
Neurobiological Mechanisms of Electroconvulsive Therapy: Molecular Perspectives of Brain Stimulation

 , , , ,
, , , ,
Abstract
1. Introduction
2. Understanding the Mechanisms of ECT: Key Theories
2.1. Memory Disruption and the Abandoned Amnesia Hypothesis
2.2. The Anticonvulsant Hypothesis
2.3. The Neurogenesis Hypothesis
2.4. The Neuroendocrine Hypothesis
2.5. The Neuroplasticity Hypothesis
2.6. The Neurotransmitter Hypothesis
2.7. The Receptor Hypothesis
2.8. The Cytokine Hypothesis
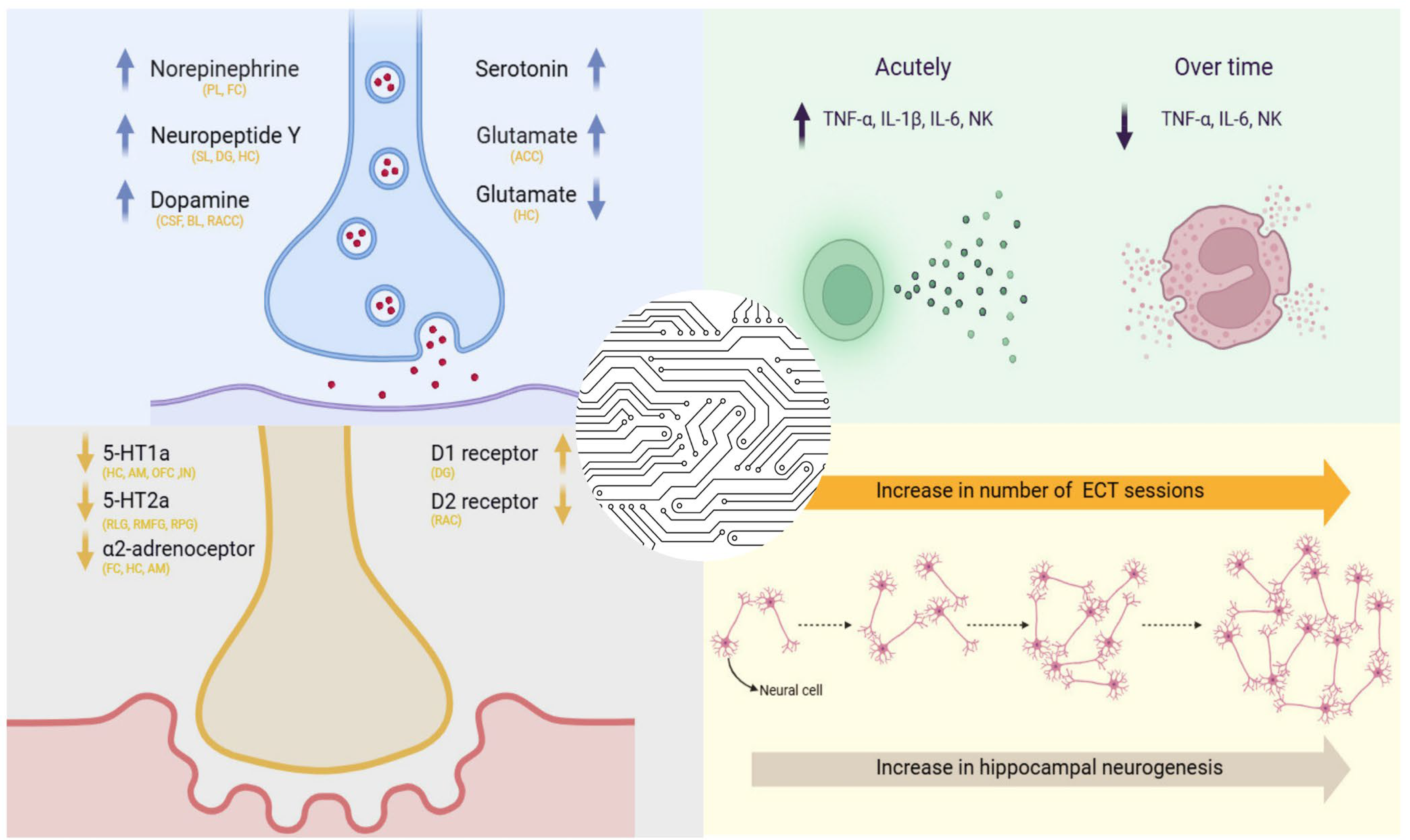
3. Neurotransmitter and Neuropeptide Modulation by ECT
3.1. Modulation of Neurotransmitter Systems Following Electroconvulsive Therapy
3.2. Alterations in Neuropeptide Expression Associated with Electroconvulsive Therapy
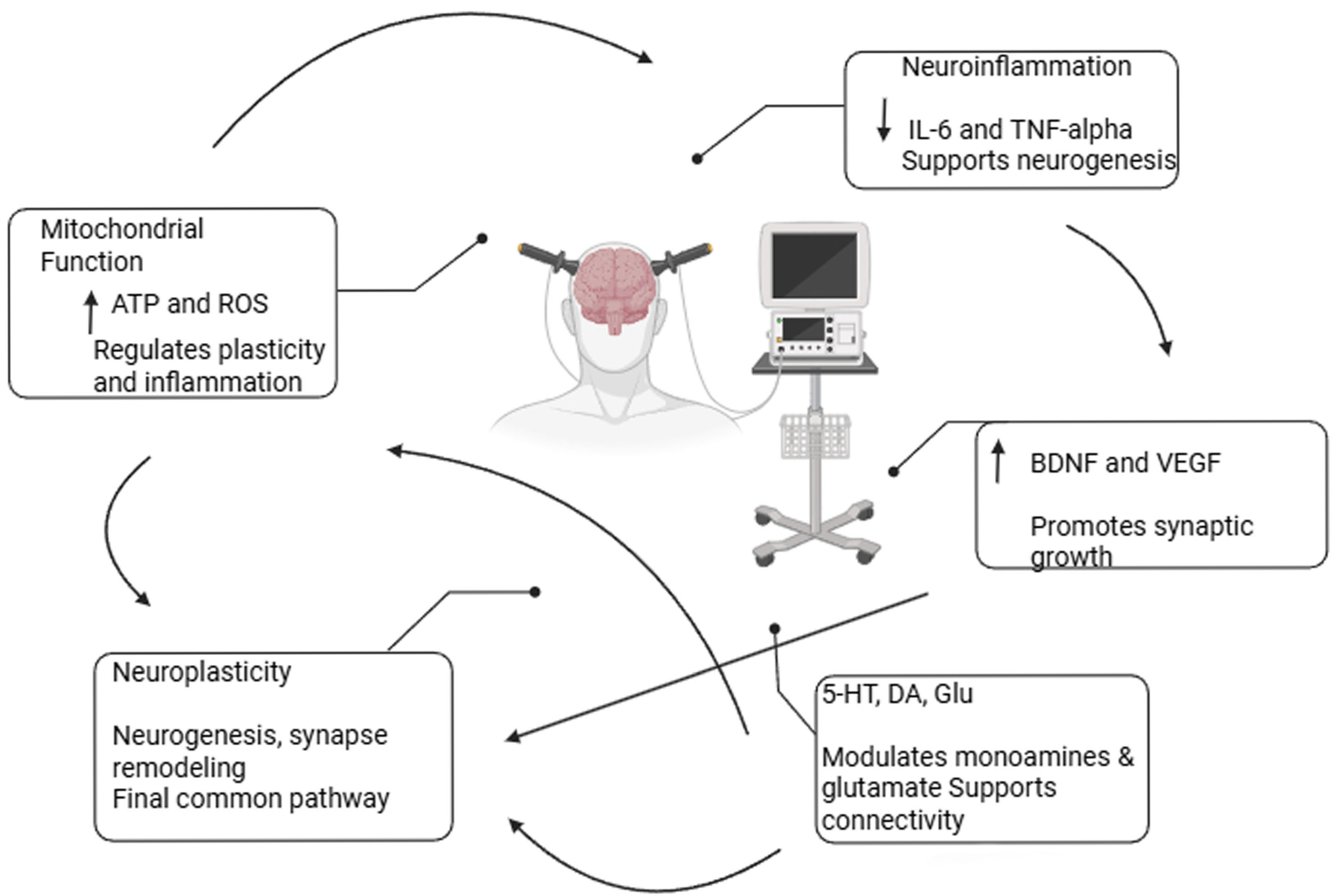
4. The Role of Neuroplasticity, Functional Network Reorganization, and Neuroanatomical Changes in the Therapeutic Effects of ECT
5. Molecular Pathways Related to ECT
5.1. Neurotrophins and ECT
5.2. ECT and Immunological Alterations
5.3. Mitochondrial Function and Energy Metabolism During ECT
5.4. Oxidative Stress and ECT
5.5. Apoptosis and ECT
6. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marx, W.; Penninx, B.W.; Solmi, M.; Furukawa, T.A.; Firth, J.; Carvalho, A.F.; Berk, M. Major depressive disorder. Nat. Rev. Dis. Primers 2023, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Institute for Health Metrics and Evaluation. Global Burden of Disease Study 2017 (GBD 2017) Data Resources [Internet]; IHME: Seattle, WA, USA, 2019. [Google Scholar]
- GBD 2019 Mental Disorders Collaborators. Global, regional, and national burden of 12 mental disorders in 204 countries and territories, 1990–2019: A systematic analysis for the global burden of disease study 2019. Lancet Psychiatry 2022, 9, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C. The Global Burden of Disease: 2004 Update; World Health Organization: Geneva, Switzerland, 2008; pp. 7–49. [Google Scholar]
- Abe, Y.; Erchinger, V.J.; Ousdal, O.T.; Oltedal, L.; Tanaka, K.F.; Takamiya, A. Neurobiological mechanisms of electroconvulsive therapy for depression: Insights into hippocampal volumetric increases from clinical and preclinical studies. J. Neurochem. 2024, 168, 1738–1750. [Google Scholar] [CrossRef] [PubMed]
- Rybak, Y.E.; Lai, K.S.; Ramasubbu, R.; Vila-Rodriguez, F.; Blumberger, D.M.; Chan, P.; Delva, N.; Giacobbe, P.; Gosselin, C.; Kennedy, S.H.; et al. Treatment-resistant major depressive disorder: Canadian expert consensus on definition and assessment. Depress. Anxiety 2021, 38, 456–467. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Alsuwaidan, M.; Baune, B.T.; Berk, M.; Demyttenaere, K.; Goldberg, J.F.; Gorwood, P.; Ho, R.; Kasper, S.; Kennedy, S.H.; et al. Treatment-resistant depression: Definition, prevalence, detection, management, and investigational interventions. World J. Psychiatry 2023, 22, 394–412. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Soczynska, J.K.; Cha, D.S.; Woldeyohannes, H.O.; Dale, R.S.; Alsuwaidan, M.T.; Gallaugher, L.A.; Mansur, R.B.; Muzina, D.J.; Carvalho, A.; et al. The prevalence and illness characteristics of DSM-5-defined “mixed feature specifier” in adults with major depressive disorder and bipolar disorder: Results from the International Mood Disorders Collaborative Project. J. Affect. Disord. 2015, 172, 259–264. [Google Scholar] [CrossRef]
- Espinoza, R.T.; Kellner, C.H. Electroconvulsive therapy. N. Engl. J. Med. 2022, 386, 667–672. [Google Scholar] [CrossRef]
- Spaans, H.P.; Sienaert, P.; Bouckaert, F.; van den Berg, J.F.; Verwijk, E.; Kho, K.H.; Stek, M.L.; Kok, R.M. Speed of remission in elderly patients with depression: Electroconvulsive therapy v. medication. Br. J. Psychiatry 2015, 206, 67–71. [Google Scholar] [CrossRef]
- Kellner, C.H.; Fink, M.; Knapp, R.; Petrides, G.; Husain, M.; Rummans, T.; Mueller, M.; Bernstein, H.; Rasmussen, K.; O’Connor, K.; et al. Relief of expressed suicidal intent by ECT: A consortium for research in ECT study. Am. J. Psychiatry 2005, 162, 977–982. [Google Scholar] [CrossRef]
- Trifu, S.; Sevcenco, A.; Stănescu, M.; Drăgoi, A.M.; Cristea, M.B. Efficacy of electroconvulsive therapy as a potential first-choice treatment in treatment-resistant depression. Exp. Ther. Med. 2021, 22, 1281. [Google Scholar] [CrossRef]
- The UK ECT Review Group. Efficacy and safety of electroconvulsive therapy in depressive disorders: A systematic review and meta-analysis. Lancet 2003, 361, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, M.D.; Peterchev, A.V.; Camprodon, J.A. Electroconvulsive therapy: Mechanisms of action, clinical considerations, and future directions. Harv. Rev. Psychiatry 2023, 31, 101–113. [Google Scholar] [CrossRef] [PubMed]
- McClintock, S.M.; Brandon, A.R.; Husain, M.M.; Jarrett, R.B. A systematic review of the combined use of electroconvulsive therapy and psychotherapy for depression. J. ECT 2011, 27, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Zhao, L.B.; Liu, Y.Y.; Fan, S.H.; Xie, P. Comparative efficacy and acceptability of electroconvulsive therapy versus repetitive transcranial magnetic stimulation for major depression: A systematic review and multiple-treatments meta-analysis. Behav. Brain Res. 2017, 320, 30–36. [Google Scholar] [CrossRef]
- Kumar, S.; Mulsant, B.H.; Liu, A.Y.; Blumberger, D.M.; Daskalakis, Z.J.; Rajji, T.K. Systematic review of cognitive effects of electroconvulsive therapy in late-life depression. Am. J. Geriatr. Psychiatry 2016, 24, 547–565. [Google Scholar] [CrossRef]
- McCall, W.V.; Lisanby, S.H.; Rosenquist, P.B.; Dooley, M.; Husain, M.M.; Knapp, R.G.; Petrides, G.; Rudorfer, M.V.; Young, R.C.; McClintock, S.M.; et al. Effects of continuation electroconvulsive therapy on quality of life in elderly depressed patients: A randomized clinical trial. J. Psychiatr. Res. 2018, 97, 65–69. [Google Scholar] [CrossRef]
- Liang, C.S.; Chung, C.H.; Tsai, C.K.; Chien, W.C. In-hospital mortality among electroconvulsive therapy recipients: A 17-year nationwide population-based retrospective study. Eur. Psychiatr. 2017, 42, 29–35. [Google Scholar] [CrossRef]
- Liang, C.S.; Chung, C.H.; Ho, P.S.; Tsai, C.K.; Chien, W.C. Superior anti-suicidal effects of electroconvulsive therapy in unipolar disorder and bipolar depression. Bipolar Disord. 2018, 20, 539–546. [Google Scholar] [CrossRef]
- Peterchev, A.V.; Rosa, M.A.; Deng, Z.D.; Prudic, J.; Lisanby, S.H. Electroconvulsive therapy stimulus parameters: Rethinking dosage. J. ECT 2010, 26, 159–174. [Google Scholar] [CrossRef]
- Ryan, K.M.; McLoughlin, D.M. From Molecules to Mind: Mechanisms of Action of Electroconvulsive Therapy. Focus Am. J. Psychiatry 2019, 17, 73–75. [Google Scholar] [CrossRef]
- Wiedemann, L.; Trumm, S.; Bajbouj, M.; Grimm, S.; Aust, S. The influence of electroconvulsive therapy on reconsolidation of autobiographical memories: A retrospective quasi-experimental study in patients with depression. Int. J. Clin. Health Psychol. 2023, 23, 100412. [Google Scholar] [CrossRef] [PubMed]
- Miller, E. Psychological theories of ECT: A review. Br. J. Psychiatry 1967, 113, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.E. Production of differential amnesia as a factor in the treatment of schizophrenia. Compr. Psychiatry 1960, 1, 26–34. [Google Scholar] [CrossRef]
- Netto, C.A.; Izquierdo, I. Amnesia as a major side effect of electroconvulsive shock: The possible involvement of hypothalamic opioid systems. Braz. J. Med. Biol. Res. 1984, 17, 349–351. [Google Scholar] [PubMed]
- Bassa, A.; Sagués, T.; Porta-Casteràs, D.; Serra, P.; Martínez-Amorós, E.; Palao, D.J.; Cano, M.; Cardoner, N. The neurobiological basis of cognitive side effects of electroconvulsive therapy: A systematic review. Brain Sci. 2021, 11, 1273. [Google Scholar] [CrossRef]
- Neuhaus, A.H.; Gallinat, J.; Bajbouj, M.; Reischies, F.M. Interictal slow-wave focus in left medial temporal lobe during bilateral electroconvulsive therapy. Neuropsychobiology 2005, 52, 183–189. [Google Scholar] [CrossRef]
- Sackeim, H.A.; Prudic, J.; Devanand, D.P.; Kiersky, J.E.; Fitzsimons, L.; Moody, B.J.; McElhiney, M.C.; Coleman, E.A.; Settembrino, J.M. Effects of stimulus intensity and electrode placement on the efficacy and cognitive effects of electroconvulsive therapy. N. Engl. J. Med. 1993, 328, 839–846. [Google Scholar] [CrossRef]
- Sienaert, P.; Vansteelandt, K.; Demyttenaere, K.; Peuskens, J. Randomized comparison of ultra-brief bifrontal and unilateral electroconvulsive therapy for major depression: Cognitive side-effects. J. Affect. Disord. 2010, 122, 60–67. [Google Scholar] [CrossRef]
- Farzan, F.; Boutros, N.N.; Blumberger, D.M.; Daskalakis, Z.J. What does the electroencephalogram tell us about the mechanisms of action of ECT in major depressive disorders? J. ECT 2014, 30, 98–106. [Google Scholar] [CrossRef]
- Duthie, A.C.; Perrin, J.S.; Bennett, D.M.; Currie, J.; Reid, I.C. Anticonvulsant mechanisms of electroconvulsive therapy and relation to therapeutic efficacy. J. ECT 2015, 31, 173–178. [Google Scholar] [CrossRef]
- Knudsen, M.K.; Near, J.; Blicher, A.B.; Videbech, P.; Blicher, J.U. Magnetic resonance (MR) spectroscopic measurement of γ-aminobutyric acid (GABA) in major depression before and after electroconvulsive therapy. Acta Neuropsychiatr. 2019, 31, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.D.; Robins, P.L.; Regenold, W.; Rohde, P.; Dannhauer, M.; Lisanby, S.H. How electroconvulsive therapy works in the treatment of depression: Is it the seizure, the electricity, or both? Neuropsychopharmacology 2024, 49, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Altar, C.A.; Laeng, P.; Jurata, L.W.; Brockman, J.A.; Lemire, A.; Bullard, J.; Bukhman, Y.V.; Young, T.A.; Charles, V.; Palfreyman, M.G. Electroconvulsive seizures regulate gene expression of distinct neurotrophic signaling pathways. J. Neurosci. 2004, 24, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Perera, T.D.; Coplan, J.D.; Lisanby, S.H.; Lipira, C.M.; Arif, M.; Carpio, C.; Spitzer, G.; Santarelli, L.; Scharf, B.; Hen, R.; et al. Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J. Neurosci. 2007, 27, 4894–4901. [Google Scholar] [CrossRef]
- Haskett, R.F. Electroconvulsive therapy’s mechanism of action: Neuroendocrine hypotheses. J. ECT 2014, 30, 107–110. [Google Scholar] [CrossRef]
- Eşel, E.; Baştürk, M.; Kula, M.; Reyhancan, M.; Turan, M.T.; Sofuoğlu, S. Effects of electroconvulsive therapy on pituitary hormones in depressed patients. Klin. Psikofarmakol. Bülteni 2003, 13, 109–117. [Google Scholar]
- Burgese, D.F.; Bassitt, D.P. Variation of plasma cortisol levels in patients with depression after treatment with bilateral electroconvulsive therapy. Trends Psychiatry Psychother. 2015, 37, 27–36. [Google Scholar] [CrossRef]
- Shahin, O.; Gohar, S.M.; Ibrahim, W.; El-Makawi, S.M.; Fakher, W.; Taher, D.B.; Abdel Samie, M.; Khalil, M.A.; Saleh, A.A. Brain-Derived neurotrophic factor (BDNF) plasma level increases in patients with resistant schizophrenia treated with electroconvulsive therapy (ECT). Int. J. Psychiatry Clin. 2022, 26, 370–375. [Google Scholar] [CrossRef]
- Xie, X.H.; Xu, S.X.; Yao, L.; Chen, M.M.; Zhang, H.; Wang, C.; Nagy, C.; Liu, Z. Altered in vivo early neurogenesis traits in patients with depression: Evidence from neuron-derived extracellular vesicles and electroconvulsive therapy. Brain Stimul. 2024, 17, 19–28. [Google Scholar] [CrossRef]
- Schurgers, G.; Walter, S.; Pishva, E.; Guloksuz, S.; Peerbooms, O.; Incio, L.R.; Arts, B.M.; Kenis, G.; Rutten, B.P. Longitudinal alterations in mRNA expression of the BDNF neurotrophin signaling cascade in blood correlate with changes in depression scores in patients undergoing electroconvulsive therapy. Eur. Neuropsychopharmacol. 2022, 63, 60–70. [Google Scholar] [CrossRef]
- Ramnauth, A.D.; Maynard, K.R.; Kardian, A.S.; Phan, B.N.; Tippani, M.; Rajpurohit, S.; Hobbs, J.W.; Page, S.C.; Jaffe, A.E.; Martinowich, K. Induction of Bdnf from promoter I following electroconvulsive seizures contributes to structural plasticity in neurons of the piriform cortex. Brain Stimul. 2022, 15, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Meyers, K.T.; Damphousse, C.C.; Ozols, A.B.; Campbell, J.M.; Newbern, J.M.; Hu, C.; Marrone, D.F.; Gallitano, A.L. Serial electroconvulsive Seizure alters dendritic complexity and promotes cellular proliferation in the mouse dentate gyrus; a role for Egr3. Brain Stimul. 2023, 16, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Ledesma-Corvi, S.; García-Fuster, M.J. Electroconvulsive seizures regulate various stages of hippocampal cell genesis and mBDNF at different times after treatment in adolescent and adult rats of both sexes. Front. Mol. Neurosci. 2023, 16, 1275783. [Google Scholar] [CrossRef] [PubMed]
- Pollak, C.; Maier, H.B.; Moschny, N.; Jahn, K.; Bleich, S.; Frieling, H.; Neyazi, A. Epinephrine levels decrease in responders after electroconvulsive therapy. J. Neural Transm. 2021, 128, 1917–1921. [Google Scholar] [CrossRef]
- Erchinger, V.J.; Craven, A.R.; Ersland, L.; Oedegaard, K.J.; Bartz-Johannessen, C.A.; Hammar, Å.; Haavik, J.; Riemer, F.; Kessler, U.; Oltedal, L. Electroconvulsive therapy triggers a reversible decrease in brain N-acetylaspartate. Front. Psychiatry 2023, 14, 1155689. [Google Scholar] [CrossRef]
- Lillethorup, T.P.; Iversen, P.; Fontain, J.; Wegener, G.; Doudet, D.J.; Landau, A.M. Electroconvulsive shocks decrease α2-adrenoceptor binding in the Flinders rat model of depression. Eur. Neuropsychopharmacol. 2015, 25, 404–412. [Google Scholar] [CrossRef]
- Biedermann, S.; Weber-Fahr, W.; Zheng, L.; Hoyer, C.; Vollmayr, B.; Gass, P.; Ende, G.; Sartorius, A. Increase of hippocampal glutamate after electroconvulsive treatment: A quantitative proton MR spectroscopy study at 9.4 T in an animal model of depression. World J. Biol. Psychiatry 2012, 13, 447–457. [Google Scholar] [CrossRef]
- Kobayashi, K.; Imoto, Y.; Yamamoto, F.; Kawasaki, M.; Ueno, M.; Segi-Nishida, E.; Suzuki, H. Rapid and lasting enhancement of dopaminergic modulation at the hippocampal mossy fiber synapse by electroconvulsive treatment. J. Neurophysiol. 2017, 117, 284–289. [Google Scholar] [CrossRef]
- Lanzenberger, R.; Baldinger, P.; Hahn, A.; Ungersboeck, J.; Mitterhauser, M.; Winkler, D.; Micskei, Z.; Stein, P.; Karanikas, G.; Wadsak, W.; et al. Global decrease of serotonin-1A receptor binding after electroconvulsive therapy in major depression measured by PET. Mol. Psychiatry 2013, 18, 93–100. [Google Scholar] [CrossRef]
- Yatham, L.N.; Liddle, P.F.; Lam, R.W.; Zis, A.P.; Stoessl, A.J.; Sossi, V.; Adam, M.J.; Ruth, T.J. Effect of electroconvulsive therapy on brain 5-HT2 receptors in major depression. Br. J. Psychiatry 2010, 196, 474–479. [Google Scholar] [CrossRef]
- Saijo, T.; Takano, A.; Suhara, T.; Arakawa, R.; Okumura, M.; Ichimiya, T.; Ito, H.; Okubo, Y. Effect of electroconvulsive therapy on 5-HT1A receptor binding in patients with depression: A PET study with [11C] WAY 100635. Int. J. Neuropsychopharmacol. 2010, 13, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Strome, E.M.; Clark, C.M.; Zis, A.P.; Doudet, D.J. Electroconvulsive shock decreases binding to 5-HT2 receptors in nonhuman primates: An in vivo positron emission tomography study with [18F] setoperone. Biol. Psychiatry 2005, 57, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Landau, A.M.; Chakravarty, M.M.; Clark, C.M.; Zis, A.P.; Doudet, D.J. Electroconvulsive therapy alters dopamine signaling in the striatum of non-human primates. Neuropsychopharmacology 2011, 36, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Pasini, M.; Sartorius, A.; Scherer, R.; Racagni, G.; Riva, M.A.; Gass, P. Repeated electroconvulsive shock (ECS) alters the phosphorylation of glutamate receptor subunits in the rat hippocampus. Int. J. Neuropsychopharmacol. 2010, 13, 1255–1260. [Google Scholar] [CrossRef]
- Lehtimäki, K.; Keränen, T.; Huuhka, M.; Palmio, J.; Hurme, M.; Leinonen, E.; Peltola, J. Increase in plasma proinflammatory cytokines after electroconvulsive therapy in patients with depressive disorder. J. ECT 2008, 24, 88–91. [Google Scholar] [CrossRef]
- Yoshimura, R.; Mitoma, M.; Sugita, A.; Hori, H.; Okamoto, T.; Umene, W.; Ueda, N.; Nakamura, J. Effects of paroxetine or milnacipran on serum brain-derived neurotrophic factor in depressed patients. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2007, 31, 1034–1037. [Google Scholar] [CrossRef]
- Järventausta, K.; Sorri, A.; Kampman, O.; Björkqvist, M.; Tuohimaa, K.; Hämäläinen, M.; Moilanen, E.; Leinonen, E.; Peltola, J.; Lehtimäki, K. Changes in interleukin-6 levels during electroconvulsive therapy may reflect the therapeutic response in major depression. Acta Psychiatr. Scand. 2017, 135, 87–92. [Google Scholar] [CrossRef]
- Goldfarb, S.; Fainstein, N.; Ganz, T.; Vershkov, D.; Lachish, M.; Ben-Hur, T. Electric neurostimulation regulates microglial activation via retinoic acid receptor α signaling. Brain Behav. Immun. 2021, 96, 40–53. [Google Scholar] [CrossRef]
- Madsen, T.M.; Yeh, D.D.; Valentine, G.W.; Duman, R.S. Electroconvulsive seizure treatment increases cell proliferation in rat frontal cortex. Neuropsychopharmacology 2005, 30, 27–34. [Google Scholar] [CrossRef]
- Goldfarb, S.; Fainstein, N.; Ben-Hur, T. Electroconvulsive stimulation attenuates chronic neuroinflammation. JCI Insight 2020, 5, e137028. [Google Scholar] [CrossRef]
- Bouckaert, F.; Sienaert, P.; Obbels, J.; Dols, A.; Vandenbulcke, M.; Stek, M.; Bolwig, T. ECT: Its brain enabling effects: A review of electroconvulsive therapy–induced structural brain plasticity. J. ECT 2014, 30, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Madsen, T.M.; Treschow, A.; Bengzon, J.; Bolwig, T.G.; Lindvall, O.; Tingström, A. Increased neurogenesis in a model of electroconvulsive therapy. Biol. Psychiatry 2000, 47, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Kato, N. Neurophysiological mechanisms of electroconvulsive therapy for depression. Neurosci. Res. 2009, 64, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Willard, S.S.; Koochekpour, S. Glutamate, glutamate receptors, and downstream signaling pathways. Int. J. Biol. Sci. 2013, 9, 948. [Google Scholar] [CrossRef]
- Gilbert, J.R.; Yarrington, J.S.; Wills, K.E.; Nugent, A.C.; Zarate, C.A., Jr. Glutamatergic signaling drives ketamine-mediated response in depression: Evidence from dynamic causal modeling. Int. J. Neuropsychopharmacol. 2018, 21, 740–747. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, N.; Yang, C.; Li, X.M.; Zhou, Z.Q.; Yang, J.J. Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur. Psychiatry 2014, 29, 419–423. [Google Scholar] [CrossRef]
- Li, N.; Lee, B.; Liu, R.J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef]
- Sanacora, G.; Zarate, C.A.; Krystal, J.H.; Manji, H.K. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat. Rev. Drug Discov. 2008, 7, 426–437. [Google Scholar] [CrossRef]
- De Jager, J.E.; Boesjes, R.; Roelandt, G.H.; Koliaki, I.; Sommer, I.E.; Schoevers, R.A.; Nuninga, J.O. Shared effects of electroconvulsive shocks and ketamine on neuroplasticity: A systematic review of animal models of depression. Neurosci. Biobehav. Rev. 2024, 164, 105796. [Google Scholar] [CrossRef]
- Njau, S.; Joshi, S.H.; Espinoza, R.; Leaver, A.M.; Vasavada, M.; Marquina, A.; Woods, R.P.; Narr, K.L. Neurochemical correlates of rapid treatment response to electroconvulsive therapy in patients with major depression. J. Psychiatry Neurosci. 2017, 42, 6–16. [Google Scholar] [CrossRef]
- Baldinger, P.; Lotan, A.; Frey, R.; Kasper, S.; Lerer, B.; Lanzenberger, R. Neurotransmitters and electroconvulsive therapy. J. ECT 2014, 30, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, A.M.; Vasile, A.I.; Trifu, S.C. Neurobiological mechanisms and therapeutic impact of electroconvulsive therapy (ECT). Rom. J. Morphol. Embryol. 2024, 65, 13. [Google Scholar] [CrossRef] [PubMed]
- Landau, A.M.; Phan, J.A.; Iversen, P.; Lillethorup, T.P.; Simonsen, M.; Wegener, G.; Jakobsen, S.; Doudet, D.J. Decreased in vivo α2 adrenoceptor binding in the Flinders Sensitive Line rat model of depression. Neuropharmacology 2015, 91, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Lillethorup, T.P.; Iversen, P.; Wegener, G.; Doudet, D.J.M.; Landau, A.M. α2-adrenoceptor binding in Flinders-sensitive line compared with Flinders-resistant line and Sprague-Dawley rats. Acta Neuropsyciatr. 2015, 27, 345–352. [Google Scholar] [CrossRef]
- Dai, X.; Zhang, R.; Deng, N.; Tang, L.; Zhao, B. Anesthetic Influence on Electroconvulsive Therapy: A Comprehensive Review. Neuropsychiatr. Dis. Treat. 2024, 20, 1491–1502. [Google Scholar] [CrossRef]
- Markianos, M.; Hatzimanolis, J.; Lykouras, L. Relationship between prolactin responses to ECT and dopaminergic and serotonergic responsivity in depressed patients. Eur. Arch. Psychiatry Clin. Neurosci. 2002, 252, 166–171. [Google Scholar] [CrossRef]
- Burnet, P.W.J.; Mead, A.; Eastwood, S.L.; Lacey, K.; Harrison, P.J.; Sharp, T. Repeated ECS differentially affects rat brain 5-HT1A and 5-HT2A receptor expression. Neuroreport 1995, 6, 901–904. [Google Scholar] [CrossRef]
- Saijo, T.; Takano, A.; Suhara, T.; Arakawa, R.; Okumura, M.; Ichimiya, T.; Ito, H.; Okubo, Y. Electroconvulsive therapy decreases dopamine D2 receptor binding in the anterior cingulate in patients with depression: A controlled study using positron emission tomography with radioligand [11C] FLB 457. J. Clin. Psychiatry 2010, 71, 793. [Google Scholar] [CrossRef]
- Lammers, C.H.; Diaz, J.; Schwartz, J.C.; Sokoloff, P. Selective increase of dopamine D3 receptor gene expression as a common effect of chronic antidepressant treatments. Mol. Psychiatry 2000, 5, 378–388. [Google Scholar] [CrossRef]
- Werstiuk, E.S.; Coote, M.; Griffith, L.; Shannon, H.; Steiner, M. Effects of electroconvulsive therapy on peripheral adrenoceptors, plasma, noradrenaline, MHPG and cortisol in depressed patients. Br. J. Psychiatry 1996, 169, 758–765. [Google Scholar] [CrossRef]
- Mann, J.J.; Manevitz, A.Z.; Chen, J.S.; Johnson, K.S.; Adelsheimer, E.F.; Azima-Heller, R.; Massina, A.; Wilner, P.J. Acute effects of single and repeated electroconvulsive therapy on plasma catecholamines and blood pressure in major depressive disorder. Psychiatry Res. 1990, 34, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.B.; Cooper, S.J. Plasma noradrenaline response to electroconvulsive therapy in depressive illness. Br. J. Psychiatry 1997, 171, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Ebstein, R.P.; Lerer, B.; Shlaufman, M.; Belmaker, R.H. The effect of repeated electroconvulsive shock treatment and chronic lithium feeding on the release of norepinephrine from rat cortical vesicular preparations. Cell. Mol. Neurobiol. 1983, 3, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Narr, K.L.; Woods, R.P.; Phillips, O.R.; Alger, J.R.; Espinoza, R.T. Glutamate normalization with ECT treatment response in major depression. Mol. Psychiatry 2013, 18, 268–270. [Google Scholar] [CrossRef]
- Pfleiderer, B.; Michael, N.; Erfurth, A.; Ohrmann, P.; Hohmann, U.; Wolgast, M.; Fiebich, M.; Arolt, V.; Heindel, W. Effective electroconvulsive therapy reverses glutamate/glutamine deficit in the left anterior cingulum of unipolar depressed patients. Psychiatry Res. Neuroimaging 2003, 122, 185–192. [Google Scholar] [CrossRef]
- Dong, J.; Min, S.; Wei, K.; Li, P.; Cao, J.; Li, Y. Effects of electroconvulsive therapy and propofol on spatial memory and glutamatergic system in hippocampus of depressed rats. J. ECT 2010, 26, 126–130. [Google Scholar] [CrossRef]
- Burnet, P.W.; Sharp, T.; LeCorre, S.M.; Harrison, P.J. Expression of 5-HT receptors and the 5-HT transporter in rat brain after electroconvulsive shock. Neurosci. Lett. 1999, 277, 79–82. [Google Scholar] [CrossRef]
- Newman, M.E.; Gur, E.; Shapira, B.; Lerer, B. Neurochemical mechanisms of action of ECS: Evidence from in vivo studies. J. ECT 1998, 14, 153–171. [Google Scholar] [CrossRef]
- Chaput, Y.; de Montigny, C.; Blier, P. Presynaptic and postsynaptic modifications of the serotonin system by long-term administration of antidepressant treatments. An in vivo electrophysiologic study in the rat. Neuropsychopharmacology 1991, 5, 219–229. [Google Scholar]
- Gray, J.A.; Roth, B.L. Paradoxical trafficking and regulation of 5-HT2A receptors by agonists and antagonists. Brain Res. Bull. 2001, 56, 441–451. [Google Scholar] [CrossRef]
- Meyer, J.H.; Kapur, S.; Eisfeld, B.; Brown, G.M.; Houle, S.; DaSilva, J.; Wilson, A.A.; Rafi-Tari, S.; Mayberg, H.S.; Kennedy, S.H. The effect of paroxetine on 5-HT2A receptors in depression: An [18F] setoperone PET imaging study. Am. J. Psychiatry 2001, 158, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Nikisch, G.; Mathé, A.A. CSF monoamine metabolites and neuropeptides in depressed patients before and after electroconvulsive therapy. Eur. Psychiatr. 2008, 23, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Yoshimura, R.; Ikenouchi-Sugita, A.; Hori, H.; Umene-Nakano, W.; Inoue, Y.; Ueda, N.; Nakamura, J. Efficacy of electroconvulsive therapy is associated with changing blood levels of homovanillic acid and brain-derived neurotrophic factor (BDNF) in refractory depressed patients: A pilot study. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Huuhka, K.; Anttila, S.; Huuhka, M.; Hietala, J.; Huhtala, H.; Mononen, N.; Lehtimäki, T.; Leinonen, E. Dopamine 2 receptor C957T and catechol-o-methyltransferase Val158Met polymorphisms are associated with treatment response in electroconvulsive therapy. Neurosci. Lett. 2008, 448, 79–83. [Google Scholar] [CrossRef]
- Ambade, V.; Arora, M.M.; Singh, P.; Somani, B.L.; Basannar, D. Adrenaline, noradrenaline and dopamine level estimation in depression: Does it help? Med. J. Armed Forces India 2009, 65, 216–220. [Google Scholar] [CrossRef]
- El Mansari, M.; Guiard, B.P.; Chernoloz, O.; Ghanbari, R.; Katz, N.; Blier, P. Relevance of norepinephrine–dopamine interactions in the treatment of major depressive disorder. CNS Neurosci. Ther. 2010, 16, e1–e17. [Google Scholar] [CrossRef]
- Sartorius, A.; Mahlstedt, M.M.; Vollmayr, B.; Henn, F.A.; Ende, G. Elevated spectroscopic glutamate/γ-amino butyric acid in rats bred for learned helplessness. Neuroreport 2007, 18, 1469–1473. [Google Scholar] [CrossRef]
- Hashimoto, K.; Sawa, A.; Iyo, M. Increased levels of glutamate in brains from patients with mood disorders. Biol. Psychiatry 2007, 62, 1310–1316. [Google Scholar] [CrossRef]
- Hasler, G.; van der Veen, J.W.; Tumonis, T.; Meyers, N.; Shen, J.; Drevets, W.C. Reduced prefrontal glutamate/glutamine and γ-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry 2007, 64, 193–200. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflug. Arch. Eur. J. Physiol. 2010, 460, 525–542. [Google Scholar] [CrossRef]
- Kupcova, I.; Danisovic, L.; Grgac, I.; Harsanyi, S. Anxiety and depression: What do we know of neuropeptides? Behav. Sci. 2022, 12, 262. [Google Scholar] [CrossRef] [PubMed]
- Kask, A.; Harro, J.; von Hörsten, S.; Redrobe, J.P.; Dumont, Y.; Quirion, R. The neurocircuitry and receptor subtypes mediating anxiolytic-like effects of neuropeptide Y. Neurosci. Biobehav. Rev. 2002, 26, 259–283. [Google Scholar] [CrossRef] [PubMed]
- Ozsoy, S.; Eker, O.O.; Abdulrezzak, U. The effects of antidepressants on neuropeptide Y in patients with depression and anxiety. Pharmacopsychiatry 2016, 49, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Stenfors, C.; Mathé, A.A.; Theodorsson, E. Repeated electroconvulsive stimuli: Changes in neuropeptide Y, neurotensin and tachykinin concentrations in time. Prog. Neuropsychopharmacol. Biol. Psychiatry 1994, 18, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, J.D.; Woldbye, D.P. Accumulated increase in neuropeptide Y and somatostatin gene expression of the rat in response to repeated electroconvulsive stimulation. J. Psychiatr. Res. 2006, 40, 153–159. [Google Scholar] [CrossRef]
- Stengaard-Pedersen, K.; Schou, M. Opioid receptors in the brain of the rat following chronic treatment with desipramine and electroconvulsive shock. Neuropharmacology 1986, 25, 1365–1371. [Google Scholar] [CrossRef]
- Weizman, A.; Gil-Ad, I.; Grupper, D.; Tyano, S.; Laron, Z. The effect of acute and repeated electroconvulsive treatment on plasma β-endorphin, growth hormone, prolactin and cortisol secretion in depressed patients. Psychopharmacology 1987, 93, 122–126. [Google Scholar] [CrossRef]
- Kolb, B.; Whishaw, I.Q. Brain plasticity and behavior. Annu. Rev. Psychol. 1998, 49, 43–64. [Google Scholar] [CrossRef]
- Zatorre, R.J.; Fields, R.D.; Johansen-Berg, H. Plasticity in gray and white: Neuroimaging changes in brain structure during learning. Nat. Neurosci. 2012, 15, 528–536. [Google Scholar] [CrossRef]
- Puderbaugh, M.; Emmady, P.D. Neuroplasticity; StatPearls Publishing: Treasure Island, FL, USA, 2023; pp. 3–9. [Google Scholar]
- Bouckaert, F.; Dols, A.; Emsell, L.; De Winter, F.L.; Vansteelandt, K.; Claes, L.; Sunaert, S.; Stek, M.; Sienaert, P.; Vandenbulcke, M. Relationship between hippocampal volume, serum BDNF, and depression severity following electroconvulsive therapy in late-life depression. Neuropsychopharmacology 2016, 41, 2741–2748. [Google Scholar] [CrossRef]
- Sartorius, A.; Demirakca, T.; Böhringer, A.; von Hohenberg, C.C.; Aksay, S.S.; Bumb, J.M.; Kranaster, L.; Nickl-Jockschat, T.; Grözinger, M.; Thomann, P.A.; et al. Electroconvulsive therapy induced gray matter increase is not necessarily correlated with clinical data in depressed patients. Brain Stimul. 2019, 12, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, J.A.; Hoffstaedter, F.; Zavorotny, M.; Zöllner, R.; Wolf, R.C.; Thomann, P.; Redlich, R.; Opel, N.; Dannlowski, U.; Groezinger, M.; et al. Electroconvulsive therapy modulates grey matter increase in a hub of an affect processing network. Neuroimage Clin. 2020, 25, 102114. [Google Scholar] [CrossRef] [PubMed]
- van Oostrom, I.; van Eijndhoven, P.; Butterbrod, E.; van Beek, M.H.; Janzing, J.; Donders, R.; Schene, A.; Tendolkar, I. Decreased cognitive functioning after electroconvulsive therapy is related to increased hippocampal volume: Exploring the role of brain plasticity. J. ECT 2018, 34, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Lyden, H.; Espinoza, R.T.; Pirnia, T.; Clark, K.; Joshi, S.H.; Leaver, A.M.; Woods, R.P.; Narr, K.L. Electroconvulsive therapy mediates neuroplasticity of white matter microstructure in major depression. Transl. Psychiatry 2014, 4, e380. [Google Scholar] [CrossRef]
- Abbott, C.C.; Lemke, N.T.; Gopal, S.; Thoma, R.J.; Bustillo, J.; Calhoun, V.D.; Turner, J.A. Electroconvulsive therapy response in major depressive disorder: A pilot functional network connectivity resting state FMRI investigation. Front. Psychiatry 2013, 4, 10. [Google Scholar] [CrossRef]
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 2000, 20, 9104–9110. [Google Scholar] [CrossRef]
- Scott, B.W.; Wojtowicz, J.M.; Burnham, W.M. Neurogenesis in the dentate gyrus of the rat following electroconvulsive shock seizures. Exp. Neurol. 2000, 165, 231–236. [Google Scholar] [CrossRef]
- Newton, S.S.; Collier, E.F.; Hunsberger, J.; Adams, D.; Terwilliger, R.; Selvanayagam, E.; Duman, R.S. Gene profile of electroconvulsive seizures: Induction of neurotrophic and angiogenic factors. J. Neurosci. 2003, 23, 10841–10851. [Google Scholar] [CrossRef]
- Müller, H.K.; Orlowski, D.; Bjarkam, C.R.; Wegener, G.; Elfving, B. Potential roles for Homer1 and Spinophilin in the preventive effect of electroconvulsive seizures on stress-induced CA3c dendritic retraction in the hippocampus. Eur. Neuropsychopharmacol. 2015, 25, 1324–1331. [Google Scholar] [CrossRef]
- Joshi, S.H.; Espinoza, R.T.; Pirnia, T.; Shi, J.; Wang, Y.; Ayers, B.; Leaver, A.; Woods, R.P.; Narr, K.L. Structural plasticity of the hippocampus and amygdala induced by electroconvulsive therapy in major depression. Biol. Psychiatry 2016, 79, 282–292. [Google Scholar] [CrossRef]
- Nordanskog, P.; Dahlstrand, U.; Larsson, M.R.; Larsson, E.M.; Knutsson, L.; Johanson, A. Increase in hippocampal volume after electroconvulsive therapy in patients with depression: A volumetric magnetic resonance imaging study. J. ECT 2010, 26, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, A.; Magnusson, P.; Hanson, L.G.; Kirkegaard, T.; Benveniste, H.; Lee, H.; Svarer, C.; Mikkelsen, J.D.; Fink-Jensen, A.; Knudsen, G.M.; et al. Regional brain volumes, diffusivity, and metabolite changes after electroconvulsive therapy for severe depression. Acta Psychiatr. Scand. 2016, 133, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Argyelan, M.; Lencz, T.; Kang, S.; Ali, S.; Masi, P.J.; Moyett, E.; Joanlanne, A.; Watson, P.; Sanghani, S.; Petrides, G.; et al. ECT-induced cognitive side effects are associated with hippocampal enlargement. Transl. Psychiatry 2021, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, S.; Kennedy, M.; Guinan, B.; O’Mara, S.; McLoughlin, D.M. A comparison of brief pulse and ultrabrief pulse electroconvulsive stimulation on rodent brain and behaviour. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 37, 147–152. [Google Scholar] [CrossRef]
- Ousdal, O.T.; Brancati, G.E.; Kessler, U.; Erchinger, V.; Dale, A.M.; Abbott, C.; Oltedal, L. The neurobiological effects of electroconvulsive therapy studied through magnetic resonance: What have we learned, and where do we go? Biol. Psychiatry 2022, 91, 540–549. [Google Scholar] [CrossRef]
- Pirnia, T.; Joshi, S.H.; Leaver, A.M.; Vasavada, M.; Njau, S.; Woods, R.P.; Espinoza, R.; Narr, K.L. Electroconvulsive therapy and structural neuroplasticity in neocortical, limbic and paralimbic cortex. Transl. Psychiatry 2016, 6, e832. [Google Scholar] [CrossRef]
- Sorri, A.; Järventausta, K.; Kampman, O.; Lehtimäki, K.; Björkqvist, M.; Tuohimaa, K.; Hämäläinen, M.; Moilanen, E.; Leinonen, E. Effect of electroconvulsive therapy on brain-derived neurotrophic factor levels in patients with major depressive disorder. Brain Behav. 2018, 8, e01101. [Google Scholar] [CrossRef]
- Kyeremanteng, C.; MacKay, J.C.; James, J.S.; Kent, P.; Cayer, C.; Anisman, H.; Merali, Z. Effects of electroconvulsive seizures on depression-related behavior, memory and neurochemical changes in Wistar and Wistar–Kyoto rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 54, 170–178. [Google Scholar] [CrossRef]
- Luo, J.; Min, S.; Wei, K.; Cao, J.; Wang, B.; Li, P.; Dong, J.; Liu, Y. Behavioral and molecular responses to electroconvulsive shock differ between genetic and environmental rat models of depression. Psychiatry Res. 2015, 226, 451–460. [Google Scholar] [CrossRef]
- Shiraishi-Yamaguchi, Y.; Furuichi, T. The Homer family proteins. Genome Biol. 2007, 8, 206. [Google Scholar] [CrossRef]
- Hu, J.H.; Park, J.M.; Park, S.; Xiao, B.; Dehoff, M.H.; Kim, S.; Hayashi, T.; Schwarz, M.K.; Huganir, R.L.; Seeburg, P.H.; et al. Homeostatic scaling requires group I mGluR activation mediated by Homer1a. Neuron 2010, 68, 1128–1142. [Google Scholar] [CrossRef] [PubMed]
- Serchov, T.; Clement, H.W.; Schwarz, M.K.; Iasevoli, F.; Tosh, D.K.; Idzko, M.; Jacobson, K.A.; de Bartolomeis, A.; Normann, C.; Biber, K.; et al. Increased signaling via adenosine A1 receptors, sleep deprivation, imipramine, and ketamine inhibit depressive-like behavior via induction of Homer1a. Neuron 2015, 87, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.V.; Hartmann, J.; Labermaier, C.; Häusl, A.S.; Zhao, G.; Harbich, D.; Schmid, B.; Wang, X.D.; Santarelli, S.; Kohl, C.; et al. Homer1/mGluR5 activity moderates vulnerability to chronic social stress. Neuropsychopharmacology 2015, 40, 1222–1233. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yao, X.; Sun, L.; Zhao, L.; Xu, W.; Zhao, H.; Zhao, F.; Zou, X.; Cheng, Z.; Li, B.; et al. Effects of electroconvulsive therapy on depression and its potential mechanism. Front. Psychol. 2020, 11, 80. [Google Scholar] [CrossRef]
- Pang, Y.; Wei, Q.; Zhao, S.; Li, N.; Li, Z.; Lu, F.; Pang, J.; Zhang, R.; Wang, K.; Chu, C.; et al. Enhanced default mode network functional connectivity links with electroconvulsive therapy response in major depressive disorder. J. Affect. Disord. 2022, 306, 47–54. [Google Scholar] [CrossRef]
- Jelovac, A.; Kolshus, E.; McLoughlin, D.M. Relapse following successful electroconvulsive therapy for major depression: A meta-analysis. Neuropsychopharmacology 2013, 38, 2467–2474. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef]
- Nordenskjöld, A.; von Knorring, L.; Ljung, T.; Carlborg, A.; Brus, O.; Engström, I. Continuation electroconvulsive therapy with pharmacotherapy versus pharmacotherapy alone for prevention of relapse of depression: A randomized controlled trial. J. ECT 2013, 29, 86–92. [Google Scholar] [CrossRef]
- Sackeim, H.A.; Haskett, R.F.; Mulsant, B.H.; Thase, M.E.; Mann, J.J.; Pettinati, H.M.; Greenberg, R.M.; Crowe, R.R.; Cooper, T.B.; Prudic, J. Continuation pharmacotherapy in the prevention of relapse following electroconvulsive therapy: A randomized controlled trial. JAMA 2001, 285, 1299–1307. [Google Scholar] [CrossRef]
- Freire, T.F.V.; da Rocha, N.S.; de Almeida Fleck, M.P. The association of electroconvulsive therapy to pharmacological treatment and its influence on cytokines. J. Psychiatr. Res. 2017, 92, 205–211. [Google Scholar] [CrossRef]
- Van Den Bossche, M.J.; Emsell, L.; Dols, A.; Vansteelandt, K.; De Winter, F.L.; Van den Stock, J.; Sienaert, P.; Stek, M.L.; Bouckaert, F.; Vandenbulcke, M. Hippocampal volume change following ECT is mediated by rs699947 in the promotor region of VEGF. Transl. Psychiatry 2019, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.J.; Boulle, F.; Steinbusch, H.W.; van den Hove, D.L.; Kenis, G.; Lanfumey, L. Neurotrophic factors and neuroplasticity pathways in the pathophysiology and treatment of depression. Psychopharmacology 2018, 235, 2195–2220. [Google Scholar] [CrossRef] [PubMed]
- Pardossi, S.; Fagiolini, A.; Cuomo, A. Variations in BDNF and Their Role in the Neurotrophic Antidepressant Mechanisms of Ketamine and Esketamine: A Review. Int. J. Mol. Sci. 2024, 25, 13098. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Seki, T.; Liu, J.; Nakamura, K.; Namba, T.; Matsubara, Y.; Suzuki, T.; Arai, H. Effects of repeated electroconvulsive seizure on cell proliferation in the rat hippocampus. Synapse 2010, 64, 814–821. [Google Scholar] [CrossRef]
- Ricken, R.; Adli, M.; Lange, C.; Krusche, E.; Stamm, T.J.; Gaus, S.; Koehler, S.; Nase, S.; Bschor, T.; Richter, C.; et al. Brain-derived neurotrophic factor serum concentrations in acute depressive patients increase during lithium augmentation of antidepressants. J. Clin. Psychopharmacol. 2013, 33, 806–809. [Google Scholar] [CrossRef]
- Mikoteit, T.; Beck, J.; Eckert, A.; Hemmeter, U.; Brand, S.; Bischof, R.; Holsboer-Trachsler, E.; Delini-Stula, A. High baseline BDNF serum levels and early psychopathological improvement are predictive of treatment outcome in major depression. Psychopharmacology 2014, 231, 2955–2965. [Google Scholar] [CrossRef]
- Nase, S.; Köhler, S.; Jennebach, J.; Eckert, A.; Schweinfurth, N.; Gallinat, J.; Lang, U.E.; Kühn, S. Role of serum brain derived neurotrophic factor and central n-acetylaspartate for clinical response under antidepressive pharmacotherapy. Neurosignals 2018, 24, 1–14. [Google Scholar] [CrossRef]
- Yoshimura, R.; Kishi, T.; Hori, H.; Katsuki, A.; Sugita-Ikenouchi, A.; Umene-Nakano, W.; Atake, K.; Iwata, N.; Nakamura, J. Serum levels of brain-derived neurotrophic factor at 4 weeks and response to treatment with SSRIs. Psychiatry Investig. 2014, 11, 84. [Google Scholar] [CrossRef]
- Tadić, A.; Wagner, S.; Schlicht, K.F.; Peetz, D.; Borysenko, L.; Dreimüller, N.; Hiemke, C.; Lieb, K. The early non-increase of serum BDNF predicts failure of antidepressant treatment in patients with major depression: A pilot study. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 415–420. [Google Scholar] [CrossRef]
- Dreimüller, N.; Schlicht, K.F.; Wagner, S.; Peetz, D.; Borysenko, L.; Hiemke, C.; Lieb, K.; Tadić, A. Early reactions of brain-derived neurotrophic factor in plasma (pBDNF) and outcome to acute antidepressant treatment in patients with Major Depression. Neuropharmacology 2012, 62, 264–269. [Google Scholar] [CrossRef]
- Karege, F.; Perret, G.; Bondolfi, G.; Schwald, M.; Bertschy, G.; Aubry, J.M. Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res. 2002, 109, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Haile, C.N.; Murrough, J.W.; Iosifescu, D.V.; Chang, L.C.; Al Jurdi, R.K.; Foulkes, A.; Iqbal, S.; Mahoney, J.J., III; De La Garza, R.; Charney, D.S.; et al. Plasma brain derived neurotrophic factor (BDNF) and response to ketamine in treatment-resistant depression. Int. J. Neuropsychopharmacol. 2014, 17, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Piccinni, A.; Del Debbio, A.; Medda, P.; Bianchi, C.; Roncaglia, I.; Veltri, A.; Zanello, S.; Massimetti, E.; Origlia, N.; Domenici, L.; et al. Plasma Brain-Derived Neurotrophic Factor in treatment-resistant depressed patients receiving electroconvulsive therapy. Eur. Neuropsychopharmacol. 2009, 19, 349–355. [Google Scholar] [CrossRef]
- Maffioletti, E.; Gennarelli, M.; Gainelli, G.; Bocchio-Chiavetto, L.; Bortolomasi, M.; Minelli, A. BDNF genotype and baseline serum levels in relation to electroconvulsive therapy effectiveness in treatment-resistant depressed patients. J. ECT 2019, 35, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M.; Dunne, R.; McLoughlin, D.M. BDNF plasma levels and genotype in depression and the response to electroconvulsive therapy. Brain Stimul. 2018, 11, 1123–1131. [Google Scholar] [CrossRef]
- Fernandes, B.; Gama, C.S.; Massuda, R.; Torres, M.; Camargo, D.; Kunz, M.; Belmonte-de-Abreu, P.S.; Kapczinski, F.; de Almeida Fleck, M.P.; Lobato, M.I. Serum brain-derived neurotrophic factor (BDNF) is not associated with response to electroconvulsive therapy (ECT): A pilot study in drug resistant depressed patients. Neurosci. Lett. 2009, 453, 195–198. [Google Scholar] [CrossRef]
- van Zutphen, E.M.; Rhebergen, D.; van Exel, E.; Oudega, M.L.; Bouckaert, F.; Sienaert, P.; Vandenbulcke, M.; Stek, M.; Dols, A. Brain-derived neurotrophic factor as a possible predictor of electroconvulsive therapy outcome. Transl. Psychiatry 2019, 9, 155. [Google Scholar] [CrossRef]
- Vanicek, T.; Kranz, G.S.; Vyssoki, B.; Fugger, G.; Komorowski, A.; Höflich, A.; Saumer, G.; Milovic, S.; Lanzenberger, R.; Eckert, A.; et al. Acute and subsequent continuation electroconvulsive therapy elevates serum BDNF levels in patients with major depression. Brain Stimul. 2019, 12, 1041–1050. [Google Scholar] [CrossRef]
- Zelada, M.I.; Garrido, V.; Liberona, A.; Jones, N.; Zúñiga, K.; Silva, H.; Nieto, R.R. Brain-derived neurotrophic factor (BDNF) as a predictor of treatment response in major depressive disorder (MDD): A systematic review. Int. J. Mol. Sci. 2023, 24, 14810. [Google Scholar] [CrossRef]
- Maynard, K.R.; Hobbs, J.W.; Rajpurohit, S.K.; Martinowich, K. Electroconvulsive seizures influence dendritic spine morphology and BDNF expression in a neuroendocrine model of depression. Brain Stimul. 2018, 11, 856–859. [Google Scholar] [CrossRef]
- Ryan, K.M.; O’Donovan, S.M.; McLoughlin, D.M. Electroconvulsive stimulation alters levels of BDNF-associated microRNAs. Neurosci. Lett. 2013, 549, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Liu, Y.; Gui, S.; Tian, L.; Xu, S.; Song, X.; Zhong, X.; Chen, Y.; Chen, X.; Yu, Y.; et al. Vascular endothelial growth factor in major depressive disorder, schizophrenia, and bipolar disorder: A network meta-analysis. Psychiatry Res. 2020, 292, 113319. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.N.; Soares, J.C.; Carvalho, A.F.; Quevedo, J. Role of trophic factors GDNF, IGF-1 and VEGF in major depressive disorder: A comprehensive review of human studies. J. Affect. Disord. 2016, 197, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Minelli, A.; Zanardini, R.; Abate, M.; Bortolomasi, M.; Gennarelli, M.; Bocchio-Chiavetto, L. Vascular Endothelial Growth Factor (VEGF) serum concentration during electroconvulsive therapy (ECT) in treatment resistant depressed patients. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 1322–1325. [Google Scholar] [CrossRef]
- Minelli, A.; Maffioletti, E.; Bortolomasi, M.; Conca, A.; Zanardini, R.; Rillosi, L.; Abate, M.; Giacopuzzi, M.; Maina, G.; Gennarelli, M.; et al. Association between baseline serum vascular endothelial growth factor levels and response to electroconvulsive therapy. Acta Psychiatr. Scand. 2014, 129, 461–466. [Google Scholar] [CrossRef]
- Grønli, O.; Stensland, G.Ø.; Wynn, R.; Olstad, R. Neurotrophic factors in serum following ECT: A pilot study. World J. Biol. Psychiatry 2009, 10, 295–301. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Z.; Sha, W.; Xie, C.; Xi, G.; Zhou, H.; Zhang, Y. Electroconvulsive therapy increases glial cell-line derived neurotrophic factor (GDNF) serum levels in patients with drug-resistant depression. Psychiatry Res. 2009, 170, 273–275. [Google Scholar] [CrossRef]
- Angelucci, F.; Aloe, L.; Jiménez-Vasquez, P.; Mathé, A.A. Electroconvulsive stimuli alter the regional concentrations of nerve growth factor, brain-derived neurotrophic factor, and glial cell line-derived neurotrophic factor in adult rat brain. J. ECT 2002, 18, 138–143. [Google Scholar] [CrossRef]
- Enomoto, S.; Shimizu, K.; Nibuya, M.; Suzuki, E.; Nagata, K.; Kondo, T. Activated brain-derived neurotrophic factor/TrkB signaling in rat dorsal and ventral hippocampi following 10-day electroconvulsive seizure treatment. Neurosci. Lett. 2017, 660, 45–50. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef]
- Mössner, R.; Mikova, O.; Koutsilieri, E.; Saoud, M.; Ehlis, A.C.; Müller, N.; Fallgatter, A.J.; Riederer, P. Consensus paper of the WFSBP Task Force on Biological Markers: Biological markers in depression. World J. Biol. Psychiatry 2007, 8, 141–174. [Google Scholar] [CrossRef] [PubMed]
- Silverman, M.N.; Macdougall, M.G.; Hu, F.; Pace, T.W.W.; Raison, C.L.; Miller, A.H. Endogenous glucocorticoids protect against TNF-alpha-induced increases in anxiety-like behavior in virally infected mice. Mol. Psychiatry 2007, 12, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Simen, B.B.; Duman, C.H.; Simen, A.A.; Duman, R.S. TNFα signaling in depression and anxiety: Behavioral consequences of individual receptor targeting. Biol. Psychiatry 2006, 59, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Tyring, S.; Gottlieb, A.; Papp, K.; Gordon, K.; Leonardi, C.; Wang, A.; Lalla, D.; Woolley, M.; Jahreis, A.; Zitnik, R.; et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: Double-blind placebo-controlled randomised phase III trial. Lancet 2006, 367, 29–35. [Google Scholar] [CrossRef]
- Wang, C.; Zong, S.; Cui, X.; Wang, X.; Wu, S.; Wang, L.; Liu, Y.; Lu, Z. The effects of microglia-associated neuroinflammation on Alzheimer’s disease. Front. Immunol. 2023, 14, 1117172. [Google Scholar] [CrossRef]
- Yirmiya, R.; Rimmerman, N.; Reshef, R. Depression as a microglial disease. Trends Neurosci. 2015, 38, 637–658. [Google Scholar] [CrossRef]
- Kronfol, Z.A.; Lemay, L.; Nair, M.P.; Kluger, M.J. Electroconvulsive Therapy Increases Plasma Levels of Interleukin-6a. In Neuropeptides and Immunopeptides: Messengers in a Neuroinunune Axis; The New York Academy of Sciences: New York, NY, USA, 1990. [Google Scholar]
- Hestad, K.A.; Tønseth, S.; Støen, C.D.; Ueland, T.; Aukrust, P. Raised plasma levels of tumor necrosis factor α in patients with depression: Normalization during electroconvulsive therapy. J. ECT 2003, 19, 183–188. [Google Scholar] [CrossRef]
- Kranaster, L.; Hoyer, C.; Aksay, S.S.; Bumb, J.M.; Müller, N.; Zill, P.; Schwarz, M.J.; Sartorius, A. Antidepressant efficacy of electroconvulsive therapy is associated with a reduction of the innate cellular immune activity in the cerebrospinal fluid in patients with depression. World J. Biol. Psychiatry 2018, 19, 379–389. [Google Scholar] [CrossRef]
- Fluitman, S.B.; Heijnen, C.J.; Denys, D.A.; Nolen, W.A.; Balk, F.J.; Westenberg, H.G. Electroconvulsive therapy has acute immunological and neuroendocrine effects in patients with major depressive disorder. J. Affect. Disord. 2011, 131, 388–392. [Google Scholar] [CrossRef]
- Kronfol, Z.; Nair, M.P.; Weinberg, V.; Young, E.A.; Aziz, M. Acute effects of electroconvulsive therapy on lymphocyte natural killer cell activity in patients with major depression. J. Affect. Disord. 2002, 71, 211–215. [Google Scholar] [CrossRef]
- Moschny, N.; Jahn, K.; Maier, H.B.; Khan, A.Q.; Ballmaier, M.; Liepach, K.; Sack, M.; Skripuletz, T.; Bleich, S.; Frieling, H.; et al. Electroconvulsive therapy, changes in immune cell ratios, and their association with seizure quality and clinical outcome in depressed patients. Eur. Neuropsychopharmacol. 2020, 36, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Donato, R. S100: A multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int. J. Biochem. Cell Biol. 2001, 33, 637–668. [Google Scholar] [CrossRef] [PubMed]
- Marenholz, I.; Heizmann, C.W.; Fritz, G. S100 proteins in mouse and man: From evolution to function and pathology (including an update of the nomenclature). Biochem. Biophys. Res. Commun. 2004, 322, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Winningham-Major, F.; Staecker, J.L.; Barger, S.W.; Coats, S.; Van Eldik, L.J. Neurite extension and neuronal survival activities of recombinant S100 beta proteins that differ in the content and position of cysteine residues. J. Cell Biol. 1989, 109, 3063–3071. [Google Scholar] [CrossRef]
- Arts, B.; Peters, M.; Ponds, R.; Honig, A.; Menheere, P.; van Os, J. S100 and impact of ECT on depression and cognition. J. ECT 2006, 22, 206–212. [Google Scholar] [CrossRef]
- Aziz, N.; Nishanian, P.; Mitsuyasu, R.; Detels, R.; Fahey, J.L. Variables that affect assays for plasma cytokines and soluble activation markers. Clin. Diagn. Lab. Immunol. 1999, 6, 89–95. [Google Scholar] [CrossRef]
- Khan, M.; Baussan, Y.; Hebert-Chatelain, E. Connecting Dots between Mitochondrial Dysfunction and Depression. Biomolecules 2023, 13, 695. [Google Scholar] [CrossRef]
- Mailloux, R.J. An update on mitochondrial reactive oxygen species production. Antioxidants 2020, 9, 472. [Google Scholar] [CrossRef]
- Andrés Juan, C.; Pérez de Lastra, J.M.; Plou Gasca, F.J.; Pérez-Lebeña, E. The chemistry of reactive oxygen species (ROS) revisited: Outlining their role in biological macromolecules (DNA, lipids and proteins) and induced pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef]
- Petschner, P.; Gonda, X.; Baksa, D.; Eszlari, N.; Trivaks, M.; Juhasz, G.; Bagdy, G. Genes linking mitochondrial function, cognitive impairment and depression are associated with endophenotypes serving precision medicine. Neuroscience 2018, 370, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wang, L.; Sheng, H. Mitochondria in depression: The dysfunction of mitochondrial energy metabolism and quality control systems. CNS Neurosci. Ther. 2024, 30, e14576. [Google Scholar] [CrossRef] [PubMed]
- Gebara, E.; Zanoletti, O.; Ghosal, S.; Grosse, J.; Schneider, B.L.; Knott, G.; Astori, S.; Sandi, C. Mitofusin-2 in the nucleus accumbens regulates anxiety and depression-like behaviors through mitochondrial and neuronal actions. Biol. Psychiatry 2021, 89, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Huang, Y.; Gong, Y.; Xu, Y.; Lu, J.; Sheng, H.; Ni, X. Treadmill exercise ameliorates depression-like behavior in the rats with prenatal dexamethasone exposure: The role of hippocampal mitochondria. Front. Neurosci. 2019, 13, 264. [Google Scholar] [CrossRef]
- Gong, Y.; Chai, Y.; Ding, J.H.; Sun, X.L.; Hu, G. Chronic mild stress damages mitochondrial ultrastructure and function in mouse brain. Neurosci. Lett. 2011, 488, 76–80. [Google Scholar] [CrossRef]
- Caruso, G.; Benatti, C.; Blom, J.M.; Caraci, F.; Tascedda, F. The many faces of mitochondrial dysfunction in depression: From pathology to treatment. Front. Pharmacol. 2019, 10, 995. [Google Scholar] [CrossRef]
- Li, W.; Zhu, L.; Chen, Y.; Zhuo, Y.; Wan, S.; Guo, R. Association between mitochondrial DNA levels and depression: A systematic review and meta-analysis. BMC Psychiatry 2023, 23, 866. [Google Scholar] [CrossRef]
- Fattal, O.; Link, J.; Quinn, K.; Cohen, B.H.; Franco, K. Psychiatric comorbidity in 36 adults with mitochondrial cytopathies. CNS Spectr. 2007, 12, 429–438. [Google Scholar] [CrossRef]
- Gardner, A.; Johansson, A.; Wibom, R.; Nennesmo, I.; von Döbeln, U.; Hagenfeldt, L.; Hällström, T. Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J. Affect. Disord. 2003, 76, 55–68. [Google Scholar] [CrossRef]
- Brymer, K.J.; Fenton, E.Y.; Kalynchuk, L.E.; Caruncho, H.J. Peripheral etanercept administration normalizes behavior, hippocampal neurogenesis, and hippocampal reelin and GABAA receptor expression in a preclinical model of depression. Front. Pharmacol. 2018, 9, 121. [Google Scholar] [CrossRef]
- Wang, J.; Hodes, G.E.; Zhang, H.; Zhang, S.; Zhao, W.; Golden, S.A.; Bi, W.; Menard, C.; Kana, V.; Leboeuf, M.; et al. Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat. Commun. 2018, 9, 477. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, A.; Rollins, B.; Magnan, C.; van Oven, M.; Baldi, P.; Myers, R.M.; Barchas, J.D.; Schatzberg, A.F.; Watson, S.J.; Akil, H.; et al. Mitochondrial mutations in subjects with psychiatric disorders. PLoS ONE 2015, 10, e0127280. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, T.; Kubota, M.; Miyauchi, T.; Ishiwata, M.; Kato, T. A marked effect of electroconvulsive stimulation on behavioral aberration of mice with neuron-specific mitochondrial DNA defects. PLoS ONE 2008, 3, e1877. [Google Scholar] [CrossRef] [PubMed]
- Búrigo, M.; Roza, C.A.; Bassani, C.; Fagundes, D.A.; Rezin, G.T.; Feier, G.; Dal-Pizzol, F.; Quevedo, J.; Streck, E.L. Effect of electroconvulsive shock on mitochondrial respiratory chain in rat brain. Neurochem. Res. 2006, 31, 1375–1379. [Google Scholar] [CrossRef]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef]
- Yoshikawa, T.; You, F. Oxidative stress and bio-regulation. Int. J. Mol. Sci. 2024, 25, 3360. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Oršolić, N.; Karlović, D.; Peitl, V. Flavonols in action: Targeting oxidative stress and neuroinflammation in major depressive disorder. Int. J. Mol. Sci. 2023, 24, 6888. [Google Scholar] [CrossRef]
- Gadoth, N.; Göbel, H.H. Oxidative Stress in Applied Basic Research and Clinical Practice. Humana Press: Totowa, NJ, USA, 2011; pp. 19–27. [Google Scholar]
- Bakunina, N.; Pariante, C.M.; Zunszain, P.A. Immune mechanisms linked to depression via oxidative stress and neuroprogression. Immunology 2015, 144, 365–373. [Google Scholar] [CrossRef]
- Maes, M.; Galecki, P.; Chang, Y.S.; Berk, M. A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro) degenerative processes in that illness. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 676–692. [Google Scholar]
- Bhatt, S.; Nagappa, A.N.; Patil, C.R. Role of oxidative stress in depression. Drug Discov. Today 2020, 25, 1270–1276. [Google Scholar] [CrossRef]
- Ait Tayeb, A.E.K.; Poinsignon, V.; Chappell, K.; Bouligand, J.; Becquemont, L.; Verstuyft, C. Major depressive disorder and oxidative stress: A review of peripheral and genetic biomarkers according to clinical characteristics and disease stages. Antioxidants 2023, 12, 942. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Fernández, S.; Gurpegui, M.; Garrote-Rojas, D.; Gutiérrez-Rojas, L.; Carretero, M.D.; Correll, C.U. Oxidative stress parameters and antioxidants in adults with unipolar or bipolar depression versus healthy controls: Systematic review and meta-analysis. J. Affect. Disord. 2022, 314, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Atagün, M.İ.; Canbek, Ö.A. A systematic review of the literature regarding the relationship between oxidative stress and electroconvulsive therapy. Alpha Psychiatry 2022, 23, 47. [Google Scholar] [CrossRef] [PubMed]
- Bader, M.; Abdelwanis, M.; Maalouf, M.; Jelinek, H.F. Detecting depression severity using weighted random forest and oxidative stress biomarkers. Sci. Rep. 2024, 14, 16328. [Google Scholar] [CrossRef]
- Barichello, T.; Bonatto, F.; Feier, G.; Martins, M.R.; Moreira, J.C.F.; Dal-Pizzol, F.; Izquierdo, I.; Quevedo, J. No evidence for oxidative damage in the hippocampus after acute and chronic electroshock in rats. Brain Res. 2004, 1014, 177–183. [Google Scholar] [CrossRef]
- Şahin, Ş.; Aybastı, Ö.; Elboğa, G.; Altındağ, A.; Tamam, L. Major depresyonda elektrokonvulsif terapinin oksidatif metabolizma üzerine etkisi. Çukurova Med. J. 2017, 42, 513–517. [Google Scholar]
- Lv, Q.; Hu, Q.; Zhang, W.; Huang, X.; Zhu, M.; Geng, R.; Cheng, X.; Bao, C.; Wang, Y.; Zhang, C.; et al. Disturbance of oxidative stress parameters in treatment-resistant bipolar disorder and their association with electroconvulsive therapy response. Int. J. Neuropsychopharmacol. 2020, 23, 207–216. [Google Scholar] [CrossRef]
- Karayağmurlu, E.; Elboğa, G.; Şahin, Ş.K.; Karayağmurlu, A.; Taysı, S.; Ulusal, H.; Altındağ, A. Effects of electroconvulsive therapy on nitrosative stress and oxidative DNA damage parameters in patients with a depressive episode. Int. J. Psychiatry 2022, 26, 259–268. [Google Scholar] [CrossRef]
- Barichello, T.; Bonatto, F.; Agostinho, F.R.; Reinke, A.; Moreira, J.C.F.; Dal-Pizzol, F.; Izquierdo, I.; Quevedo, J. Structure-related oxidative damage in rat brain after acute and chronic electroshock. Neurochem. Res. 2004, 29, 1749–1753. [Google Scholar] [CrossRef]
- Župan, G.; Pilipović, K.; Hrelja, A.; Peternel, S. Oxidative stress parameters in different rat brain structures after electroconvulsive shock-induced seizures. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 771–777. [Google Scholar] [CrossRef]
- Eraković, V.; Župan, G.; Varljen, J.; Radošević, S.; Simonić, A. Electroconvulsive shock in rats: Changes in superoxide dismutase and glutathione peroxidase activity. Mol. Brain Res. 2000, 76, 266–274. [Google Scholar] [CrossRef]
- Nielsen, B.; Cejvanovic, V.; Wörtwein, G.; Hansen, A.R.; Marstal, K.K.; Weimann, A.; Bjerring, P.N.; Dela, F.; Poulsen, H.E.; Jørgensen, M.B. Increased oxidation of RNA despite reduced mitochondrial respiration after chronic electroconvulsive stimulation of rat brain tissue. Neurosci. Lett. 2019, 690, 1–5. [Google Scholar] [CrossRef]
- Hollville, E.; Romero, S.E.; Deshmukh, M. Apoptotic cell death regulation in neurons. FEBS Lett. 2019, 286, 3276–3298. [Google Scholar] [CrossRef]
- Erekat, N.S. Apoptosis and its therapeutic implications in neurodegenerative diseases. Clin. Anat. 2022, 35, 65–78. [Google Scholar] [CrossRef]
- Lucassen, P.J.; Heine, V.M.; Muller, M.B.; van der Beek, E.M.; Wiegant, V.M.; De Kloet, E.R.; Joels, M.; Fuchs, E.; Swaab, D.F.; Czeh, B. Stress, depression and hippocampal apoptosis. CNS Neurol. Disord. Drug Targets 2006, 5, 531–546. [Google Scholar] [CrossRef]
- Kondratyev, A.; Sahibzada, N.; Gale, K. Electroconvulsive shock exposure prevents neuronal apoptosis after kainic acid-evoked status epilepticus. Mol. Brain Res. 2001, 91, 1–13. [Google Scholar] [CrossRef]
- Zarubenko, I.I.; Yakovlev, A.A.; Stepanichev, M.Y.; Gulyaeva, N.V. Electroconvulsive shock induces neuron death in the mouse hippocampus: Correlation of neurodegeneration with convulsive activity. Neurosci. Behav. Physiol. 2005, 35, 715–721. [Google Scholar] [CrossRef]
- Sigström, R.; Göteson, A.; Joas, E.; Pålsson, E.; Liberg, B.; Nordenskjöld, A.; Blennow, K.; Zetterberg, H.; Landén, M. Blood biomarkers of neuronal injury and astrocytic reactivity in electroconvulsive therapy. Mol. Psychiatry 2024, 30, 1601–1609. [Google Scholar] [CrossRef]
- McGrory, C.L.; Ryan, K.M.; Kolshus, E.; McLoughlin, D.M. Peripheral blood E2F1 mRNA in depression and following electroconvulsive therapy. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 89, 380–385. [Google Scholar] [CrossRef]
- Jeon, W.J.; Kim, S.H.; Seo, M.S.; Kim, Y.; Kang, U.G.; Juhnn, Y.S.; Kim, Y.S. Repeated electroconvulsive seizure induces c-Myc down-regulation and Bad inactivation in the rat frontal cortex. Exp. Mol. Med. 2008, 40, 435–444. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Hypothesis | Evidence Type | Population/Model | ECT Characteristics | Main Findings | Ref. |
|---|---|---|---|---|---|
| Neuroplasticity/Neurotrophic | Clinical | TRS patients (ECT or antipschycotics) | * 4–10 ECT sessions over 4 weeks * bitemporal electrode placement Parameters: pulse width 0.5 ms, frequency 80 Hz, duration 1 s, current 800 mA (individually adjusted and gradually increased) | Post-ECT: * ↑ plasma BDNF levels along with clinical improvement | [40] |
| Clinical | TRD patients and HC | * 3 times per week, 12 ECT sessions * bitemporal electrode placement * stimulus dose was initially set at 50 mC, titrated upward until ≥15 s seizure achieved * stimulation dose for subsequent sessions: 1.5 × seizure threshold | Baseline: * ↓ plasma BDNF levels and the ratio of DCX to CD81 in NDEVs in TRD group * ↑ levels of CD81 in the TRD group Post-ECT: *↑ plasma BDNF levels and the ratio of DCX to CD81 in NDEVs * ↔ CD81 in NDEVs | [41] | |
| Clinical | TRD patients | * twice a week, 6.1 (range 3–11) ECT in total * bitemporal electrode placement * stimulus dose was set at 350 mC and further adjusted according to EEG characteristics and seizure duration | During ECT period: * ↑ mRNA expression of BDNF and ERK1 * ↔ mRNA expression of CREB Follow-up: * ↔mRNA expression of BDNF, ERK1 and CREB ** ↔ mRNA expression of AKT1, NR3C1 and IGF1 during ECT period and follow-up ** ↑ plasma BDNF levels during study | [42] | |
| Preclinical | Bdnf-e1 −/+ and Bdnf-e1 −/− mice receiving sham or ECS | * 7 ECT sessions during 15 days Parameters: pulse width 3 ms, frequency 100 pulse/s, duration 1 s, current 50 mA | 48 h after the final ECS: * ↓ in spine density in piriform cortex neurons in Bdnf-e1 −/− mice * ↔ spine lenght * ↓ % of smaller spines in Bdnf-e1 −/− | [43] | |
| Preclinical | C57BL/6 male wildtype (WT) and Egr3−/− littermate mice | * ECS once daily during 10 consecutive days * ocular electrode placement Parameters: pulse width 0.3 ms, frequency 260 Hz, duration 100 ms, current 80 mA | 11 days after the final ECS: * ↔ dendritic intersections in dorsal/ventral DG of WT and Egr3−/− mice, ↓ dendritic intersections in dorsal DG of Egr3−/− vs. WT, ↑ proximal and ↓ distal dendritic intersections in ventral DG of Egr3−/− vs. WT * ↑ dendritic branches in hippocampus (significant in ventral), ↑ dendritic spines in dorsal and ventral DG (all layers) * ↑ BrdU+ cells in all subregions of dorsal and ventral DG ** serial ECS doubles proliferating cells in dorsal/ventral hippocampus, independent of Egr3 | [44] | |
| Preclinical | Adolescent and adult Sprague–Dawley rats, males and females | * ECS treatment started 4 days after the final BrdU injection * applied once daily during 5 days * earclip electrode placement Baseline parameters: pulse width 0.6 ms, frequency 100 Hz, duration 0.6 ms, current 95 mA | * ↑ BrdU+ cells in adolescent females (1-day) and adult males (all time points); transient in adult females; no effect in adolescent males * ↑ Ki-67+ cell proliferation at 1 day; normalized/reduced later; NeuroD+ differentiation peaked at 8 days, elevated at 15, baseline by 30 (adult females) * ↑ mBDNF in hippocampus of all groups (peak at 1 day, sustained to 15–30 days, age-/sex-dependent); ECS had no effect on p-ERK1/2 or p-mTOR in adolescents; ↑ p-ERK1/2 at 15 days in adults | [45] | |
| Neurotransmitter | Clinical | MDD or patients with depressive episode of bipolar disorder and HC | * 3 times per week * majority recived right unilateral ECT * stimulus intensity was age-based and adjusted per session to achieve 20–25 s of EEG seizure activity | Baseline: * ↔ prefrontal GABA levels, ↔ GABA/NAA ratios and NAA/Cr ratios, ↔ GM content, ↔ Glu/Cr, Gln/Cr, Cho/Cr, GSH/Cr or Glu/GABA ratios,↔ GABA/Cr ratios and NAA/Cr ratios in OCC During ECT: * ↓ NAA/Cr ratios in the PFC Post-ECT: * ↔GABA/Cr levels * ↓ NAA/Cr ratios in the PFC * NAA/Cr ratios decreasing as the number of ECT sessions increased *↔ Glu/Cr, Gln/Cr, Cho/Cr or GSH/Cr levels | [33] |
| Clinical | TR-MDD patients | * brief pulse ECT three times per week usually for 12 treatment sessions * right unilateral electrode placement (bitemporal if non-responsive by 6th session or seizures inadequate) | * ↑ basal (T1) epinephrine in responders who later showed ↓ post-first ECT * ↑ epinephrine in all non-responders (ΔT1–T2) * ↓ epinephrine in 7 of 13 responders (ΔT1–T2) * ↓ epinephrine at T3 vs. T1 in responders with initial T1–T2 decrease * ↑ cortisol after first ECT (T1→T2), ↓ at T3 to baseline * ↑ norepinephrine at T2; no further significant changes * ↔ no significant ΔT1–T3 in epinephrine, norepinephrine, or cortisol | [46] | |
| Clinical | Patients with depression and HC | * right unilateral electrode placement * initial stimulus charge was age-based and increased over sessions to account for rising seizure threshold | Post-ECT: * ↓ tNAA in ACC (~10.6 sessions): 6% (creatine ratio), 3% (water referenced) Follow-up: * tNAA returned to baseline after 6 months * ↑ tNAA at 6-month follow-up: +6% (creatine ratio), +7% (water referenced) | [47] | |
| Preclinical | Adult female FSL rats and FRL rats with SD rats. | * ECS once daily for 10 consecutive days * earclip electrode placement Parameters: pulse width 0.5 ms, frequency 100 Hz, current 55–70 mA | Post-ECT: * ↓ α2-adrenoceptor binding post-ECS * ↓ α2-adrenoceptor binding by 9% in FC of FSL * ↔ α2-adrenoceptor binding in IN | [48] | |
| Preclinical | Male SD rats, cLH and SD WT rats | * 6 WT and 6 cLH rats received daily ECS for 5 days * earclip electrode placement Parameters: pulse width 1 ms, frequency 80 Hz, duration 1 ms | Post-ECT: * ↑ glutamate in ECS-treated cLH vs. naive cLH * ↑ glutamate trend in ECS-treated WT vs. naive WT * ↔ glutamine in hippocampus * no baseline differences between untreated cLH and WT in hippocampal glutamate, glutamine, or GABA | [49] | |
| Preclinical | Male C57BL/6J mice | * ECS was administered 4 times a week for up to 3 weeks * bilateral ECS * earclip electrode placement Parameters: pulse width 0.5 ms, frequency 100 Hz, duration 1 s, current 25 mA | * enhanced synaptic potentiation induced by dopamine, remained during 4 weeks of follow up, but suppressed by D1-like receptor antagonist * graded effect of electrical stimulation | [50] | |
| Receptor Hypothesis | Clinical | MDD patients | * ECT sessions 4–13, 3 times per week * right unilateral ECT (bilateral approach used in patients with minimal or no improvement from sixth ECT onward) | Baseline: * ↔ no change in 5-HT1A BPND between PET1 and PET2 in any brain region Post-ECT: * ↓ 5-HT1A BPND (PET2 vs. PET3), with a large cortical cluster (436 cm3) showing reductions, peaking in ACC (including sgACC), OFC, IN, hippocampus, and amygdala * no regions showed significant ↑ 5-HT1A BPND post-ECT * no significant effects of sex, ECT laterality, anticonvulsants, treatment outcome, age, or session number on 5-HT1A BPND changes * no hemispheric differences, No correlation between 5-HT1A BPND changes and HAM-D score changes | [51] |
| Clinical | TR-MDD patients | * three times a week * right unilateral ECT * seizure threshold was determined during the first session; subsequent treatments used three times this threshold, with 10–20% adjustments to elicit ≥ 20 s seizures measured by single-strip EEG | Post-ECT: * ↓ 5-HT2 receptor binding in bilateral OC, medial PC (peak: LG), limbic cortex (peak: right PHG), bilateral PFC (peak: right inferomedial PFC) * reduction in 5-HT2 binding in right medial PFC, right LG, and right PHG showed trend-level correlation with HRSD improvement | [52] | |
| Clinical | MDD patients and HC | * 6–7 bilateral ECTs, 2–3 per week * bifrontal electrode placement | * ↓ 5-HT1A BP in midbrain by 32% in MDD patients before ECT vs. controls * ↓ 5-HT1A BP in midbrain raphe by 31% in MDD patients after ECT vs. controls * ↔ no significant change in 5-HT1A BP in MDD patients pre- vs. post-ECT | [53] | |
| Preclinical | Adult male rhesus monkeys | * ESC administered twice weekly for 3 weeks * bitemporal electrode placement Parameters: pulse width 0.5 ms, frequency 70 Hz, current 0.9 mA | * ↓ [18F] setoperone binding potential at 24 h and 1 week post-treatment, ↔ at 4–6 weeks * ↓ 5-HT2 binding in all regions at 24 h and 1 week post-ECS, returning to baseline by 4–6 weeks * ↓ 5-HT2 binding observed after 2–3 ECS treatments in 3 of 5 animals at 24 h post-treatment across most brain regions | [54] | |
| Preclinical | Male rhesus monkeys | * ECT was administered twice a week over a 3-week period * bilateral temporal electrode placement Parameters: pulse width 0.5 ms, frequency 70 Hz, current 0.9 mA | Post-ECT: * ↑ MP and DTBZ binding in all striatal regions; returned toward baseline by 6–8 weeks * ↑ SCH23390 binding in striatum at 24–48 h post-ECT; ↔ at later time points | [55] | |
| Preclinical | SD rats | * 10 or five consecutive ECS treatments (one every other day) * earclip electrode placement Parameters: pulse width 500 ms, frequency 50 Hz, duration 1 s, current 100 mA | Post-10 ECT: * ↑ GluR-A (Ser831) by +68% in whole homogenate (+50% in TIF) * ↑ NR2B (Ser1303) by +72% in homogenate (+78% in TIF) * ↔ in phosphorylation of NR1 (Ser896) or total protein levels of any glutamate receptor subunits Post-5 ECS: * ↔ in phosphorylation or protein levels. * Effects in Triton Insoluble Fraction (TIF) confirm synaptic localization of phosphorylation changes | [56] | |
| Cytokine (Inflammation) | Clinical | MDD patients and HC | * 1–7 ECT session (3.7 on average) * bilateral electrode placement * initial stimulus was ~5× patient’s age, adjusted for demographics and medications, and modified during ECT based on seizure quality | Post-ECT: * ↑ IL-1A (transient) * ↑ IL-6 (↑ at 3 h & 6 h, ↓ by 24 h) * ↔ IL-1RA | [57] |
| Clinical | Patients with severe TR depressive episode | * 2–3 ECT sessions per week * right unilateral brief pulse ECT, bilateral in case of non-responsiveness * seizure threshold was titrated at the first session, with dose increases if seizures were inadequate or no clinical response occurred | Post-ECT (CSF): * ↔ IL-6, neopterin, sCD14, sCD163, MIF, MCP-1 * baseline ↑ sCD14 predicted ↓ HDRS scores * Δ MIF differed between remitters vs. non-remitters | [58] | |
| Clinical | MDD patients | * total ECT sessions: 10.4 ± 3.6, 3 times per week with a brief pulse Parameters: pulse width 0.5–1 s, frequency 20–50 Hz | Post-ECT: * ↑ IL-6 vs. baseline * ↓ IL-6 (baseline) from first to final ECT in remitters only * ↔ IL-1RA * ↔ IL levels and ECT parameters | [59] | |
| Preclinical | Biozzi ABH mice (Envigo) | * earclip electrode placement Parameters: pulse width 0.5 ms, frequency 100 Hz, duration 1 s, current charging at 2 mA, starting at 8 Ma | * ↔ spinal microglia count in naïve mice * ↔ microglia count with ECS pre-treatment, but → altered morphology & ↑ RARα pathway activation in response to LPS | [60] | |
| Preclinical | Male SD rats | * once daily ESC for 10 days * earclip electrode placement Parameters: current 55 mA, duration 0.3 s | * ↑ BrdU+ cells (~2×) in mPFC after 10 daily ECS vs. sham * ↔ BrdU+ cells differentiating into astrocytes in FC * ↑ Rip+ (oligodendrocytes) in FC post-ECS * ↑ endothelial cell number after ECS | [61] | |
| Preclinical | Biozzi mice with first relapse of EAE | * ECS initiation at first day of clinical signs and 4 additional ECS sessions on alternating days * ECS was applied with twice the average threshold | * ↓ T cell infiltration (−59%) and IBA1+ microglia/macrophages (−44%) in SC WM after ECS * ↔ oligodendrocyte numbers between groups * ↑ NG2+ cells in control EAE (3.8× vs. naive), marginal ↑ in ECS-treated EAE | [62] |
| Neurotransmitter | Receptor/Target | Study | Brain Region(s)/Blood Components | ECT Effect | Ref. |
|---|---|---|---|---|---|
| Serotonin | 5HT1A | Clinical | subgenual part of ACC, OFC, AMY, hippocampus, IN midbrain raphe | ↓ ↔ | [51] [53] |
| Preclinical | CA3c DG | ↓ ↑ | [79] | ||
| 5HT2A | Clinical | all cortical areas with changes slightly more prominent in the right hemisphere | ↓ | [52] | |
| Preclinical | cingulate and frontoparietal cortex, FC cortical areas | ↑ ↓ | [79] [52,54] | ||
| Dopamine | D1 | Preclinical | hippocampal mossy fiber (MF)-CA3 excitatory synapse striatum | ↑ ↑ | [50] [55] |
| D2 | Clinical | ACC | ↓ | [80] | |
| Preclinical | striatum | ↔ | [55] | ||
| D3 | Preclinical | D3 receptor mRNA and binding shell of nucleus accumbens D3 receptor mRNA islands of Calleja | ↑ | [81] | |
| Norepinephrine | α2-adrenoceptors | Clinical | platelet α2-adrenoceptor numbers leukocyte α2-adrenoceptor densities | ↓ ↑ | [82] |
| Preclinical | cortical regions and amygdaloid regions | ↓ | [48] | ||
| NE | Clinical | plasma NE plasma NE plasma level in patients responding to ECT | ↑ ↓ ↓ | [83] [84] [46] | |
| Preclinical | presynaptic release of [3H] norepinephrine from rat cortical vesicular preparation | ↔ | [85] | ||
| Glutamate | Glu (overall) | Clinical | ACC and connected prefrontal and subcortical centers left hippocampus and right hippocampus the subgenual ACC left ACC | ↑ ↓ ↑ ↑ | [86] [72] [87] |
| Preclinical | hippocampus | ↑ | [88] |
| Study Type | Subjects | ECT Protocol | Brain Region | Main Findings | Ref. |
|---|---|---|---|---|---|
| Clinical | Depressive patients | * twice a week with a constant-current brief-pulse device * mostly right unilateral ECT | Hippocampus | * ↑ bilateral hippocampal volume one week post-ECT; not detectable at 6 months. | [113] |
| Clinical | Depressive patients | * right unilaterally or bilaterally | Hippocampus and Amygdala | * ↑ whole gray matter (particularly right-sided); not correlated with outcomes. | [114] |
| Clinical | Patients with unipolar depression vs. HC | * right unilateral electrode placement, later bilateral due to insufficient response | Right hippocampus and Amygdala | * ↑ gray matter volume increased post-ECT; clinical outcomes not assessed. | [115] |
| Clinical | Depressive patients | * twice a week, bitemporally with a brief pulse | Hippocampus | * ↑ hippocampal volumes; linked to decrease in cognitive functioning. | [116] |
| Clinical | MDD patients vs. HC | * mostly right unilateral | White matter tracts (DTI) | * ↑ FA, ↓ reduced RD and MD—improved fiber integrity. | [117] |
| Clinical | Depressive patients vs. HC | * mostly right unilateral | DMN, CEN (functional networks) | * ↓ DMN hyperconnectivity and ↑ CEN connectivity; linked to improvement. | [118] |
| Preclinical | Rodents and non-human primates | * brief pulse, bilateral frontotemporal electrode placement, 3 times weekly, 4 weeks * earclip electrode placement | DG (hippocampus) | * ↑ progenitor cell proliferation; * ↑ BrdU+ cell increase. | [36,64,119,120] |
| Preclinical | Male Sprague–Dawley rats | * bilateral ECS via moistened pads on spring-loaded earclip electrodes | Hippocampus and choroid plexus | * ↑ BDNF and VEGF genes expression; associated with synaptic plasticity and recovery. | [121] |
| Preclinical | Male Wistar rats | * once daily for 10 days * earclip electrodes | Hippocampus (CA1 and CA3 regions) | * ↑ spine density in CA1 neurons in non-stressed animals; * ↔ CA3c spine densities. | [122] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fetahovic, E.; Janjic, V.; Muric, M.; Jovicic, N.; Radmanovic, B.; Rosic, G.; Selakovic, D.; Filipovic, M.; Muric, N. Neurobiological Mechanisms of Electroconvulsive Therapy: Molecular Perspectives of Brain Stimulation. Int. J. Mol. Sci. 2025, 26, 5905. https://doi.org/10.3390/ijms26125905
Fetahovic E, Janjic V, Muric M, Jovicic N, Radmanovic B, Rosic G, Selakovic D, Filipovic M, Muric N. Neurobiological Mechanisms of Electroconvulsive Therapy: Molecular Perspectives of Brain Stimulation. International Journal of Molecular Sciences. 2025; 26(12):5905. https://doi.org/10.3390/ijms26125905
Chicago/Turabian StyleFetahovic, Ermin, Vladimir Janjic, Maja Muric, Nemanja Jovicic, Branimir Radmanovic, Gvozden Rosic, Dragica Selakovic, Milos Filipovic, and Nemanja Muric. 2025. "Neurobiological Mechanisms of Electroconvulsive Therapy: Molecular Perspectives of Brain Stimulation" International Journal of Molecular Sciences 26, no. 12: 5905. https://doi.org/10.3390/ijms26125905
APA StyleFetahovic, E., Janjic, V., Muric, M., Jovicic, N., Radmanovic, B., Rosic, G., Selakovic, D., Filipovic, M., & Muric, N. (2025). Neurobiological Mechanisms of Electroconvulsive Therapy: Molecular Perspectives of Brain Stimulation. International Journal of Molecular Sciences, 26(12), 5905. https://doi.org/10.3390/ijms26125905