
Host–Virus Interface in Persistent SARS-CoV-2 Infections: Viral Characteristic Evolution and Gene Expression Profiling Analysis

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. Clinical Patient Data and the Characteristics of SARS-CoV-2 Isolates
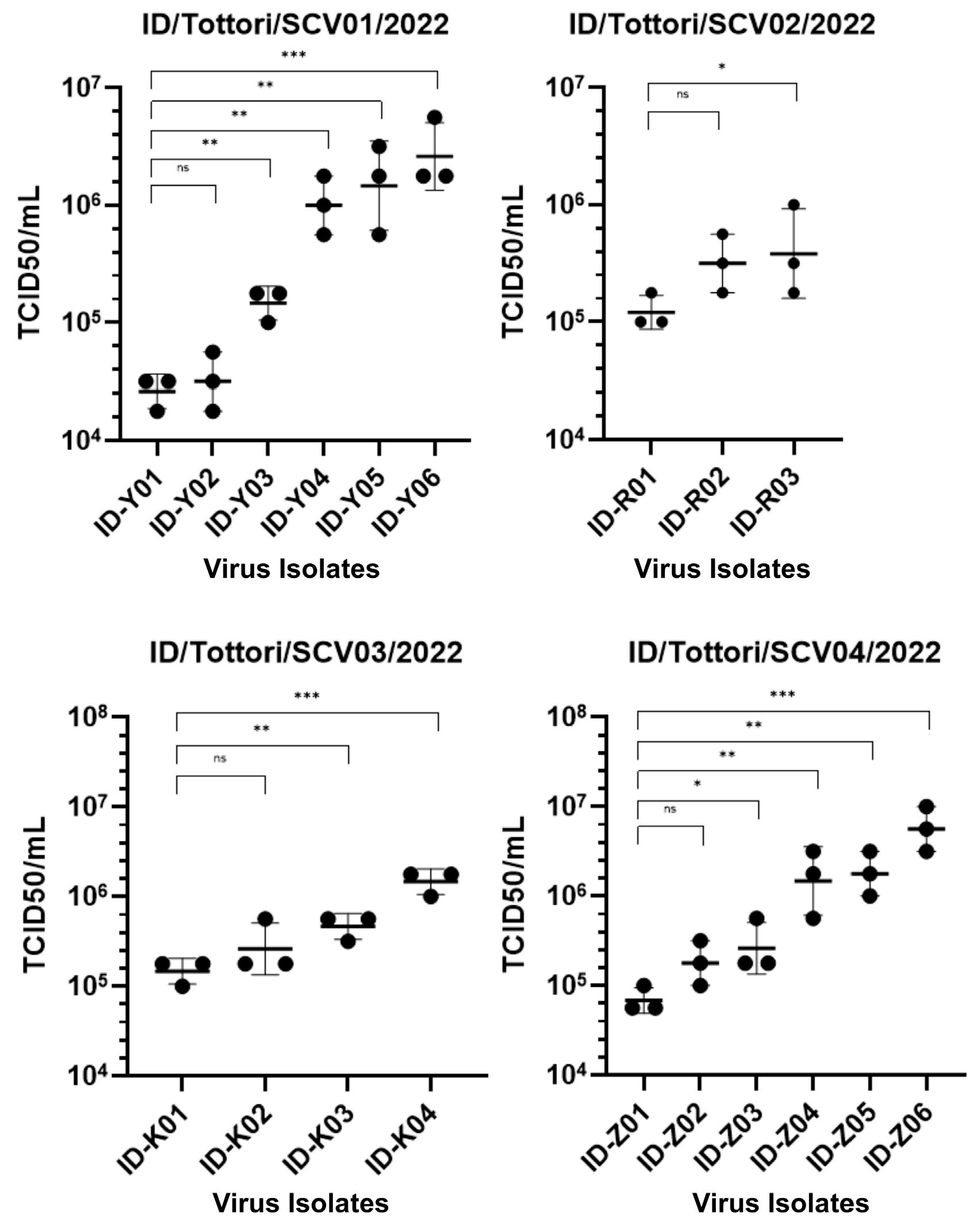
2.2. Dynamic Changes in Infectivity Across Persistent COVID-19 Infection
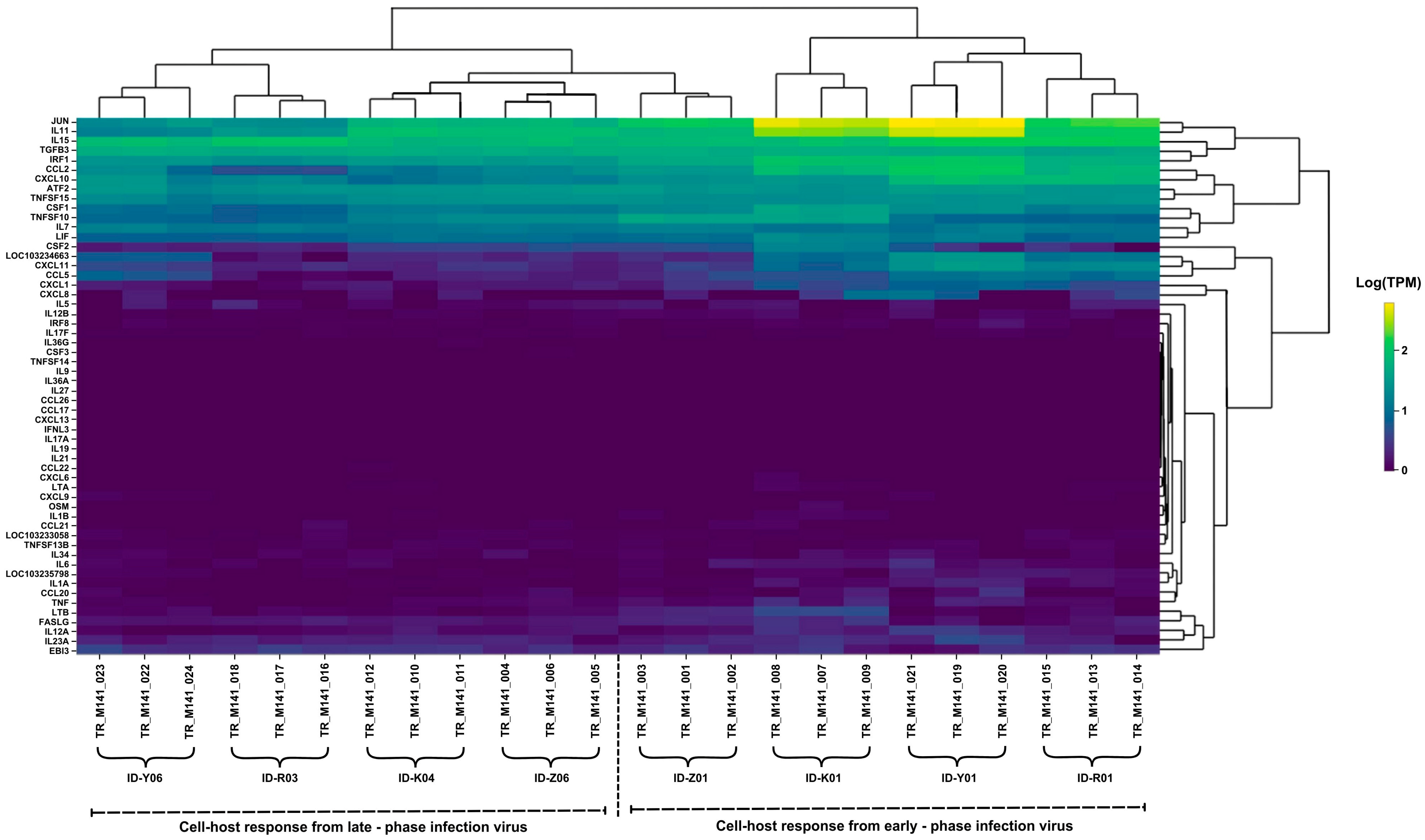
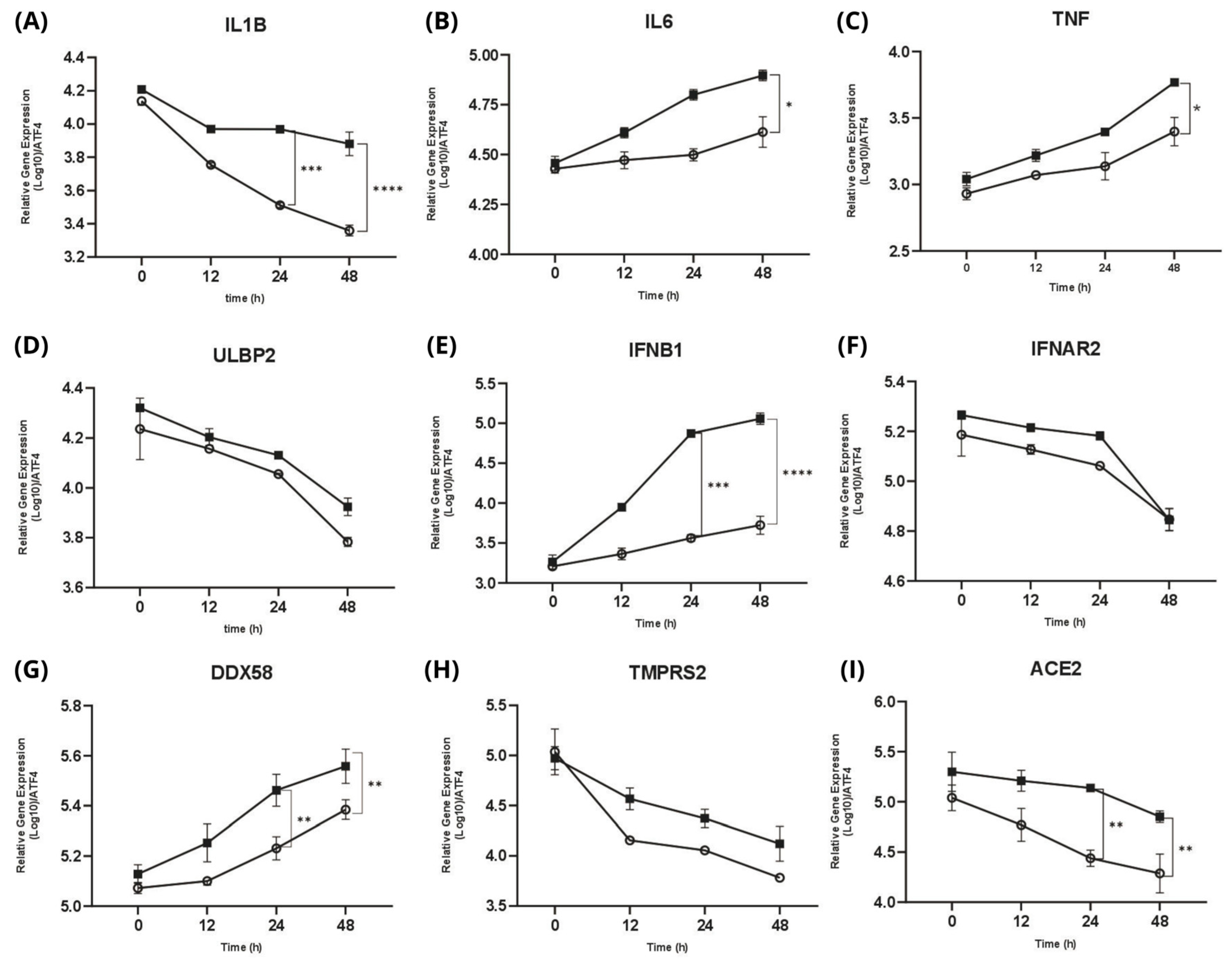
2.3. Comparison of Inflammatory Gene Expression Patterns in Host Cells
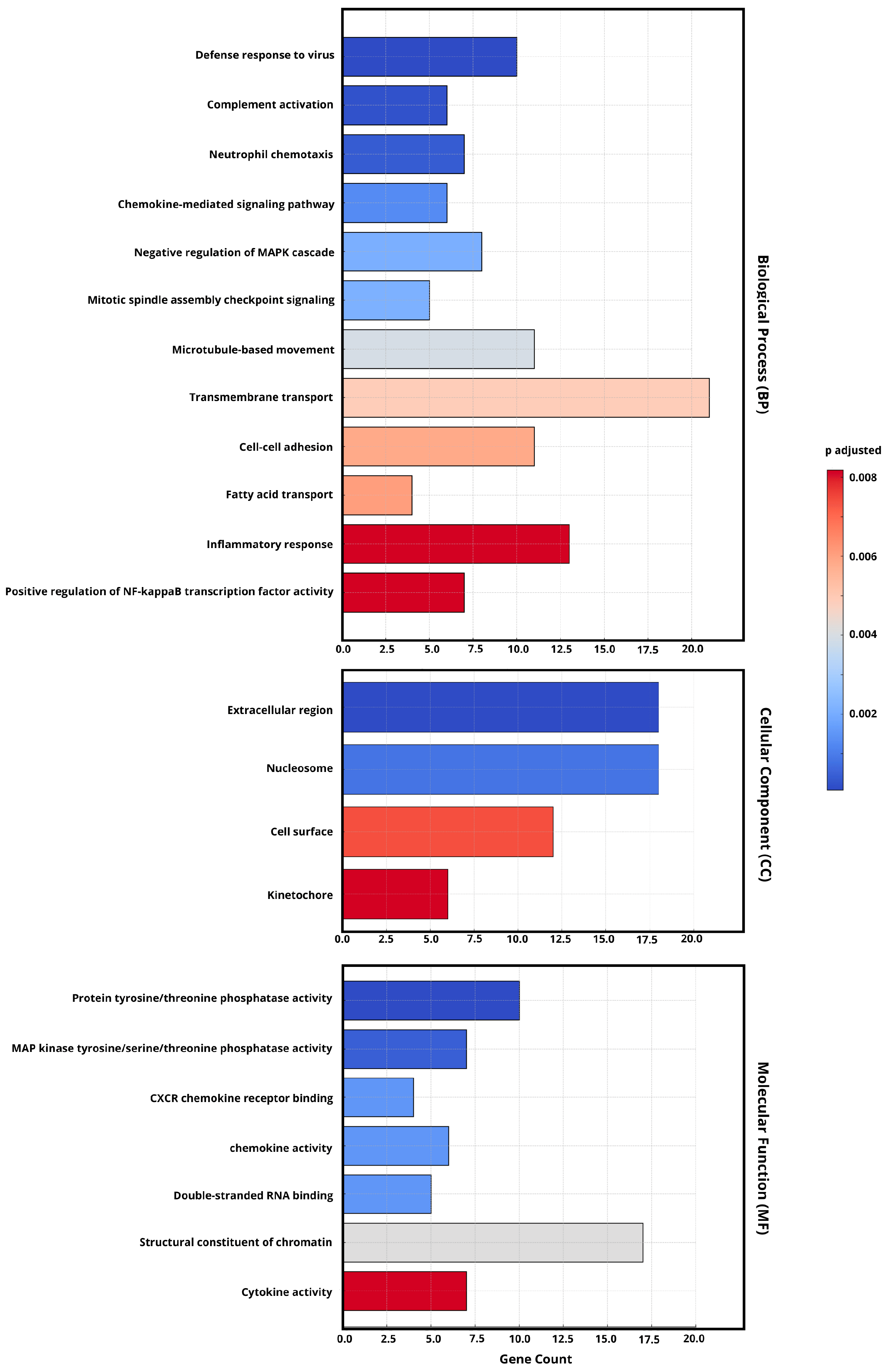
2.4. Gene Ontology (GO) Enrichment Analysis of DEGs
2.5. Gene Expression Profiling of Human Host Cell Response
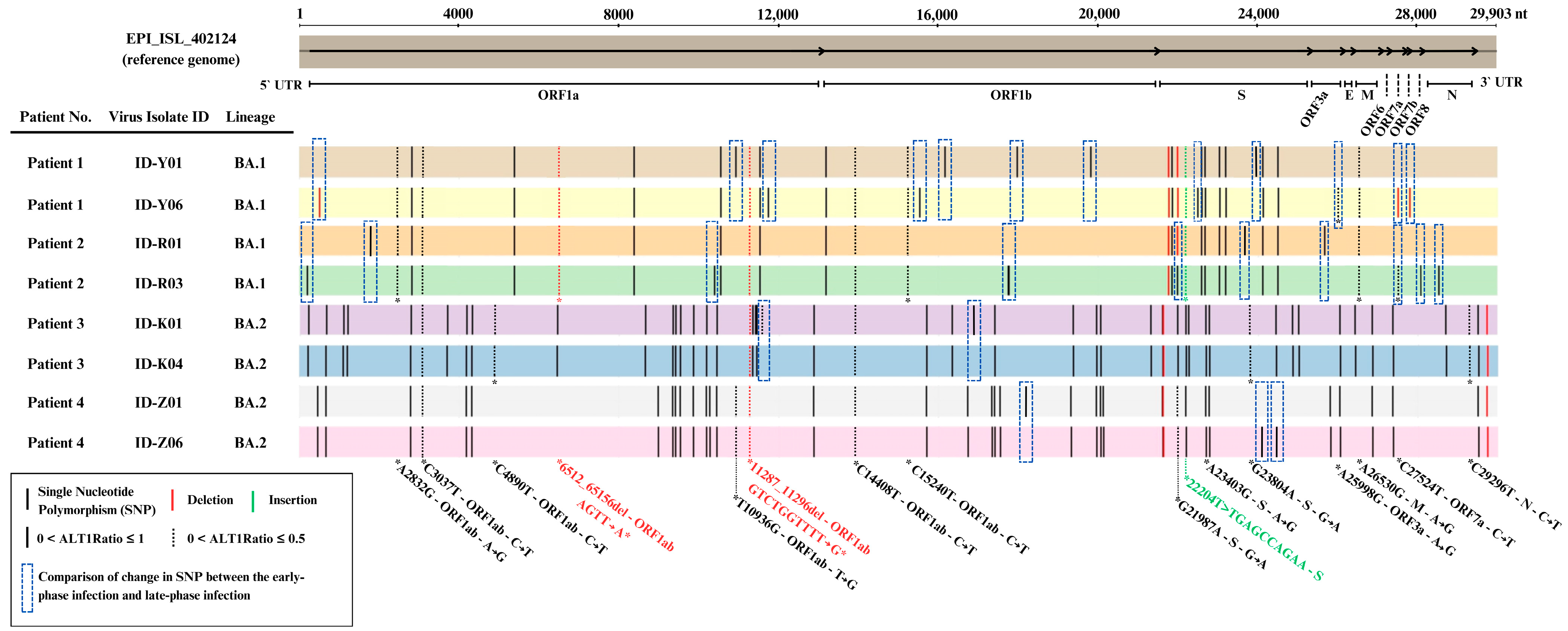
2.6. Distinct Genomic Analysis Between Early-Phase and Late-Phase Infection
3. Discussion
4. Materials and Methods
4.1. Clinical Sample Collection
4.2. SARS-CoV-2 Isolates Using VERO-E6/TMPRSS2 Cells
4.3. Calu-3 Human Airway Epithelial Cell Infection
4.4. Assessment of Viral Growth Kinetics
4.5. RNA Extraction and Viral Quantification qPCR
4.6. Viral Infectivity Assessment
4.7. Gene Expression Assay Using Quantitative Real-Time PCR
4.8. Next-Generation Sequencing
4.9. Genomic Sequencing Analysis
4.10. RNA Sequencing
4.11. RNA-Sequencing Analysis
4.12. Gene Ontology (GO) Enrichment Analysis
4.13. Statistical Analysis
4.14. Generative AI Disclosure
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baloch, S.; Baloch, M.A.; Zheng, T.; Pei, X. The Coronavirus Disease 2019 (COVID-19) Pandemic. Tohoku J. Exp. Med. 2020, 250, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Tiwari, S.; Deb, M.K.; Marty, J.L. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): A global pandemic and treatment strategies. Int. J. Antimicrob. Agents 2020, 56, 106054. [Google Scholar] [CrossRef] [PubMed]
- Nesteruk, I. Endemic characteristics of SARS-CoV-2 infection. Sci. Rep. 2023, 13, 14841. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Bussani, R.; Zentilin, L.; Correa, R.; Colliva, A.; Silvestri, F.; Zacchigna, S.; Collesi, C.; Giacca, M. Persistent SARS-CoV-2 infection in patients seemingly recovered from COVID-19. J. Pathol. 2023, 259, 254–263. [Google Scholar] [CrossRef]
- Cevik, M.; Tate, M.; Lloyd, O.; Maraolo, A.E.; Schafers, J.; Ho, A. SARS-CoV-2, SARS-CoV, and MERS-CoV viral load dynamics, duration of viral shedding, and infectiousness: A systematic review and meta-analysis. Lancet Microbe 2021, 2, e13–e22. [Google Scholar] [CrossRef] [PubMed]
- Sweis, J.J.G.; Alnaimat, F.; Esparza, V.; Prasad, S.; Azam, A.; Modi, Z.; Al-Awqati, M.; Jetanalin, P.; Sweis, N.J.; Ascoli, C.; et al. From Acute Infection to Prolonged Health Consequences: Understanding Health Disparities and Economic Implications in Long COVID Worldwide. Int. J. Environ. Res. Public Health 2024, 21, 325. [Google Scholar] [CrossRef]
- Desimmie, B.A.; Raru, Y.Y.; Awadh, H.M.; He, P.; Teka, S.; Willenburg, K.S. Insights into SARS-CoV-2 Persistence and Its Relevance. Viruses 2021, 13, 1025. [Google Scholar] [CrossRef]
- Machkovech, H.M.; Hahn, A.M.; Wang, J.G.; Grubaugh, N.D.; Halfman, P.J.; Johnson, M.C.; Lemieux, J.E.; O’Connor, H.; Piantadosi, A.; Wei, W.; et al. Persistent SARS-CoV-2 infection: Significance and implications. Lancet Infect. Dis. 2024, 24, E453–E462. [Google Scholar] [CrossRef]
- Huot, N.; Planchais, C.; Rosenbaum, P.; Contreras, V.; Jacquelin, B.; Petitdemange, C.; Lazzerini, M.; Beaumont, E.; Orta-Resendiz, A.; Reeves, R.K.; et al. SARS-CoV-2 viral persistence in lung alveolar macrophages is controlled by IFN-γ and NK cells. Nat. Immunol. 2023, 24, 2068–2079. [Google Scholar] [CrossRef]
- Wilkinson, S.A.J.; Richter, A.; Casey, A.; Osman, H.; Mirza, J.D.; Stockton, J.; Quick, J.; Ratcliffe, L.; Sparks, N.; Cumley, N.; et al. Recurrent SARS-CoV-2 mutations in immunodeficient patients. Virus Evol. 2022, 8, veac050. [Google Scholar] [CrossRef]
- Killigley, B.; Mann, A.J.; Kalinova, M.; Boyers, A.; Goonawardane, N.; Zhou, J.; Lindsell, K.; Hare, S.S.; Brown, J.; Frise, R.; et al. Safety, tolerability and viral kinetics during SARS-CoV-2 human challenge in young adults. Nat. Med. 2022, 28, 1031–1041. [Google Scholar] [CrossRef]
- Ghafari, M.; Hall, M.; Golubchik, T.; Ayoubkhani, D.; House, T.; Maclntyre-Cockett, G.; Fryer, H.R.; Thomson, L.; Nurtay, A.; Kemp, S.A.; et al. Prevalence of persistent SARS-CoV-2 in a large community surveillance study. Nature 2024, 626, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Wölfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Müller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ye, F.; Wu, A.; Yang, R.; Pan, M.; Sheng, J.; Zhu, W.; Mao, L.; Wang, M.; Xia, Z.; et al. Comparative Transcriptome Analysis Reveals the Intensive Early Stage Responses of Host Cells to SARS-CoV-2 Infection. Front. Microbiol. 2020, 11, 593857. [Google Scholar] [CrossRef]
- Peacock, T.P.; Brown, J.C.; Zhou, J.; Thakur, N.; Sukhova, K.; Newman, J.; Kugathasan, R.; Yan, A.W.C.; Furnon, W.; De Lorenzo, G.; et al. The altered entry pathway and antigenic distance of the SARS-CoV-2 Omicron variant map to separate domains of spike protein. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hui, K.P.Y.; Ho, J.C.W.; Cheung, M.-c.; Ng, K.-c.; Ching, R.H.H.; Lai, K.-l.; Kam, T.T.; Gu, H.; Sit, K.-Y.; Hsin, M.K.Y.; et al. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature 2022, 603, 715–720. [Google Scholar] [CrossRef]
- Chaguza, C.; Hahn, A.M.; Petrone, M.E.; Zhou, S.; Ferguson, D.; Breban, M.I.; Pham, K.; Peña-Hernández, M.A.; Castaldi, C.; Hill, V. Accelerated SARS-CoV-2 intrahost evolution leading to distinct genotypes during chronic infection. Cell Rep. Med. 2023, 4, 100943. [Google Scholar] [CrossRef] [PubMed]
- Voloch, C.M.; da Silva Francisco, R., Jr.; de Almeida, L.G.P.; Brustolini, O.J.; Cardoso, C.C.; Gerber, A.L.; de C Guimarães, A.P.; de Carvalho Leitão, I.; Mariani, D.; Ota, V.K.; et al. Intra-host evolution during SARS-CoV-2 prolonged infection. Virus Evol. 2021, 7, veab078. [Google Scholar] [CrossRef]
- Tripathy, A.S.; Vishwakarma, S.; Trimbake, D.; Gurav, Y.K.; Potdar, V.A.; Mokashi, N.D.; Patsute, S.D.; Kaushal, H.; Choudhary, M.L.; Tilekar, B.N.; et al. Pro-inflammatory CXCL-10, TNF-α, IL-1β, and IL-6: Biomarkers of SARS-CoV-2 infection. Arch. Virol. 2021, 166, 3301–3310. [Google Scholar] [CrossRef]
- Liu, B.; Li, M.; Zhou, Z.; Guan, X.; Xiang, Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J. Autoimmun. 2020, 111, 102452. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-E.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Lowery, S.A.; Sariol, A.; Perlman, S. Innate immune and inflammatory responses to SARS-CoV-2: Implications for COVID-19. Cell Host Microbe 2021, 29, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Iwasaki, A. Type I and Type III Interferons—Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef]
- Gudowska-Sawczuk, M.; Mroczko, B. The Role of Nuclear Factor Kappa B (NF-κB) in Development and Treatment of COVID-19: Review. Int. J. Mol. Sci. 2022, 23, 5283. [Google Scholar] [CrossRef]
- Shah, V.K.; Firmal, P.; Alam, A.; Ganguly, D.; Chattopadhyay, S. Overview of Immune Response During SARS-CoV-2 Infection: Lessons From the Past. Front. Immunol. 2020, 11, 1949. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Moore, J.B.; June, C.H. Cytokine release syndrome in severe COVID-19. Science 2020, 368, 473–474. [Google Scholar] [CrossRef]
- Hashizume, M. Outlook of IL-6 signaling blockade for COVID-19 pneumonia. Inflamm. Regen. 2020, 40, 24. [Google Scholar] [CrossRef]
- Kouwaki, T.; Nishimura, T.; Wang, G.; Oshiumi, H. RIG-I-Like Receptor-Mediated Recognition of Viral Genomic RNA of Severe Acute Respiratory Syndrome Coronavirus-2 and Viral Escape From the Host Innate Immune Responses. Front. Immunol. 2021, 12, 700926. [Google Scholar] [CrossRef] [PubMed]
- Busnadiego, I.; Fernbach, S.; Pohl, M.O.; Karakus, U.; Huber, M.; Trkola, A.; Stertz, S.; Hale, B.G. Antiviral Activity of Type I, II, and III Interferons Counterbalances ACE2 Inducibility and Restricts SARS-CoV-2. mBio 2020, 11, e01928-20. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Immunotherapeutic Implications of Il-6 Blockade for Cytokine Storm. Immunotherapy 2016, 8, 959–970. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.-E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000. [Google Scholar] [CrossRef]
- Cobar, O.; Cóbar, S. SARS-CoV-2 Orf1ab Genome Mutations, the Driving Force for Virus Pathogenicity. Med. Res. Arch. 2013, 11. [Google Scholar] [CrossRef]
- Magazine, N.; Zhang, T.; Wu, Y.; McGee, M.C.; Veggiani, G.; Huang, W. Mutations and Evolution of the SARS-CoV-2 Spike Protein. Viruses 2022, 14, 640. [Google Scholar] [CrossRef]
- Kumar, S.; Karuppanan, K.; Subramaniam, G. Omicron (BA.1) and sub-variants (BA.1.1, BA.2, and BA.3) of SARS-CoV-2 spike infectivity and pathogenicity: A comparative sequence and structural-based computational assessment. J. Med. Virol. 2022, 94, 4780–4791. [Google Scholar] [CrossRef]
- Heyer, A.; Günther, T.; Robitaille, A.; Lütgehetmann, M.; Addo, M.M.; Jarczak, D.; Kluge, S.; Aepfelbacher, M.; zur Wiesch, J.S.; Fischer, N.; et al. Remdesivir-induced emergence of SARS-CoV2 variants in patients with prolonged infection. Cell Rep. Med. 2022, 3, 100735. [Google Scholar] [CrossRef]
- Szemiel, A.M.; Mertis, A.; Orton, R.J.; MacLean, O.A.; Pinto, R.M.; Wickenhangen, A.; Lieber, G.; Turnbull, M.L.; Wang, S.; Furnon, W.; et al. In vitro selection of Remdesivir resistance suggests evolutionary predictability of SARS-CoV-2. PLoS Pathog. 2021, 17, e1009929. [Google Scholar] [CrossRef]
- Wang, L.; Guzman, M.; Muñoz-Santos, D.; Honrubia, J.S.; Ripoll-Gomez, J.; Delgado, R.; Sola, I.; Enjuanes, L.; Zuñiga, S. Cell type dependent stability and virulence of a recombinant SARS-CoV-2, and engineering of a propagation deficient RNA replicon to analyze virus RNA synthesis. Front. Cell Infect. Microbiol. 2023, 13, 1268227. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Sakai-Tagawa, Y.; Koga, M.; Akasaka, O.; Nakachi, I.; Koh, H.; Maeda, K.; Adachi, E.; Saito, M.; Nagai, H.; et al. Comparison of Rapid Antigen Tests for COVID-19. Viruses 2020, 12, 1420. [Google Scholar] [CrossRef]
- Oba, J.; Taniguchi, H.; Sato, M.; Takamatsu, R.; Morikawa, S.; Nakagawa, T.; Takaishi, H.; Saya, H.; Matsuo, K.; Nishihara, H. RT-PCR Screening Tests for SARS-CoV-2 with Saliva Samples in Asymptomatic People: Strategy to Maintain Social and Economic Activities while Reducing the Risk of Spreading the Virus. Keio. J. Med. 2021, 70, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Mautner, L.; Hoyos, M.; Dangel, A.; Berger, C.; Ehrhardt, A.; Baiker, A. Replication kinetics and infectivity of SARS-CoV-2 variants of concern in common cell culture models. Virol. J. 2022, 19, 76. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Nao, N.; Matsuyama, S.; Takeda, M.; Kageyama, T.; Shirabe, K.; Shinomiya, H. Manual for the Detection of Pathogen 2019-nCoV Ver.2.6. 2020. Available online: https://id-info.jihs.go.jp/diseases/sa/covid-19/2019-nCoVmanual20200217-en.pdf (accessed on 20 April 2025).
- Keiser, P.T.; Anantpadma, M.; Staples, H.; Carrion, R.; Davey, R.A. Automation of Infectious Focus Assay for Determination of Filovirus Titers and Direct Comparison to Plaque and TCID50 Assays. Microorganisms 2021, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Cardona, M.I.; Flórez-Álvarez, L.; Gómez-Gallego, D.M.; Moncada-Díaz, M.J.; Hernandez, J.C.; Díaz, F.; Rugeles, M.T.; Aguilar-Jiménez, W.; Zapata, W. Comparison among plaque assay, tissue culture infectious dose (TCID) and real-time RT-PCR for SARS-CoV-2 variants quantification. Iran. J. Microbiol. 2022, 14, 291–299. [Google Scholar] [CrossRef]
- Lei, C.; Yang, J.; Hu, J.; Sun, X. On the Calculation of TCID50 for Quantitation of Virus Infectivity. Virol. Sin. 2021, 36, 141–144. [Google Scholar] [CrossRef]
- Itokawa, K.; Sekizuka, T.; Hashino, M.; Kogino, A.; Tanaka, R.; Eto, S. New Coronavirus Genome Decoding Protocol (Edited by Qiagen QiaSEQ FX) v1.4. Available online: https://www.niid.go.jp/niid/images/lab-manual/SARS-CoV2_genome_analysis_manual_QIASEQFX_ver_1_4_220127.pdf (accessed on 4 December 2024).
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Age/Gender | Duration of Infection | Comorbidities | Symptoms | Vaccination Status | Immuno-Suppressive Medication | Complication | ICU (Yes/No) | Treatment Drug (D/R/M) * | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | 79/M | 57 Days | Esophageal cancer, rheumatoid arthritis, pneumonia | Fever | No | - | Pneumonia. ARDS | No | D, R (total 12 days) | Discharged (transferred to previous hospital for rehab) |
| Patient 2 | 37/F | 37 Days | Pregnancy | Fever, cough, nausea | Vaccinated 2x | - | - | No | No | Discharged |
| Patient 3 | 66/F | 46 Days | Lung transplant, pneumonia, CKD, Bowen’s disease | Fever, sore throat | No | Prednisolone, mycophenolate mofetil | pneumonia | No | M (total 5 days) | Discharged |
| Patient 4 | 76/M | 58 Days | Lung cancer, hypertension | Fever | Unknown | - | - | No | R (total 5 days) | Discharged |
| No. | Gene Region | Protein | Nucleotide | Type | Change | ||
|---|---|---|---|---|---|---|---|
| Start | End | Reference | Alternate | ||||
| 1 | ORF1ab | Nsp2 | 465 | 465 | SNP | C | T |
| 2 | Nsp2 | 515 | 518 | DEL | GTTA | G | |
| 3 | Nsp3 | 2485 | 2485 | SNP | C | T | |
| 4 | Nsp12 | 10,401 | 10,401 | SNP | C | T | |
| 5 | Nsp13 | 10,936 | 10,936 | SNP | T | G | |
| 6 | Nsp13 | 11,414 | 11,414 | SNP | C | T | |
| 7 | Nsp13 | 11,750 | 11,750 | SNP | C | T | |
| 8 | Nsp14 | 15,526 | 15,526 | SNP | A | G | |
| 9 | Nsp15 | 16,178 | 16,178 | SNP | C | T | |
| 10 | Nsp16 | 17,762 | 17,762 | SNP | C | T | |
| 11 | Nsp16 | 17,766 | 17,766 | SNP | C | T | |
| 12 | Nsp16 | 17,977 | 17,977 | SNP | C | T | |
| 13 | Nsp16 | 19,380 | 19,380 | SNP | C | T | |
| 14 | Nsp16 | 19,824 | 19,824 | SNP | G | A | |
| 15 | S | Spike | 21,986 | 21,995 | DEL | GGTGTTTATT | G |
| 16 | Spike | 21,987 | 21,987 | SNP | G | A | |
| 17 | Spike | 22,498 | 22,498 | SNP | C | T | |
| 18 | Spike | 23,804 | 23,804 | SNP | G | A | |
| 19 | Spike | 24,062 | 24,062 | SNP | A | T | |
| 20 | Spike | 24,452 | 24,452 | SNP | A | G | |
| 21 | ORF3a | Ap3a | 25,677 | 25,677 | SNP | G | T |
| 22 | Ap3a | 25,998 | 25,998 | SNP | A | G | |
| 23 | ORF7a | Ap7a | 27,505 | 27,510 | DEL | GGAACA | G |
| 24 | Ap7a | 27,524 | 27,524 | SNP | C | T | |
| 25 | ORF7b | Ap7b | 27,794 | 27,797 | DEL | TTTA | T |
| 26 | ORF8 | Ap8 | 28,077 | 28,077 | SNP | G | T |
| 27 | N | Nucleoprotein | 28,539 | 28,539 | SNP | G | T |
| Name | Sequence (5′-3′) |
|---|---|
| 2019 nCoV N F2 primer | AAATTTTGGGGACCAGGAAC |
| 2019 nCoV N R2 primer | TGGCAGCTGTGTAGGTCAAC |
| 2019 nCoV N P2 probe | FAM-ATGTCGCGCATTGGCATGGA-QSY |
| Gene Expression | Primer Sequence Number | Base Pairs |
|---|---|---|
| ATF4 | Hs00909569_g1 | 68 |
| TMPRSS2 | Hs01122322_m1 | 62 |
| ACE2 | Hs01085333_m1 | 141 |
| DDX58 | Hs01061436_m1 | 65 |
| IFNB1 | Hs01077958_s1 | 73 |
| IFNAR2 | Hs01022059_m1 | 88 |
| IL6 | Hs00174131_m1 | 95 |
| IL1B | Hs01555410_m1 | 91 |
| TNF | Hs00174128_m1 | 80 |
| ULBP2 | Hs01127964_m1 | 93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maarif, A.S.; Nishikawa, Y.; Takata, M.; Kanai, K.; Riyandani, E.; Mukuda, K.; Mimura, M.; Yamaguchi, K.; Kato, H.; Okamoto, R.; et al. Host–Virus Interface in Persistent SARS-CoV-2 Infections: Viral Characteristic Evolution and Gene Expression Profiling Analysis. Int. J. Mol. Sci. 2025, 26, 6221. https://doi.org/10.3390/ijms26136221
Maarif AS, Nishikawa Y, Takata M, Kanai K, Riyandani E, Mukuda K, Mimura M, Yamaguchi K, Kato H, Okamoto R, et al. Host–Virus Interface in Persistent SARS-CoV-2 Infections: Viral Characteristic Evolution and Gene Expression Profiling Analysis. International Journal of Molecular Sciences. 2025; 26(13):6221. https://doi.org/10.3390/ijms26136221
Chicago/Turabian StyleMaarif, Athok Shofiudin, Yukari Nishikawa, Miyako Takata, Kyosuke Kanai, Edo Riyandani, Kengo Mukuda, Momone Mimura, Kosuke Yamaguchi, Hiroyuki Kato, Ryo Okamoto, and et al. 2025. "Host–Virus Interface in Persistent SARS-CoV-2 Infections: Viral Characteristic Evolution and Gene Expression Profiling Analysis" International Journal of Molecular Sciences 26, no. 13: 6221. https://doi.org/10.3390/ijms26136221
APA StyleMaarif, A. S., Nishikawa, Y., Takata, M., Kanai, K., Riyandani, E., Mukuda, K., Mimura, M., Yamaguchi, K., Kato, H., Okamoto, R., Okada, K., Kitaura, T., Nakamoto, M., Yamasaki, A., Kageyama, S., & Chikumi, H. (2025). Host–Virus Interface in Persistent SARS-CoV-2 Infections: Viral Characteristic Evolution and Gene Expression Profiling Analysis. International Journal of Molecular Sciences, 26(13), 6221. https://doi.org/10.3390/ijms26136221