
Protein Kinase CK2 Inhibition Represents a Pharmacological Chance for the Treatment of Skin Diseases

 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and
Abstract

1. Introduction
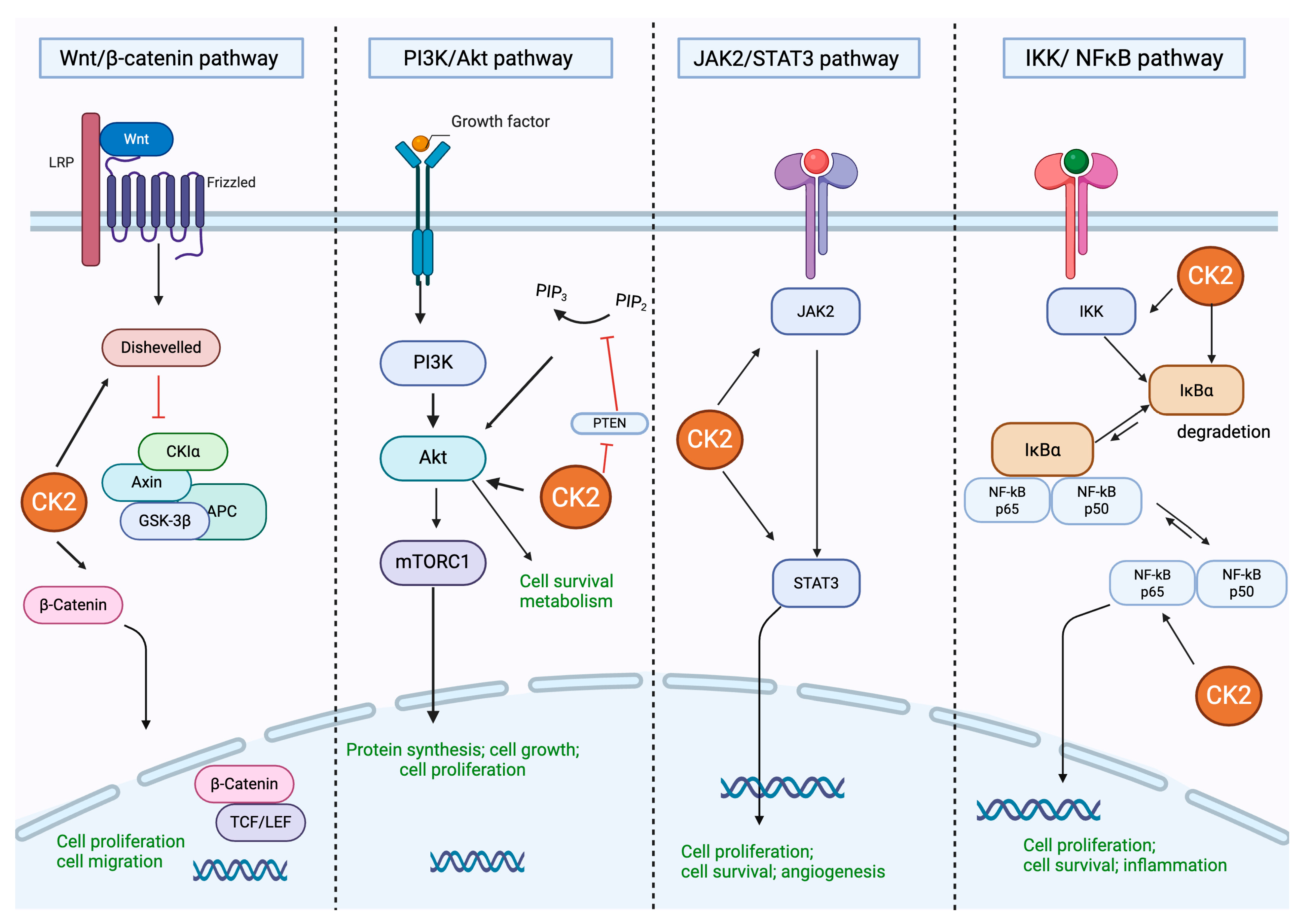
2. Protein Kinase CK2: Structure and Regulation of Different Cell Signaling
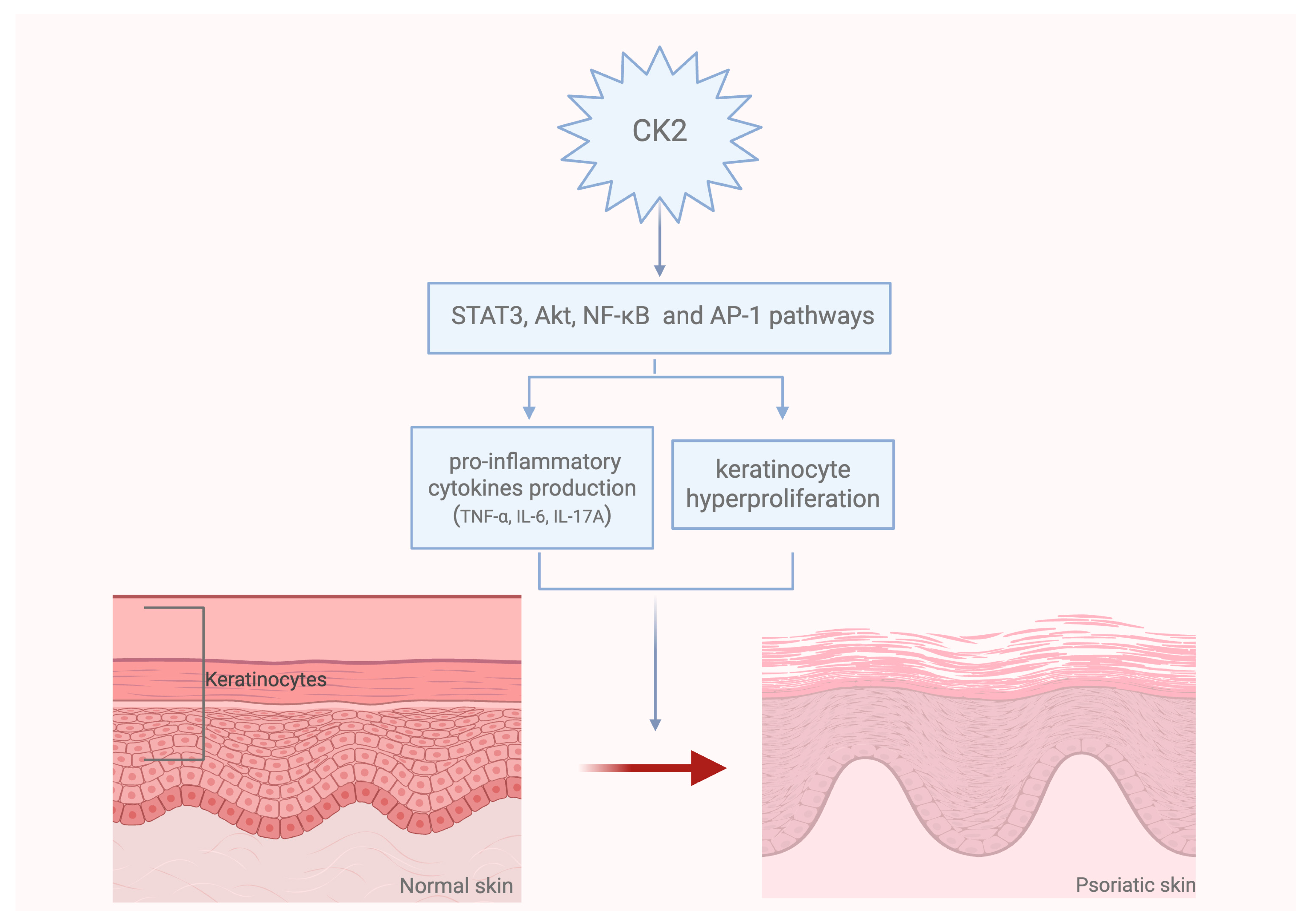
3. Role of CK2 in the Pathogenesis of Skin Diseases
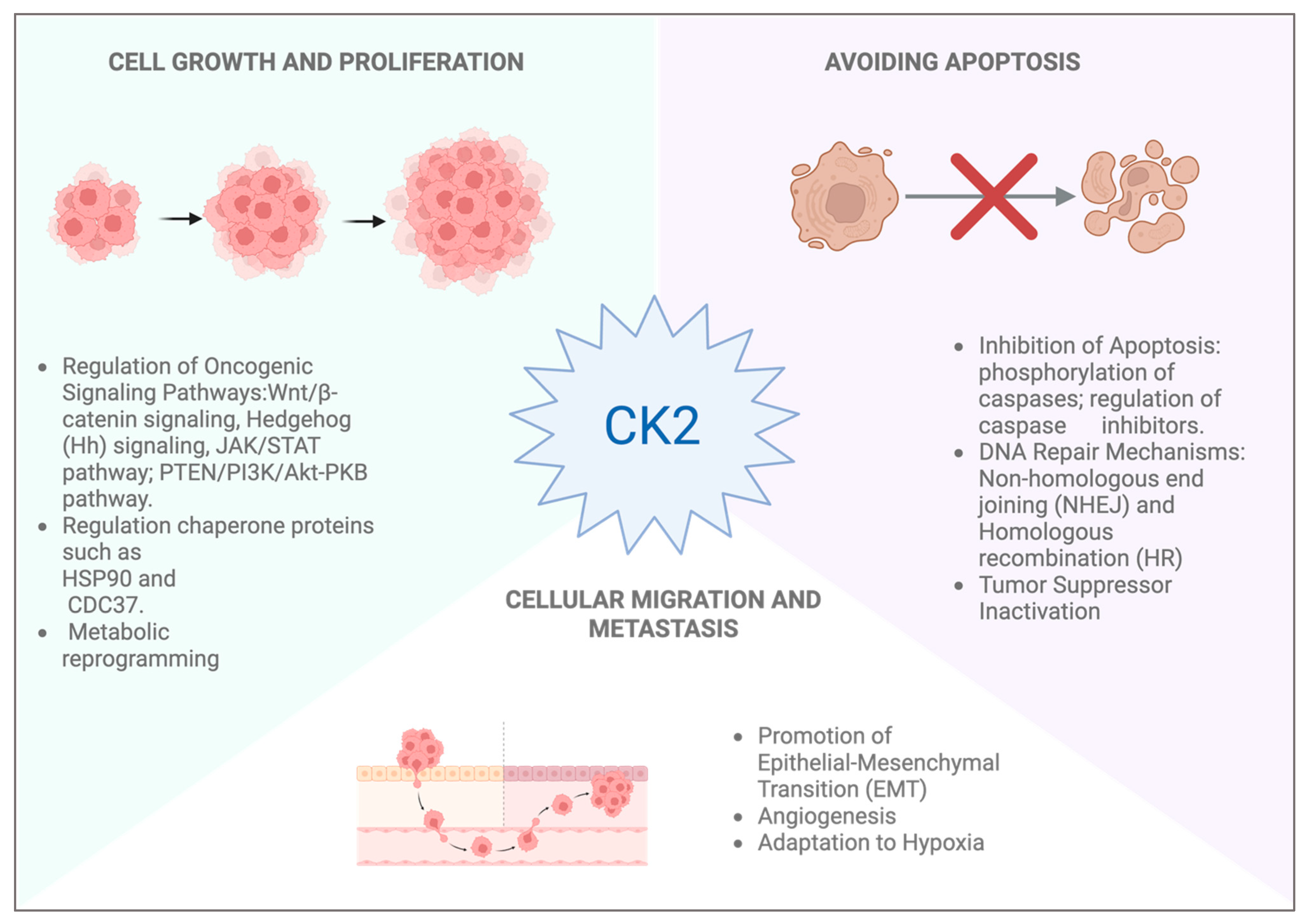
4. CK2 Role in Skin Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AKT | Ak strain transforming/Protein kinase B |
| ALM | Acral lentiginous melanoma |
| ATP | Adenosine Triphosphate |
| α-TAT1 | α-tubulin acetyltransferase |
| AP-1 | Activator protein 1 |
| ARC | Apoptosis repressor with caspase recruitment domain |
| BCC | Basal cell carcinoma |
| BCL9 | B-cell lymphoma 9 protein |
| Bcl-xL | B-cell lymphoma-extra large |
| BCRP | Breast cancer resistance protein |
| BRAF | V-Raf Murine Sarcoma Viral Oncogene Homolog B |
| CAPj | Cold atmospheric plasma jet |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| Ci | Cubitus interruptus |
| CM | Cutaneous melanoma |
| CK2 | Casein kinase 2 |
| CX-4945 | Silmitasertib |
| CREB | cyclic AMP response element-binding protein |
| dcSSc | Diffuse cutaneous systemic sclerosis |
| DUSP6 | Dual specificity phosphatase 6 |
| Dvl | Dishevelled |
| ECM | Extracellular Matrix |
| ERK | Extracellular signal-regulated kinase |
| EMT | Epithelial-mesenchymal transition |
| Fzd | Frizzled receptor |
| GSK3 | Glycogen Synthase Kinase 3 |
| IKB-α | NF-kappa-B inhibitor alpha |
| IKKγ | IκB kinase-γ |
| IFN-γ | Interferon Gamma |
| IRE1 | Inositol-requiring enzyme 1 |
| JAK | Janus kinase |
| KSR1 | Kinase suppressor of Ras 1 |
| LEF | Lymphoid enhancer-binding factor |
| lcSSc | limited cutaneous systemic sclerosis |
| LMM | Lentigo maligna melanoma |
| LRP5/6 | Low-density lipoprotein receptor-related protein 5/6 |
| MAPK | Mitogen-activated protein kinase |
| MITF | Microphthalmia-associated transcription factor |
| MMPs | Matalloproteinases |
| MRP1 | Multidrug resistance-associated protein 1 |
| NEMO | NF-κB essential modifier |
| NF1 | Neurofibromin 1 |
| NM | Nodular melanoma |
| NMSC | Non-melanoma skin cancers |
| NF-kB | Nuclear Factor-kappa B |
| NRAS | Neuroblastoma RAS viral oncogene homolog |
| PDK1 | Phosphoinositide-Dependent Kinase-1 |
| P-gp | P-glycoprotein |
| PERK | Protein kinase RNA-like endoplasmic reticulum kinase |
| PI3K | Phosphoinositide 3-kinase |
| PTEN | Phosphatase and tensin homolog |
| RIPK4 | Receptor-interacting serine/threonine-protein kinase 4 |
| RNF43 | Ring Finger Protein 43 |
| SCC | Squamous cell carcinoma |
| SMAD | Suppressor of Mothers Against Decapentaplegic |
| SMO | GPCR-like protein Smoothened |
| SSc | Systemic Sclerosis |
| SSM | Superficial spreading melanoma |
| SOCS3 | Suppressor of Cytokine Signaling 3 |
| STAT | Signal transducer and activator of transcription |
| TBB | 4,5,6,7-tetrabromobenzotriazole |
| TCF | T cell factor |
| TGF-ß | Transforming Growth Factor-beta |
| UPR | Unfolded protein response |
| UV | Ultraviolet |
| Wnt | Wingless/Integrated |
| XBP1 | X-box binding protein 1 |
References
- Griffiths, C.E.M.; Armstrong, A.W.; Gudjonsson, J.E.; Barker, J. Psoriasis. Lancet 2021, 397, 1301–1315. [Google Scholar] [CrossRef] [PubMed]
- Tasdogan, A.; Sullivan, R.J.; Katalinic, A.; Lebbe, C.; Whitaker, D.; Puig, S.; van de Poll-Franse, L.V.; Massi, D.; Schadendorf, D. Cutaneous melanoma. Nat. Rev. Dis. Primers 2025, 11, 23. [Google Scholar] [CrossRef]
- Halloran, D.; Pandit, V.; Nohe, A. The Role of Protein Kinase CK2 in Development and Disease Progression: A Critical Review. J. Dev. Biol. 2022, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Borgo, C.; D’Amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef]
- Huang, W.; Zheng, X.; Huang, Q.; Weng, D.; Yao, S.; Zhou, C.; Li, Q.; Hu, Y.; Xu, W.; Huang, K. Protein Kinase CK2 Promotes Proliferation, Abnormal Differentiation, and Proinflammatory Cytokine Production of Keratinocytes via Regulation of STAT3 and Akt Pathways in Psoriasis. Am. J. Pathol. 2023, 193, 567–578. [Google Scholar] [CrossRef]
- D’Amore, C.; Borgo, C.; Sarno, S.; Salvi, M. Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy-potential clinical relevance. Cell. Oncol. 2020, 43, 1003–1016. [Google Scholar] [CrossRef]
- Drygin, D.; Ho, C.B.; Omori, M.; Bliesath, J.; Proffitt, C.; Rice, R.; Siddiqui-Jain, A.; O’Brien, S.; Padgett, C.; Lim, J.K.C.; et al. Protein kinase CK2 modulates IL-6 expression in inflammatory breast cancer. Biochem. Biophys. Res. Commun. 2011, 415, 163–167. [Google Scholar] [CrossRef]
- Pinna, L.A. A historical view of protein kinase CK2. Cell. Mol. Biol. Res. 1994, 40, 383–390. [Google Scholar] [PubMed]
- Pinna, L.A. Protein kinase CK2: A challenge to canons. J. Cell Sci. 2002, 115 Pt 20, 3873–3878. [Google Scholar] [CrossRef]
- Montenarh, M.; Götz, C. Protein Kinase CK2α′, More than a Backup of CK2α. Cells 2023, 12, 2834. [Google Scholar] [CrossRef]
- Guerra, B.; Siemer, S.; Boldyreff, B.; Issinger, O.G. Protein kinase CK2: Evidence for a protein kinase CK2beta subunit fraction, devoid of the catalytic CK2alpha subunit, in mouse brain and testicles. FEBS Lett. 1999, 462, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, W.; Lindell, T.J. Purification of rat liver nuclear protein kinase NII. J. Biol. Chem. 1977, 252, 6660–6665. [Google Scholar] [CrossRef] [PubMed]
- Pyerin, W.; Ackermann, K. The genes encoding human protein kinase CK2 and their functional links. Prog. Nucleic Acid Res. Mol. Biol. 2003, 74, 239–273. [Google Scholar] [CrossRef]
- Yang-Feng, T.L.; Naiman, T.; Kopatz, I.; Eli, D.; Dafni, N.; Canaani, D. Assignment of the human casein kinase II alpha′ subunit gene (CSNK2A1) to chromosome 16p13.2-p13.3. Genomics 1994, 19, 173. [Google Scholar] [CrossRef]
- Pinna, L.A. The raison d’être of constitutively active protein kinases: The lesson of CK2. Acc. Chem. Res. 2003, 36, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Spinello, Z.; Fregnani, A.; Quotti Tubi, L.; Trentin, L.; Piazza, F.; Manni, S. Targeting Protein Kinases in Blood Cancer: Focusing on CK1α and CK2. Int. J. Mol. Sci. 2021, 22, 3716. [Google Scholar] [CrossRef]
- Di Maira, G.; Salvi, M.; Arrigoni, G.; Marin, O.; Sarno, S.; Brustolon, F.; Pinna, L.A.; Ruzzene, M. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005, 12, 668–677. [Google Scholar] [CrossRef]
- Ruzzene, M.; Bertacchini, J.; Toker, A.; Marmiroli, S. Cross-talk between the CK2 and AKT signaling pathways in cancer. Adv. Biol. Regul. 2017, 64, 1–8. [Google Scholar] [CrossRef]
- Guerra, B. Protein kinase CK2 subunits are positive regulators of AKT kinase. Int. J. Oncol. 2006, 28, 685–693. [Google Scholar] [CrossRef]
- Cordier, F.; Chaffotte, A.; Terrien, E.; Préhaud, C.; Theillet, F.X.; Delepierre, M.; Lafon, M.; Buc, H.; Wolff, N. Ordered phosphorylation events in two independent cascades of the PTEN C-tail revealed by NMR. J. Am. Chem. Soc. 2012, 134, 20533–20543. [Google Scholar] [CrossRef]
- Gowda, C.; Song, C.; Kapadia, M.; Payne, J.L.; Hu, T.; Ding, Y.; Dovat, S. Regulation of cellular proliferation in acute lymphoblastic leukemia by Casein Kinase II (CK2) and Ikaros. Adv. Biol. Regul. 2017, 63, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Kato, T., Jr.; Delhase, M.; Hoffmann, A.; Karin, M. CK2 Is a C-Terminal IkappaB Kinase Responsible for NF-kappaB Activation during the UV Response. Mol. Cell 2003, 12, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Westerheide, S.D.; Hanson, J.L.; Baldwin, A.S., Jr. Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J. Biol. Chem. 2000, 275, 32592–32597. [Google Scholar] [CrossRef]
- Parhar, K.; Morse, J.; Salh, B. The role of protein kinase CK2 in intestinal epithelial cell inflammatory signaling. Int. J. Color. Dis. 2007, 22, 601–609. [Google Scholar] [CrossRef]
- Qaiser, F.; Trembley, J.H.; Sadiq, S.; Muhammad, I.; Younis, R.; Hashmi, S.N.; Murtaza, B.; Rector, T.S.; Naveed, A.K.; Ahmed, K. Examination of CK2α and NF-κB p65 expression in human benign prostatic hyperplasia and prostate cancer tissues. Mol. Cell. Biochem. 2016, 420, 43–51. [Google Scholar] [CrossRef]
- Trembley, J.H.; Kren, B.T.; Abedin, M.J.; Shaughnessy, D.P.; Li, Y.; Dehm, S.M.; Ahmed, K. CK2 Pro-Survival Role in Prostate Cancer Is Mediated via Maintenance and Promotion of Androgen Receptor and NFκB p65 Expression. Pharmaceuticals 2019, 12, 89. [Google Scholar] [CrossRef] [PubMed]
- Carter-Su, C.; Argetsinger, L.S. JAKs, Stats, and CK2? Blood 2011, 118, 5–6. [Google Scholar] [CrossRef]
- Zheng, Y.; Qin, H.; Frank, S.J.; Deng, L.; Litchfield, D.W.; Tefferi, A.; Pardanani, A.; Lin, F.T.; Li, J.; Sha, B.; et al. A CK2-dependent mechanism for activation of the JAK-STAT signaling pathway. Blood 2011, 118, 156–166. [Google Scholar] [CrossRef]
- Seldin, D.C.; Landesman-Bollag, E.; Farago, M.; Currier, N.; Lou, D.; Dominguez, I. CK2 as a positive regulator of Wnt signalling and tumourigenesis. Mol. Cell. Biochem. 2005, 274, 63–67. [Google Scholar] [CrossRef]
- Filhol, O.; Leroy, D.; Valéro, E.; Loue-Mackenbach, P.; Hériché, J.K.; Goldberg, Y.; Cochet, C.; Chambaz, E.M. Has protein kinase CK2 a role in the intracellular mitogenic signalling? Comptes Rendus Seances Soc. Biol. Fil. 1995, 189, 59–69. [Google Scholar]
- Filhol-Cochet, O.; Loue-Mackenbach, P.; Cochet, C.; Chambaz, E.M. Casein kinase 2 and the cell response to growth factors. Cell. Mol. Biol. Res. 1994, 40, 529–537. [Google Scholar]
- McKendrick, L.; Meek, D.W. A novel system to investigate the phosphorylation of the p53 tumor suppressor protein by the protein kinase CK2. Cell. Mol. Biol. Res. 1994, 40, 555–561. [Google Scholar] [PubMed]
- McKendrick, L.; Milne, D.; Meek, D. Protein kinase CK2-dependent regulation of p53 function: Evidence that the phosphorylation status of the serine 386 (CK2) site of p53 is constitutive and stable. Mol. Cell. Biochem. 1999, 191, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Liu, Y.; Xia, R.; Tong, C.; Yue, T.; Jiang, J.; Jia, J. Casein kinase 2 promotes Hedgehog signaling by regulating both smoothened and Cubitus interruptus. J. Biol. Chem. 2010, 285, 37218–37226. [Google Scholar] [CrossRef] [PubMed]
- You, E.; Jeong, J.; Lee, J.; Keum, S.; Hwang, Y.E.; Choi, J.H.; Rhee, S. Casein kinase 2 promotes the TGF-β-induced activation of α-tubulin acetyltransferase 1 in fibroblasts cultured on a soft matrix. BMB Rep. 2022, 55, 192–197. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Y.; Cui, L.; Shi, Y.; Guo, C. Advances in the pathogenesis of psoriasis: From keratinocyte perspective. Cell Death Dis. 2022, 13, 81. [Google Scholar] [CrossRef]
- Ben Abdallah, H.; Johansen, C.; Iversen, L. Key Signaling Pathways in Psoriasis: Recent Insights from Antipsoriatic Therapeutics. Psoriasis 2021, 11, 83–97. [Google Scholar] [CrossRef]
- Jung, D.H.; Park, H.J.; Byun, H.E.; Park, Y.M.; Kim, T.W.; Kim, B.O.; Um, S.H.; Pyo, S. Diosgenin inhibits macrophage-derived inflammatory mediators through downregulation of CK2, JNK, NF-kappaB and AP-1 activation. Int. Immunopharmacol. 2010, 10, 1047–1054. [Google Scholar] [CrossRef]
- Yoshihisa, Y.; Andoh, T.; Rehman, M.U.; Shimizu, T. The regulation of protein kinase casein kinase II by apigenin is involved in the inhibition of ultraviolet B-induced macrophage migration inhibitory factor-mediated hyperpigmentation. Phytother. Res. 2020, 34, 1320–1328. [Google Scholar] [CrossRef]
- Mitev, V.I.; Miteva, L.; Dourmishev, A. Casein kinase II activity in psoriatic epidermis. Arch. Dermatol. Res. 1992, 284, 111–113. [Google Scholar] [CrossRef]
- Iyer, K.; Chen, Z.; Ganapa, T.; Wu, B.M.; Tawil, B.; Linsley, C.S. Keratinocyte Migration in a Three-Dimensional In Vitro Wound Healing Model Co-Cultured with Fibroblasts. Tissue Eng. Regen. Med. 2018, 15, 721–733. [Google Scholar] [CrossRef]
- LeRoy, E.C.; Black, C.; Fleischmajer, R.; Jablonska, S.; Krieg, T.; Medsger, T.A., Jr.; Rowell, N.; Wollheim, F. Scleroderma (systemic sclerosis): Classification, subsets and pathogenesis. J. Rheumatol. 1988, 15, 202–205. [Google Scholar] [PubMed]
- Mouawad, J.E.; Feghali-Bostwick, C. The Molecular Mechanisms of Systemic Sclerosis-Associated Lung Fibrosis. Int. J. Mol. Sci. 2023, 24, 2963. [Google Scholar] [CrossRef]
- Zhang, Y.; Dees, C.; Beyer, C.; Lin, N.Y.; Distler, A.; Zerr, P.; Palumbo, K.; Susok, L.; Kreuter, A.; Distler, O.; et al. Inhibition of casein kinase II reduces TGFβ induced fibroblast activation and ameliorates experimental fibrosis. Ann. Rheum. Dis. 2015, 74, 936–943. [Google Scholar] [CrossRef]
- Kim, S.; Ham, S.; Yang, K.; Kim, K. Protein kinase CK2 activation is required for transforming growth factor β-induced epithelial-mesenchymal transition. Mol. Oncol. 2018, 12, 1811–1826. [Google Scholar] [CrossRef]
- Mathew-Steiner, S.S.; Roy, S.; Sen, C.K. Collagen in Wound Healing. Bioengineering 2021, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Lee, S.Y. CK2 inhibitor CX4945 inhibits collagen degradation of HaCaT human keratinocyte cells via attenuation of MMP-1 secretion. Mol. Biol. Rep. 2023, 50, 9691–9698. [Google Scholar] [CrossRef]
- Wu, P.S.; Wong, T.H.; Hou, C.W.; Chu, T.P.; Lee, J.W.; Lou, B.S.; Lin, M.H. Cold Atmospheric Plasma Jet Promotes Wound Healing Through CK2-Coordinated PI3K/AKT and MAPK Signaling Pathways. Mol. Cell. Proteom. MCP 2025, 24, 100962. [Google Scholar] [CrossRef] [PubMed]
- Firnau, M.B.; Brieger, A. CK2 and the Hallmarks of Cancer. Biomedicines 2022, 10, 1987. [Google Scholar] [CrossRef]
- Roky, A.H.; Islam, M.M.; Ahasan, A.M.F.; Mostaq, M.S.; Mahmud, M.Z.; Amin, M.N.; Mahmud, M.A. Overview of skin cancer types and prevalence rates across continents. Cancer Pathog. Ther. 2025, 3, 89–100. [Google Scholar] [CrossRef]
- Azimi, A.; Caramuta, S.; Seashore-Ludlow, B.; Boström, J.; Robinson, J.L.; Edfors, F.; Tuominen, R.; Kemper, K.; Krijgsman, O.; Peeper, D.S.; et al. Targeting CDK2 overcomes melanoma resistance against BRAF and Hsp90 inhibitors. Mol. Syst. Biol. 2018, 14, e7858. [Google Scholar] [CrossRef] [PubMed]
- Dhomen, N.; Reis-Filho, J.S.; da Rocha Dias, S.; Hayward, R.; Savage, K.; Delmas, V.; Larue, L.; Pritchard, C.; Marais, R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell 2009, 15, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Li Pomi, F.; Borgia, F.; Custurone, P.; Vaccaro, M.; Pioggia, G.; Gangemi, S. Role of HMGB1 in Cutaneous Melanoma: State of the Art. Int. J. Mol. Sci. 2022, 23, 9327. [Google Scholar] [CrossRef]
- Chua, M.M.; Ortega, C.E.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.L.; Dominguez, I. CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals 2017, 10, 18. [Google Scholar] [CrossRef]
- Rabalski, A.J.; Gyenis, L.; Litchfield, D.W. Molecular Pathways: Emergence of Protein Kinase CK2 (CSNK2) as a Potential Target to Inhibit Survival and DNA Damage Response and Repair Pathways in Cancer Cells. Clin. Cancer Res. 2016, 22, 2840–2847. [Google Scholar] [CrossRef]
- Xue, C.; Chu, Q.; Shi, Q.; Zeng, Y.; Lu, J.; Li, L. Wnt signaling pathways in biology and disease: Mechanisms and therapeutic advances. Signal Transduct. Target. Ther. 2025, 10, 106. [Google Scholar] [CrossRef]
- Dong, B.; Simonson, L.; Vold, S.; Oldham, E.; Barten, L.; Ahmad, N.; Chang, H. FZD6 Promotes Melanoma Cell Invasion but Not Proliferation by Regulating Canonical Wnt Signaling and Epithelial—Mesenchymal Transition. J. Investig. Dermatol. 2023, 143, 621–629.e626. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H.; Chen, Y.; Wang, H.; Tian, Y.; Yi, X.; Shi, Q.; Zhao, T.; Zhang, B.; Gao, T.; et al. Targeting Wnt/β-Catenin Signaling Exacerbates Ferroptosis and Increases the Efficacy of Melanoma Immunotherapy via the Regulation of MITF. Cells 2022, 11, 3580. [Google Scholar] [CrossRef]
- Wronski, N.; Madej, E.; Grabacka, M.; Brożyna, A.A.; Wolnicka-Glubisz, A. RIPK4 downregulation impairs Wnt3A-stimulated invasiveness via Wnt/β-catenin signaling in melanoma cells and tumor growth in vivo. Cell Signal. 2024, 113, 110938. [Google Scholar] [CrossRef]
- Damsky, W.E.; Curley, D.P.; Santhanakrishnan, M.; Rosenbaum, L.E.; Platt, J.T.; Gould Rothberg, B.E.; Taketo, M.M.; Dankort, D.; Rimm, D.L.; McMahon, M.; et al. β-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell 2011, 20, 741–754. [Google Scholar] [CrossRef]
- Qin, Z.; Zheng, M. Advances in targeted therapy and immunotherapy for melanoma (Review). Exp. Ther. Med. 2023, 26, 416. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Ritt, D.A.; Morrison, D.K.; Der, C.J.; Cox, A.D. Protein Kinase CK2α Maintains Extracellular Signal-regulated Kinase (ERK) Activity in a CK2α Kinase-independent Manner to Promote Resistance to Inhibitors of RAF and MEK but Not ERK in BRAF Mutant Melanoma. J. Biol. Chem. 2016, 291, 17804–17815. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.; Clifton-Bligh, R.; Molloy, M.P. Phosphoproteomics of MAPK inhibition in BRAF-mutated cells and a role for the lethal synergism of dual BRAF and CK2 inhibition. Mol. Cancer Ther. 2014, 13, 1894–1906. [Google Scholar] [CrossRef]
- Alkassis, S.; Shatta, M.; Wong, D.J. Therapeutic Advances in Advanced Basal Cell Carcinoma. Cancers 2024, 16, 3075. [Google Scholar] [CrossRef]
- McCormick, D.; Eroglu, Z.; Cowey, C.L. 32283 A phase I study of CX-4945 administered orally twice daily to patients with advanced basal cell carcinoma. J. Am. Acad. Dermatol. 2022, 87 (Suppl. S3), AB10. [Google Scholar] [CrossRef]
- Khalifa, H.; ElHady, A.K.; Liu, T.; Elgaher, W.A.M.; Filhol-Cochet, O.; Cochet, C.; Abadi, A.H.; Hamed, M.M.; Abdel-Halim, M.; Engel, M. Discovery of a novel, selective CK2 inhibitor class with an unusual basic scaffold. Eur. J. Med. Chem. 2025, 282, 117048. [Google Scholar] [CrossRef] [PubMed]
- Borgo, C.; Ruzzene, M. Protein kinase CK2 inhibition as a pharmacological strategy. Adv. Protein Chem. Struct. Biol. 2021, 124, 23–46. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, G. Application of JAK inhibitors in paradoxical reaction through immune-related dermatoses. Front. Immunol. 2024, 15, 1341632. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Type | Phase I, multicenter, open-label clinical trial |
| Clinical Trial ID | NCT03897036 |
| Patient Population | A total of 25 patients with locally advanced BCC (laBCC) or metastatic BCC (mBCC) who had prior failure or intolerance to Smo inhibitors |
| Dosing Regimen | A total of 1000 mg orally twice daily, administered over two 28-day cycles |
| Initial Cohort | A total of 8 patients in the Treatment–Duration–Increment cohort (focused on optimizing treatment duration) |
| Expansion Cohort | A total of 17 patients (evaluated safety and efficacy at the established dosing regimen) |
| Efficacy | Among 22 evaluable patients: 3 (with laBCC) achieved partial response, 10 had stable disease |
| Disease Control Rate (DCR) | A total of 80% in mBCC, 65% in laBCC |
| Median Progression-Free Survival (PFS) | A total of 9.2 months (laBCC), 3.7 months (mBCC) |
| Long-term Response | A total of 2 patients experienced PFS > 21 months |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scuruchi, M.; Speranza, D.; Bruschetta, G.; Vaccaro, F.; Galeano, M.; Pallio, G.; Vaccaro, M.; Borgia, F.; Li Pomi, F.; Collino, M.; et al. Protein Kinase CK2 Inhibition Represents a Pharmacological Chance for the Treatment of Skin Diseases. Int. J. Mol. Sci. 2025, 26, 5404. https://doi.org/10.3390/ijms26115404
Scuruchi M, Speranza D, Bruschetta G, Vaccaro F, Galeano M, Pallio G, Vaccaro M, Borgia F, Li Pomi F, Collino M, et al. Protein Kinase CK2 Inhibition Represents a Pharmacological Chance for the Treatment of Skin Diseases. International Journal of Molecular Sciences. 2025; 26(11):5404. https://doi.org/10.3390/ijms26115404
Chicago/Turabian StyleScuruchi, Michele, Desirèe Speranza, Giuseppe Bruschetta, Federico Vaccaro, Mariarosaria Galeano, Giovanni Pallio, Mario Vaccaro, Francesco Borgia, Federica Li Pomi, Massimo Collino, and et al. 2025. "Protein Kinase CK2 Inhibition Represents a Pharmacological Chance for the Treatment of Skin Diseases" International Journal of Molecular Sciences 26, no. 11: 5404. https://doi.org/10.3390/ijms26115404
APA StyleScuruchi, M., Speranza, D., Bruschetta, G., Vaccaro, F., Galeano, M., Pallio, G., Vaccaro, M., Borgia, F., Li Pomi, F., Collino, M., & Irrera, N. (2025). Protein Kinase CK2 Inhibition Represents a Pharmacological Chance for the Treatment of Skin Diseases. International Journal of Molecular Sciences, 26(11), 5404. https://doi.org/10.3390/ijms26115404