
Expanding the Genomic Landscape of HBOC and Cancer Risk Among Mutation Carriers

 ,
,
Abstract
1. Background
2. Results
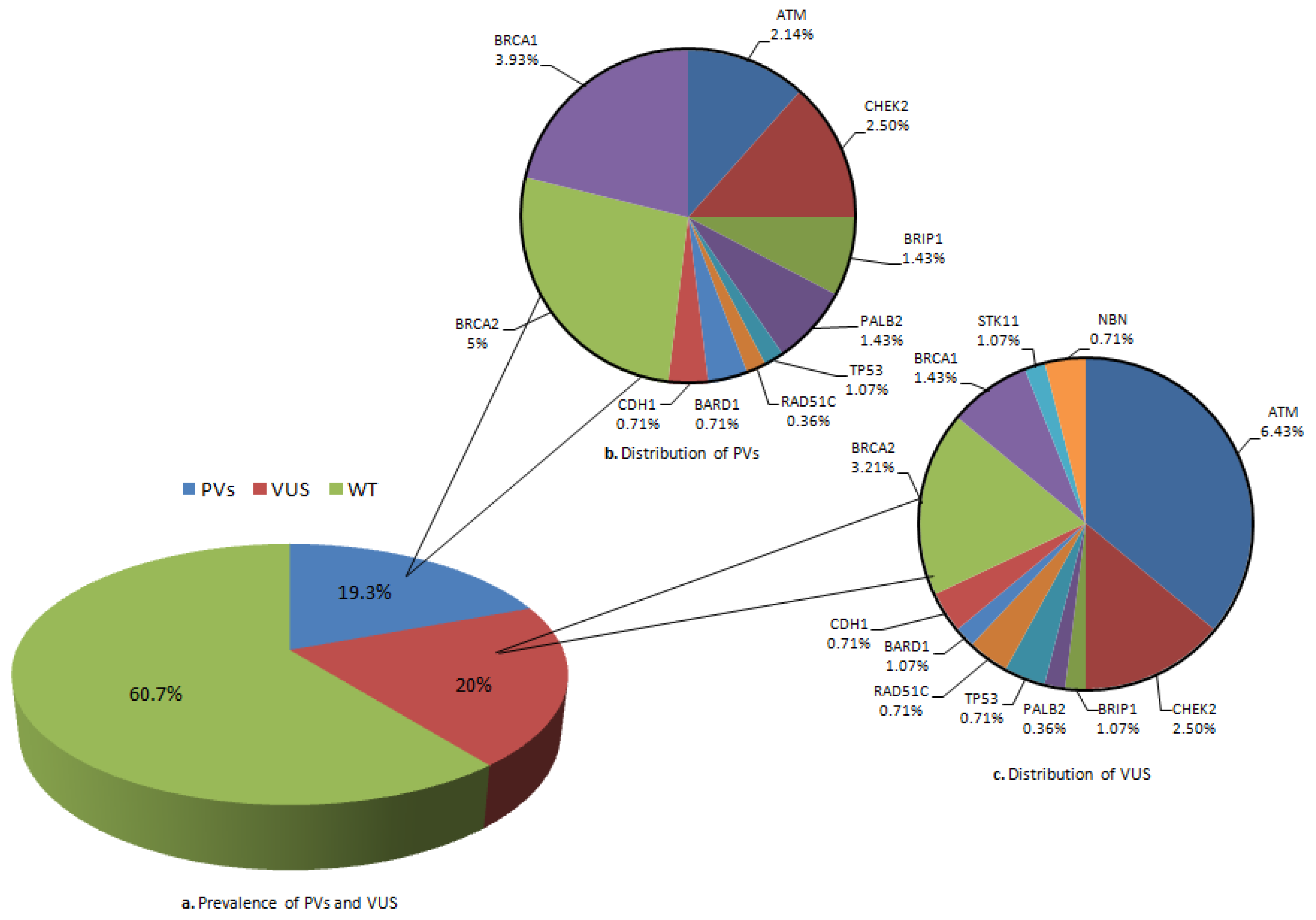
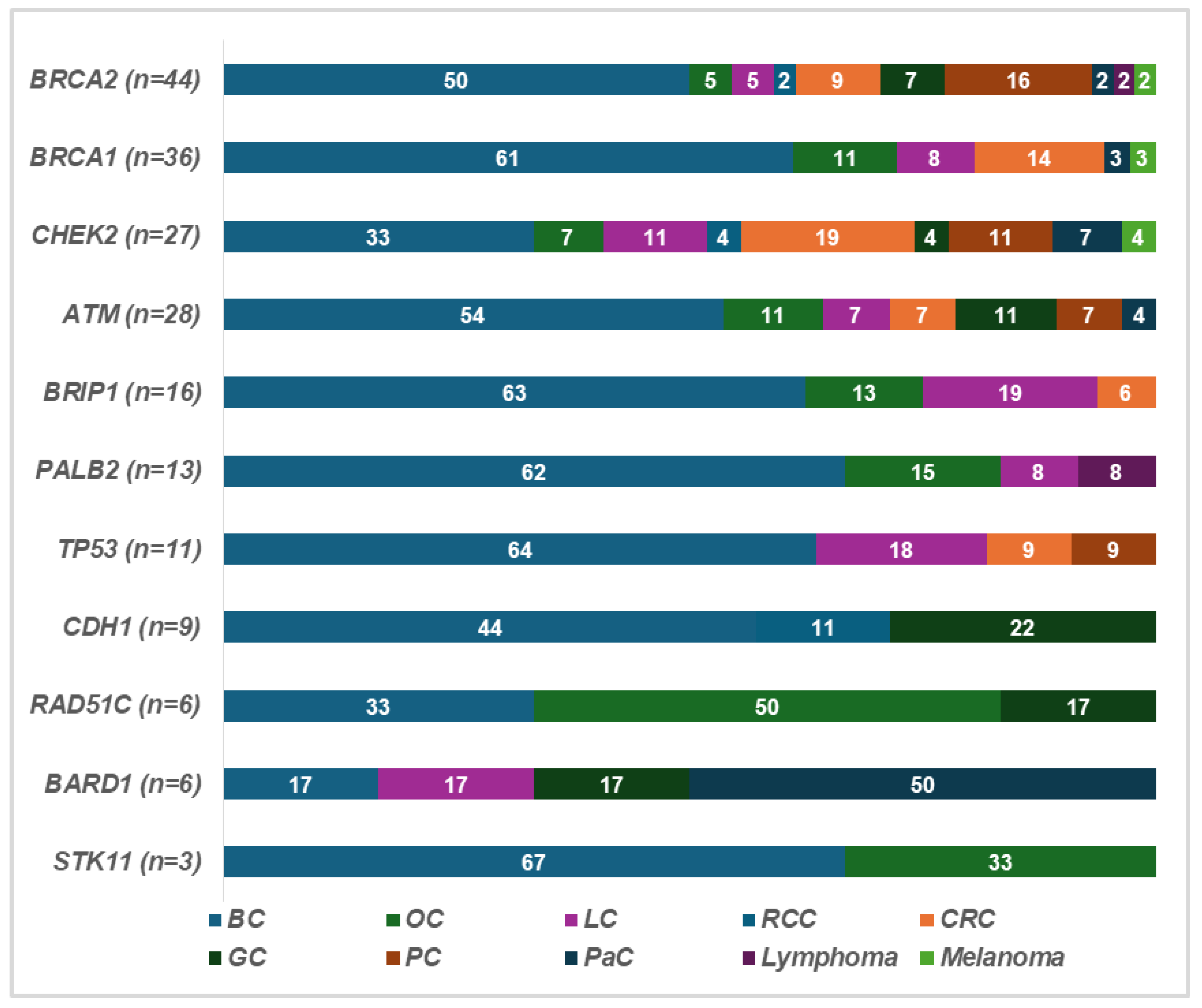
2.1. Mutation Rate and Affected Genes
2.2. In Silico Analysis
2.3. Clinical Features of Patients and Families
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Genetic Testing
4.3. In Silico Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, Y.C.; Lee, Y.L.; Li, C.Y. BRCA Genes and Related Cancers: A Meta-Analysis from Epidemiological Cohort Studies. Medicina 2021, 57, 905. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vietri, M.T.; Caliendo, G.; D’Elia, G.; Resse, M.; Casamassimi, A.; Minucci, P.B.; Dello Ioio, C.; Cioffi, M.; Molinari, A.M. Five Italian Families with Two Mutations in BRCA Genes. Genes 2020, 11, 1451. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nyberg, T.; Frost, D.; Barrowdale, D.; Evans, D.G.; Bancroft, E.; Adlard, J.; Ahmed, M.; Barwell, J.; Brady, A.F.; Brewer, C.; et al. Prostate Cancer Risks for Male BRCA1 and BRCA2 Mutation Carriers: A Prospective Cohort Study. Eur. Urol. 2020, 77, 24–35. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Katona, B.W.; Lubinski, J.; Pal, T.; Huzarski, T.; Foulkes, W.D.; Moller, P.; Eisen, A.; Randall Armel, S.; Neuhausen, S.L.; Raj, R.; et al. The incidence of pancreatic cancer in women with a BRCA1 or BRCA2 mutation. Cancer 2025, 131, e35666. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vietri, M.T.; D’Elia, G.; Caliendo, G.; Albanese, L.; Signoriello, G.; Napoli, C.; Molinari, A.M. Pancreatic Cancer with Mutation in BRCA1/2, MLH1, and APC Genes: Phenotype Correlation and Detection of a Novel Germline BRCA2 Mutation. Genes 2022, 13, 321. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thompson, D.; Easton, D.F.; Breast Cancer Linkage Consortium. Cancer Incidence in BRCA1 mutation carriers. J. Natl. Cancer Inst. 2002, 94, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Kadouri, L.; Hubert, A.; Rotenberg, Y.; Hamburger, T.; Sagi, M.; Nechushtan, C.; Abeliovich, D.; Peretz, T. Cancer risks in carriers of the BRCA1/2 Ashkenazi founder mutations. J. Med. Genet. 2007, 44, 467–471. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Navsaria, L.J.; Haas, A.M.; Corredor, J.L.; Arun, B.K.; Giordano, S.H.; Nead, K.T.; Wehner, M.R. Skin cancer risk in patients with BRCA mutations. JAAD Int. 2024, 17, 175–177. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zheng, F.; Zhang, Y.; Chen, S.; Weng, X.; Rao, Y.; Fang, H. Mechanism and current progress of Poly ADP-ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed. Pharmacother. 2020, 123, 109661. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkäs, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014, 371, 497–506. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Seca, M.; Narod, S.A. Breast cancer and ATM mutations: Treatment implications. Hered. Cancer Clin. Pract. 2024, 22, 26. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abdel-Razeq, H. Surgical options for patients with early-stage breast cancer and pathogenic germline variants: An oncologist perspectives. Front. Oncol. 2023, 13, 1265197. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mundt, E.; Mabey, B.; Rainville, I.; Ricker, C.; Singh, N.; Gardiner, A.; Manley, S.; Slavin, T., Jr. Breast and colorectal cancer risks among over 6,000 CHEK2 pathogenic variant carriers: A comparison of missense versus truncating variants. Cancer Genet. 2023, 278–279, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Amirifar, P.; Ranjouri, M.R.; Pashangzadeh, S.; Lavin, M.; Yazdani, R.; Moeini Shad, T.; Mehrmohamadi, M.; Salami, F.; Delavari, S.; Moamer, S.; et al. The spectrum of ATM gene mutations in Iranian patients with ataxia-telangiectasia. Pediatr. Allergy Immunol. 2021, 32, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; Caliendo, G.; Schiano, C.; Casamassimi, A.; Molinari, A.M.; Napoli, C.; Cioffi, M. Analysis of PALB2 in a cohort of Italian breast cancer patients: Identification of a novel PALB2 truncating mutation. Fam. Cancer 2015, 14, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Anaclerio, F.; Pilenzi, L.; Dell’Elice, A.; Ferrante, R.; Grossi, S.; Ferlito, L.M.; Marinelli, C.; Gildetti, S.; Calabrese, G.; Stuppia, L.; et al. Clinical usefulness of NGS multi-gene panel testing in hereditary cancer analysis. Front. Genet. 2023, 14, 1060504. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nagy, P.; Papp, J.; Grolmusz, V.K.; Bozsik, A.; Pócza, T.; Oláh, E.; Patócs, A.; Butz, H. Comprehensive Clinical Genetics, Molecular and Pathological Evaluation Efficiently Assist Diagnostics and Therapy Selection in Breast Cancer Patients with Hereditary Genetic Background. Int. J. Mol. Sci. 2024, 25, 12546. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rolfes, M.; Borde, J.; Möllenhoff, K.; Kayali, M.; Ernst, C.; Gehrig, A.; Sutter, C.; Ramser, J.; Niederacher, D.; Horváth, J.; et al. Prevalence of Cancer Predisposition Germline Variants in Male Breast Cancer Patients: Results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancers 2022, 14, 3292. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cruellas, M.; Papakonstantinou, A.; López-Fernández, A.; Castillo, E.; Matito, J.; Gómez, M.; Rezqallah, A.; Vega, S.; Navarro, V.; Torres, M.; et al. Identifying germline pathogenic variants in breast cancer using tumor sequencing. Breast 2025, 81, 104439. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hanson, H.; Kulkarni, A.; Loong, L.; Kavanaugh, G.; Torr, B.; Allen, S.; Ahmed, M.; Antoniou, A.C.; Cleaver, R.; Dabir, T.; et al. UK consensus recommendations for clinical management of cancer risk for women with germline pathogenic variants in cancer predisposition genes: RAD51C, RAD51D, BRIP1 and PALB2. J. Med. Genet. 2022, 60, 417–429. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hall, M.J.; Bernhisel, R.; Hughes, E.; Larson, K.; Rosenthal, E.T.; Singh, N.A.; Lancaster, J.M.; Kurian, A.W. Germline Pathogenic Variants in the Ataxia Telangiectasia Mutated (ATM) Gene are Associated with High and Moderate Risks for Multiple Cancers. Cancer Prev. Res. 2021, 14, 433–440. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Laitman, Y.; Nielsen, S.M.; Bernstein-Molho, R.; Heald, B.; Hatchell, K.E.; Esplin, E.D.; Friedman, E. Cancer risks associated with heterozygous ATM loss of function and missense pathogenic variants based on multigene panel analysis. Breast Cancer Res. Treat. 2022, 196, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Rafnar, T.; Sigurjonsdottir, G.R.; Stacey, S.N.; Halldorsson, G.; Sulem, P.; Pardo, L.M.; Helgason, H.; Sigurdsson, S.T.; Gudjonsson, T.; Tryggvadottir, L.; et al. Association of BRCA2 K3326* with Small Cell Lung Cancer and Squamous Cell Cancer of the Skin. J. Natl. Cancer Inst. 2018, 110, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Higgs, J.E.; Harkness, E.F.; Bowers, N.L.; Howard, E.; Wallace, A.J.; Lalloo, F.; Newman, W.G.; Evans, D.G. The BRCA2 polymorphic stop codon: Stuff or nonsense? J. Med. Genet. 2015, 52, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Buckley, K.H.; Niccum, B.A.; Maxwell, K.N.; Katona, B.W. Gastric Cancer Risk and Pathogenesis in BRCA1 and BRCA2 Carriers. Cancers 2022, 14, 5953. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ryan, C.E.; Fasaye, G.A.; Gallanis, A.F.; Gamble, L.A.; McClelland, P.H.; Duemler, A.; Samaranayake, S.G.; Blakely, A.M.; Drogan, C.M.; Kingham, K.; et al. Germline CDH1 Variants and Lifetime Cancer Risk. JAMA 2024, 332, 722–729. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hodan, R.; Gupta, S.; Weiss, J.M.; Axell, L.; Burke, C.A.; Chen, L.M.; Chung, D.C.; Clayback, K.M.; Felder, S.; Foda, Z.; et al. Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, and Gastric, Version 3.2024, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2024, 22, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, Q.S.; Wang, Y.J. The CHEK2 I157T variant and colorectal cancer susceptibility: A systematic review and meta-analysis. Asian Pac. J. Cancer Prev. 2012, 13, 2051–2055. [Google Scholar] [CrossRef] [PubMed]
- Randall, M.P.; Egolf, L.E.; Vaksman, Z.; Samanta, M.; Tsang, M.; Groff, D.; Evans, J.P.; Rokita, J.L.; Layeghifard, M.; Shlien, A.; et al. BARD1 germline variants induce haploinsufficiency and DNA repair defects in neuroblastoma. bioRxiv 2023. Reprinted in: J. Natl. Cancer Inst. 2024, 116, 138–148. https://doi.org/10.1093/jnci/djad182. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Weber-Lassalle, N.; Borde, J.; Weber-Lassalle, K.; Horváth, J.; Niederacher, D.; Arnold, N.; Kaulfuß, S.; Ernst, C.; Paul, V.G.; Honisch, E.; et al. Germline loss-of-function variants in the BARD1 gene are associated with early-onset familial breast cancer but not ovarian cancer. Breast Cancer Res. 2019, 21, 55. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Novelli, F.; Yoshikawa, Y.; Vitto, V.A.M.; Modesti, L.; Minaai, M.; Pastorino, S.; Emi, M.; Kim, J.H.; Kricek, F.; Bai, F.; et al. Germline BARD1 variants predispose to mesothelioma by impairing DNA repair and calcium signaling. Proc. Natl. Acad. Sci. USA 2024, 121, e2405231121. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zeng, Y.; Arisa, O.; Peer, C.J.; Fojo, A.; Figg, W.D. PARP inhibitors: A review of the pharmacology, pharmacokinetics, and pharmacogenetics. Semin. Oncol. 2024, 51, 19–24. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gruber, J.J.; Afghahi, A.; Timms, K.; DeWees, A.; Gross, W.; Aushev, V.N.; Wu, H.T.; Balcioglu, M.; Sethi, H.; Scott, D.; et al. A phase II study of talazoparib monotherapy in patients with wild-type BRCA1 and BRCA2 with a mutation in other homologous recombination genes. Nat. Cancer 2022, 3, 1181–1191. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264, Erratum in: Lancet Oncol. 2022, 23, e207. https://doi.org/10.1016/S1470-2045(22)00207-8.Erratum in: Lancet Oncol. 2022, 23, e249. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.; Blue, K.; Sommerer, K.; Shah, A.; Bottiglieri, S.; Del Cueto, A.; Berry, D.K.; Ho, T.T.; Hicks, J.K.; Kim, D.W. PARP Inhibitors in Pancreatic Cancer with Homologous Recombination Repair Gene Mutations: A Single-Institution Experience. Cancers 2024, 16, 3447. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mateo, J.; de Bono, J.S.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Agarwal, N.; Olmos, D.; Thiery-Vuillemin, A.; et al. Olaparib for the Treatment of Patients with Metastatic Castration-Resistant Prostate Cancer and Alterations in BRCA1 and/or BRCA2 in the PROfound Trial. J. Clin. Oncol. 2024, 42, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Scher, H.I.; Sandhu, S.; Efstathiou, E.; Lara, P.N., Jr.; Yu, E.Y.; George, D.J.; Chi, K.N.; Saad, F.; Ståhl, O.; et al. Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2022, 23, 362–373. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Joris, S.; Denys, H.; Collignon, J.; Rasschaert, M.; T’Kint de Roodenbeke, D.; Duhoux, F.P.; Canon, J.L.; Tejpar, S.; Mebis, J.; Decoster, L.; et al. Efficacy of olaparib in advanced cancers with germline or somatic mutations in BRCA1, BRCA2, CHEK2 and ATM, a Belgian Precision tumor-agnostic phase II study. ESMO Open 2023, 8, 102041. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nakamura, K.; Aimono, E.; Tanishima, S.; Imai, M.; Nagatsuma, A.K.; Hayashi, H.; Yoshimura, Y.; Nakayama, K.; Kyo, S.; Nishihara, H. Olaparib Monotherapy for BRIP1-Mutated High-Grade Serous Endometrial Cancer. JCO Precis. Oncol. 2020, 4, 283–290. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Khalid, A.B.; Fountzilas, C.; Burney, H.N.; Mamdani, H.; Schneider, B.P.; Fausel, C.; Perkins, S.M.; Jalal, S. A phase II study evaluating safety and efficacy of niraparib in patients with previously treated homologous recombination defective metastatic esophageal/gastroesophageal junction/proximal gastric adenocarcinoma. Front. Oncol. 2024, 14, 1435056. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Italian Society of Human Genetic. Linee di Indirizzo Sull’analisi dei Geni BRCA1 e BRCA2 in Ambito Clinico: Criteri di Accesso al Test, Aggiornamento Sulle Piattaforme Diagnostiche e Interpretazione del Test Somatico; Italian Society of Human Genetic: Candiolo, Italy, 30 March 2021; Available online: https://sigu.net/categoria-documenti/linee-guida/page/3/ (accessed on 6 May 2025).
- Associazione Italiana di Oncologia Medica. Raccomandazioni per L’implementazione del Test BRCA Predittivo E Preventivo nei Tumori Della Mammella, Dell’ovaio, Del Pancreas e Della Prostata; Associazione Italiana di Oncologia Medica: Milan, Italy, 2021; Available online: https://www.aiom.it/linee-guida-aiom/ (accessed on 6 May 2025).
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V.; et al. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vietri, M.T.; D’Elia, G.; Caliendo, G.; Casamassimi, A.; Federico, A.; Passariello, L.; Cioffi, M.; Molinari, A.M. Prevalence of mutations in BRCA and MMR genes in patients affected with hereditary endometrial cancer. Med Oncol. 2021, 38, 13. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tian, Y.; Pesaran, T.; Chamberlin, A.; Fenwick, R.B.; Li, S.; Gau, C.L.; Chao, E.C.; Lu, H.M.; Black, M.H.; Qian, D. REVEL and BayesDel outperform other in silico meta-predictors for clinical variant classification. Sci. Rep. 2019, 9, 12752. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

{kind=link}
{kind=link}
| ID | Sex (M/F) | Age at Testing | Clinical Phenotype and Age at Diagnosis | Mutation Type Gene/Name and dbSNP ID | Mutation in Other Genes | Family History | Number of Family Members Tested |
|---|---|---|---|---|---|---|---|
| Patient 1 | F | 44 | OC 44 | BRCA1: c.116G>A (p.Cys39Tyr) rs80357498 | - | Mother with PaC (42) | - |
| Patient 2 | F | 74 | BC 73 | BRCA1: c.547+2T>A rs80358047 | - | Mother with OC (49) and sister with LC (65) | 4 |
| Patient 3 | F | 55 | BC (54) and Melanoma (50) | BRCA1: c.4964_4982del (p.Ser1655Tyrfs*16) rs80359876 | MUTYH: c.1103G>A (p.Gly368Asp) rs36053993 | Two sisters with BC (35–46) | 2 |
| Patient 4 | F | 63 | CRC 61 | BRCA1: c.3756_3759del (p.Ser1253Argfs*10) rs80359876 | - | Sisters with BC (45–46) and CRC (67), brother with CRC (71), and mother with BC (84) and OC (63) | 1 |
| Patient 5 | F | 19 | BC 42 | BRCA1: c.5266dup (p.Gln1756Profs*74) rs80357906 | - | Mother with BC (60) and maternal grandmother with OC (62) and BC (63) | 3 |
| Patient 6 | F | 65 | BC 65 | BRCA1: c.5266dup (p.Gln1756Profs*74) rs80357906 | - | Father with PC (55) | 1 |
| Patient 7 | F | 49 | BC 49 | BRCA1: c.5266dup (p.Gln1756Profs*74) rs80357906 | - | Mother with CRC (65), sister with BC (42), and niece with BC (35) | 3 |
| Patient 8 | F | 18 | LC 18 | BRCA1: c.5266dup (p.Gln1756Profs*74) rs80357906 | - | Mother with LC (48) and maternal grandmother with BC (65) | 1 |
| Patient 9 | F | 50 | BC 46 | BRCA1: c.5266dup (p.Gln1756Profs*74) rs80357906 | - | Paternal aunt with CRC (69) and cousin with BC (46) | 1 |
| Patient 10 | F | 54 | BC 54 | BRCA1: c.5266dup (p.Gln1756Profs*74) rs80357906 | - | Maternal cousin with BC (58) | 1 |
| Patient 11 | F | 48 | BC 47 | BRCA1: c.5319dup (p.Asn1774Glnfs*56) rs80357823 | - | Maternal aunts with BC (57–67) | 1 |
| Patient 12 | F | 53 | OC 52 | BRCA2: c.7558C>T (p.Arg2520*) rs80357823 | - | Sister with BC (50) and maternal grandmother with CRC (70) | - |
| Patient 13 | M | 40 | PC 40 | BRCA2: c.8168A>G (p.Asp2723Gly) rs41293513 | - | Father with PC (65) | 3 |
| Patient 14 | F | 39 | BC 37 | BRCA2: c.6037A>T (p.Lys2013*) rs80358840 | - | Father with CRC (61) and paternal aunts with BC (66) and LC (60) | 1 |
| Patient 15 | F | 64 | BC 64 | BRCA2: c.67+1G>A rs81002796 | - | Father with LC (84) and paternal aunts with BC (75-35-30) | - |
| Patient 16 | M | 59 | BC 59 | BRCA2: c.67+1G>A rs81002796 | - | Father with lymphoma (74) and sister with BC (42) | - |
| Patient 17 | F | 58 | BC 58 | BRCA2: c.67+1G>A rs81002796 | - | Mother with BBC (80) | 4 |
| Patient 18 | M | 65 | GC 63 | BRCA2: c.6486_6489del (p.Lys2162Argfs*5) rs80359598 | - | Mother with melanoma (68) and brother with GC (62) | 2 |
| Patient 19 | F | 53 | BC 53 | BRCA2: c.6486_6489del (p.Lys2162Argfs*5) rs80359598 | - | Mother with BC (50) and OC (66) | - |
| Patient 20 | M | 54 | BC 47 | BRCA2: c.6486_6489del (p.Lys2162Argfs*5) rs80359598 | - | Father with RCC (75) and sister with BC (47) | 5 |
| Patient 21 | M | 63 | GC 63 | BRCA2: c.6468_6469del (p.Gln2157Argfs*18) rs80359596 | - | Mother with BC (70) and CRC (91) | - |
| Patient 22 | F | 51 | BC 49 | BRCA2: c.6468_6469del (p.Gln2157Argfs*18) rs80359596 | - | Mother with BC (58) and maternal aunt with BC (69) | 1 |
| Patient 23 | F | 43 | BC 39 | BRCA2: c.6405_6409del (p.Asn2135Lysfs*3) rs80359584 | - | Father with PC (63) and paternal uncles with PC (80) and CRC (81) | 2 |
| Patient 24 | F | 48 | PC 48 | BRCA2: c.5073dup (p.Trp1692fs*3) rs80359479 | - | Maternal uncles with PC (70–78) and brothers with PaC (52) and PC (55) | 1 |
| Patient 25 | F | 89 | BC 87 | BRCA2: c.3458dup (p.Thr1154Aspfs*4) rs80359386 | - | Mother with BC (70) | 3 |
| ID | Sex (M/F) | Age at Testing | Clinical Phenotype and Age at Diagnosis | Mutation Type Gene/Name and dbSNP ID | Mutation in Other Genes | Family History | Number of Family Members Tested |
|---|---|---|---|---|---|---|---|
| Patient 1 | F | 49 | BC 49 | ATM: c.3802del (p.Glu1267_Val1268ins*) rs587779834 | - | Father with PC (66) and paternal aunts with PC (78) and CRC (36–85) | 2 |
| Patient 2 | F | 60 | OC 60 | ATM: c.3802del (p.Glu1267_Val1268ins*) rs587779834 | - | Maternal aunt with BC (76) and sisters with BC (25–51), OC (37) and GC (47) | 1 |
| Patient 3 | F | 46 | BC 46 | ATM: c.2548G>T (p.Glu850*) rs587782280 | - | Mother with BBC (50) and maternal aunts with BC (35–50) and PaC (57) | - |
| Patient 4 | F | 54 | BC 53 | ATM: c.8833_8834del (p.Leu2945Valfs*10) rs786203030 | - | Mother with BC (82) and maternal aunts with BC (65-47-64) | 2 |
| Patient 5 | F | 53 | OC 52 | ATM: c.2250G>A (p.Lys750=) rs1137887 | - | Maternal aunts with BC (80), LC (72) and sisters with LC (49) and BC (40) | 1 |
| Patient 6 | F | 45 | BC 44 | ATM: delEX62_EX63 (Amirifar et al., 2021) [15] | - | Paternal aunts with RCC (79), BC (65), and GC (65–67) | - |
| Patient 7 | F | 51 | BC 47, TC 47, OC 45 | CHEK2: c.920dup (p.Glu308Argfs*4) rs786203053 | - | Maternal grandmother with BC (75) and sister with BC (47) | - |
| Patient 8 | F | 67 | Melanoma 67 | CHEK2: c.1427C>T (p.Thr476Met) rs142763740 | - | Brother with CRC (47) and son with RCC (42) | 2 |
| Patient 9 | F | 30 | BC 30 | CHEK2: c.1427C>T (p.Thr476Met) rs142763740 | - | Mother with BC (63), maternal grandmother with BC (65), and maternal grandfather with PC (79) | - |
| Patient 10 | F | 54 | BC 54 | CHEK2: c.1100del (p.Thr367fs*15) rs555607708 | - | Maternal aunts with LC (70–75), CRC (85–88), TC (77), and PaC (77) | 3 |
| Patient 11 | M | 78 | CRC 77 | CHEK2: c.688G>A (p.Ala230Thr) rs748636216 | - | Maternal cousin with BC (60) and daughter with OC (42) | 1 |
| Patient 12 | F | 52 | BBC 47 | CHEK2: c.470T>C (p.Ile157Thr) rs17879961 | MUTYH: c.536A>G (p.Tyr179Cys) rs34612342 | Mother with PaC (68) | 1 |
| Patient 13 | M | 59 | CRC 57 | CHEK2: c.470T>C (p.Ile157Thr) rs17879961 | - | Father with GC (80) and paternal aunts with PC (78) and LC (66) | - |
| Patient 14 | M | 81 | BC 81 | BRIP1: c.478del (p.Arg160Glufs*5) rs1555616150 | - | Paternal aunts with BC (50-54-60) | 3 |
| Patient 15 | F | 73 | OC 72 | BRIP1: c.440dup (p.Tyr147*) rs786203521 | - | Maternal cousin with BC (50) | 1 |
| Patient 16 | F | 49 | BC 37 | BRIP1: c.55dup (p.Tyr19Leufs*2) rs1567878148 | - | Mother with OC (70), maternal grandmother with CRC (72), and daughter with BC (49) | 1 |
| Patient 17 | F | 55 | BC 55 | BRIP1: c.55dup (p.Tyr19Leufs*2) rs1567878148 | MUTYH: c.1187G>A (p.Gly396Asp) rs36053993 | Father with LC (63) and paternal cousin with BC (38) | 2 |
| Patient 18 | F | 34 | BC 30 | PALB2: c.1919C>A (p.S640*) rs760094988 | - | Maternal grandmother with BC (81) | - |
| Patient 19 | F | 39 | BC 29 | PALB2: c.1285_1286delAinsTC (p.I429SfsX12) (Vietri et al., 2015) [16] | - | Father with BC (70) and sisters with BC (32–40) | - |
| Patient 20 | F | 67 | BC 43 | PALB2: c.2257C>T (p.Arg753*) rs180177110 | - | Mother with BC (60) and sister with lymphoma (39) | 2 |
| Patient 21 | F | 57 | OC (43), BC (50), TC (50), LC (51) | PALB2: c.2161_2186dup (p.Ile730fs*11) rs1567217908 | - | Maternal aunt OC (60) | 1 |
| Patient 22 | F | 46 | OC 46 | RAD51C: c.414G>C (p.Leu138Phe) rs267606999 | - | Mother with OC (52) and maternal aunt with GC (72) | 1 |
| Patient 23 | F | 61 | PaC 61 | BARD1: c.539_540del (p.Tyr180*) rs779427628 | - | Mother with PaC (69) and maternal aunt with GC (82) | 3 |
| Patient 24 | M | 76 | LC 63 | BARD1: c.1216C>T (p.Arg406*) rs377153250 | - | Sister with BC (50) and maternal aunts with PaC (78) | - |
| Patient 25 | F | 69 | BC 52 | *CDH1: c.1678_1688del (p.Thr560Profs*24) novel | - | Father with GC (39) and sister with BC (73) and TC (50) | 1 |
| Patient 26 | M | 44 | GC 70 | CDH1: c.2295+1G>C rs1596971108 | - | Maternal aunt with RCC (55) and one aunt with unspecified neoplasm | 2 |
| Patient 27 | F | 25 | BC 25 | TP53: c.1009C>T (p.Arg337Cys) rs138398778 | - | Mother with BBC (30), maternal grandmother with BBC (30), and maternal grandfather with LC (76) | - |
| Patient 28 | M | 35 | PC 35 | TP53: c.844C>T (p.Arg282Trp) rs28934574 | - | Mother with BC (46) | 2 |
| Patient 29 | F | 40 | BC 37 | TP53: c.817C>T (p.Arg273Cys) rs121913343 | - | Maternal grandmother with BC (48) | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vietri, M.T.; Della Pepa, C.; Caliendo, G.; Mignano, A.; Albanese, L.; Zitiello, M.; Stilo, M.; Molinari, A.M. Expanding the Genomic Landscape of HBOC and Cancer Risk Among Mutation Carriers. Int. J. Mol. Sci. 2025, 26, 5928. https://doi.org/10.3390/ijms26135928
Vietri MT, Della Pepa C, Caliendo G, Mignano A, Albanese L, Zitiello M, Stilo M, Molinari AM. Expanding the Genomic Landscape of HBOC and Cancer Risk Among Mutation Carriers. International Journal of Molecular Sciences. 2025; 26(13):5928. https://doi.org/10.3390/ijms26135928
Chicago/Turabian StyleVietri, Maria Teresa, Chiara Della Pepa, Gemma Caliendo, Alessia Mignano, Luisa Albanese, Marialaura Zitiello, Marianna Stilo, and Anna Maria Molinari. 2025. "Expanding the Genomic Landscape of HBOC and Cancer Risk Among Mutation Carriers" International Journal of Molecular Sciences 26, no. 13: 5928. https://doi.org/10.3390/ijms26135928
APA StyleVietri, M. T., Della Pepa, C., Caliendo, G., Mignano, A., Albanese, L., Zitiello, M., Stilo, M., & Molinari, A. M. (2025). Expanding the Genomic Landscape of HBOC and Cancer Risk Among Mutation Carriers. International Journal of Molecular Sciences, 26(13), 5928. https://doi.org/10.3390/ijms26135928