kinase as a promising therapeutic target directing multiple aspects of cancer progression and metastasis. Cancers 2022, 14, 466. [Google Scholar] [CrossRef]
- Wium, M.; Ajayi-Smith, A.F.; Paccez, J.D.; Zerbini, L.F. The Role of the receptor tyrosine kinase Axl in carcinogenesis and development of therapeutic resistance: An overview of molecular mechanisms and future applications. Cancers 2021, 13, 1521. [Google Scholar] [CrossRef]
- Gay, C.M.; Balaji, K.; Byers, L.A. Giving AXL the axe: Targeting AXL in human malignancy. Br. J. Cancer 2017, 116, 415–423. [Google Scholar] [CrossRef]
- Wimmel, A.; Glitz, D.; Kraus, A.; Roeder, J.; Schuermann, M. Axl receptor tyrosine kinase expression in human lung cancer cell lines correlates with cellular adhesion. Eur. J. Cancer 2001, 37, 2264–2274. [Google Scholar] [CrossRef]
- Melaragno, M.G.; Wuthrich, D.A.; Poppa, V.; Gill, D.; Lindner, V.; Berk, B.C.; Corson, M.A. Increased expression of Axl tyrosine kinase after vascular injury and regulation by G protein-coupled receptor agonists in rats. Circ. Res. 1998, 83, 697–704. [Google Scholar] [CrossRef]
- You, J.; Huang, R.; Zhong, R.; Shen, J.; Huang, S.; Chen, J.; Chen, F.; Kang, Y.; Chen, L. Serum AXL is a potential molecular marker for predicting COVID-19 progression. Front. Immunol. 2024, 15, 1394429. [Google Scholar] [CrossRef]
- Suzuki, Y.J. The viral protein fragment theory of COVID-19 pathogenesis. Med. Hypotheses 2020, 144, 110267. [Google Scholar] [CrossRef]
- Ayyubova, G.; Gychka, S.G.; Nikolaienko, S.I.; Alghenaim, F.A.; Teramoto, T.; Shults, N.V.; Suzuki, Y.J. The role of furin in the pathogenesis of COVID-19-associated neurological disorders. Life 2024, 14, 279. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Portales, A.E.; Mustafá, E.R.; McCarthy, C.I.; Cornejo, M.P.; Couto, P.M.; Gironacci, M.M.; Caramelo, J.J.; Perelló, M.; Raingo, J. ACE2 internalization induced by a SARS-CoV-2 recombinant protein is modulated by angiotensin II type 1 and bradykinin 2 receptors. Life Sci. 2022, 293, 120284. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 spike protein impairs endothelial function via downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef]
- Wu, Z.; Hu, R.; Zhang, C.; Ren, W.; Yu, A.; Zhou, X. Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID-19 patients. Crit. Care 2020, 24, 290. [Google Scholar] [CrossRef]
- Camargo, R.L.; Bombassaro, B.; Monfort-Pires, M.; Mansour, E.; Palma, A.C.; Ribeiro, L.C.; Ulaf, R.G.; Bernardes, A.F.; Nunes, T.A.; Agrela, M.V.; et al. Plasma angiotensin II is increased in critical coronavirus disease 2019. Front. Cardiovasc. Med. 2022, 9, 847809. [Google Scholar] [CrossRef]
- Reindl-Schwaighofer, R.; Hödlmoser, S.; Domenig, O.; Krenn, K.; Eskandary, F.; Krenn, S.; Schörgenhofer, C.; Rumpf, B.; Karolyi, M.; Traugott, M.T.; et al. The systemic renin-angiotensin system in COVID-19. Sci. Rep. 2022, 12, 20117. [Google Scholar] [CrossRef]
- Caputo, I.; Caroccia, B.; Frasson, I.; Poggio, E.; Zamberlan, S.; Morpurgo, M.; Seccia, T.M.; Calì, T.; Brini, M.; Richter, S.N.; et al. Angiotensin II promotes SARS-CoV-2 infection via upregulation of ACE2 in human bronchial cells. Int. J. Mol. Sci. 2022, 23, 5125. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, K.X.; Bablu, F.E.; Gonzales, E.S.; Izumi, T.; Suzuki, Y.J. Naturally Occurring Angiotensin Peptides Enhance the SARS-CoV-2 Spike Protein Binding to Its Receptors. Int. J. Mol. Sci. 2025, 26, 6067. https://doi.org/10.3390/ijms26136067
Oliveira KX, Bablu FE, Gonzales ES, Izumi T, Suzuki YJ. Naturally Occurring Angiotensin Peptides Enhance the SARS-CoV-2 Spike Protein Binding to Its Receptors. International Journal of Molecular Sciences. 2025; 26(13):6067. https://doi.org/10.3390/ijms26136067
Chicago/Turabian StyleOliveira, Katelin X., Fariha E. Bablu, Emily S. Gonzales, Taisuke Izumi, and Yuichiro J. Suzuki. 2025. "Naturally Occurring Angiotensin Peptides Enhance the SARS-CoV-2 Spike Protein Binding to Its Receptors" International Journal of Molecular Sciences 26, no. 13: 6067. https://doi.org/10.3390/ijms26136067
APA StyleOliveira, K. X., Bablu, F. E., Gonzales, E. S., Izumi, T., & Suzuki, Y. J. (2025). Naturally Occurring Angiotensin Peptides Enhance the SARS-CoV-2 Spike Protein Binding to Its Receptors. International Journal of Molecular Sciences, 26(13), 6067. https://doi.org/10.3390/ijms26136067