
FYCO1 Peptide Analogs: Design and Characterization of Autophagy Inhibitors as Co-Adjuvants in Taxane Chemotherapy of Prostate Cancer

 ,
,  ,
,  , , , , ,
, , , , , 
Abstract

1. Introduction
2. Results and Discussion
2.1. Computational Design of FYCO1-LIR Analogs
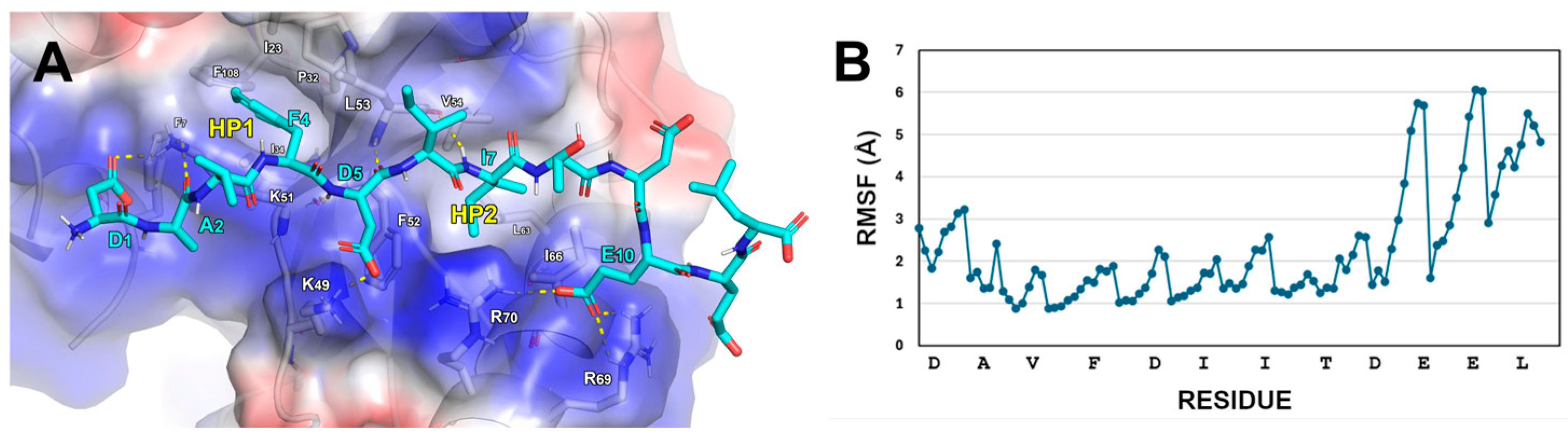
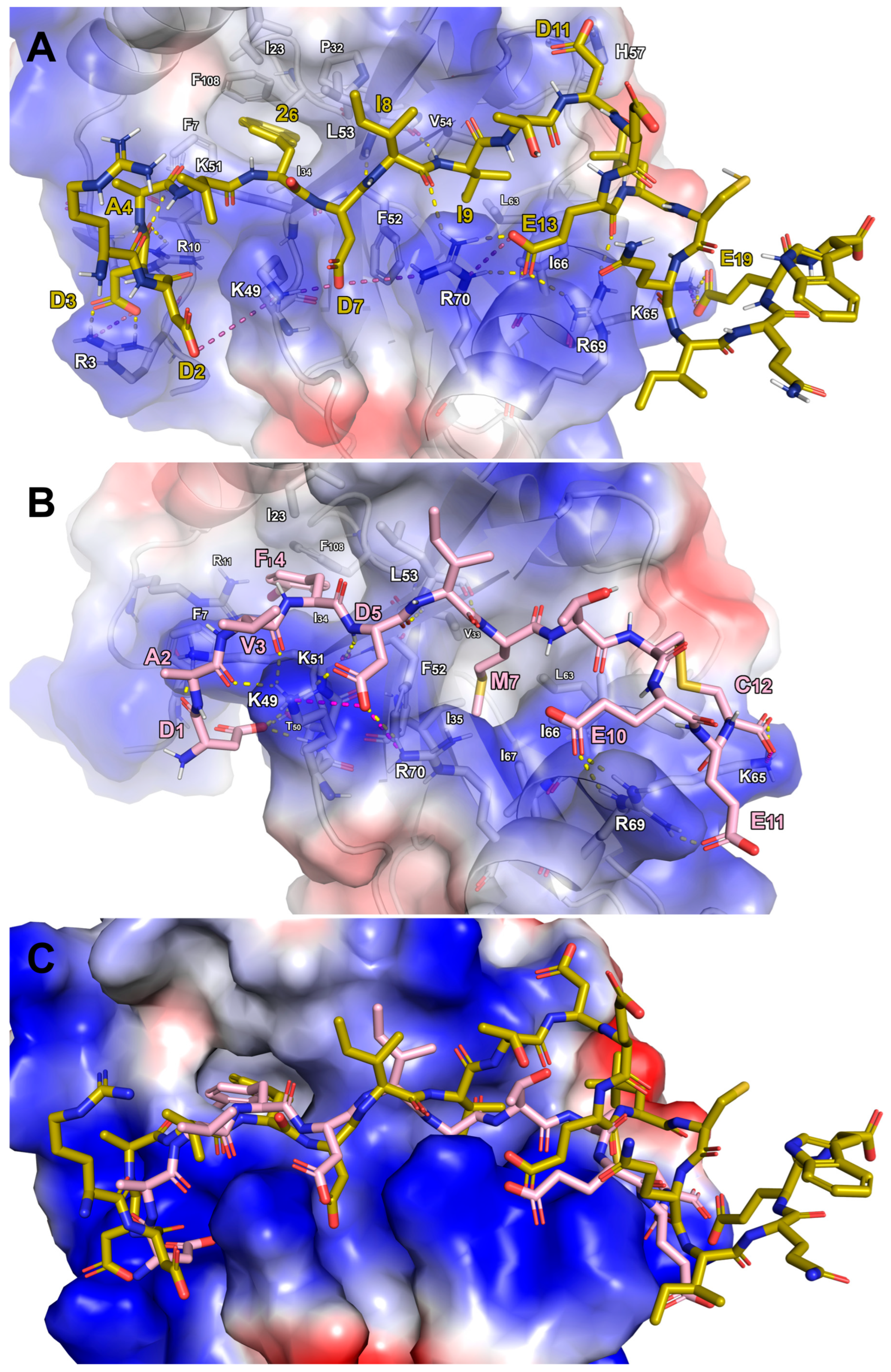
2.2. Design of FYCO1 Analogs
- -
- The D1 of FYCO1-LIR establishes contact with K51 on LC3B.
- -
- FYCO1-LIR’s F4 residue engages in cation-π stacking with the side chain of LC3B-K51. Its phenyl ring is also positioned within the hydrophobic pocket 1 (HP1) on LC3B, formed by the residues F7, I23, P32, I34, L53, and F108.
- -
- A hydrogen bond is formed between the NH group of D5 in FYCO1-LIR and the carbonyl group of LC3B-K51. Additionally, the acidic tail of D5 creates a salt bridge with LC3B-K49.
- -
- The side chain of I6 in FYCO1-LIR is solvent-exposed, while the side chain of I7 projects inward, interacting with the hydrophobic pocket 2 (HP2) on LC3B, which is composed of the amino acids I35, F52, V54, L63, I66, and I67.
- -
- The residue E10 of FYCO1-LIR interacts with the side chains of R69 and R70 on LC3B.
2.3. Design of Stapled Peptides
2.4. Synthesis of Peptides
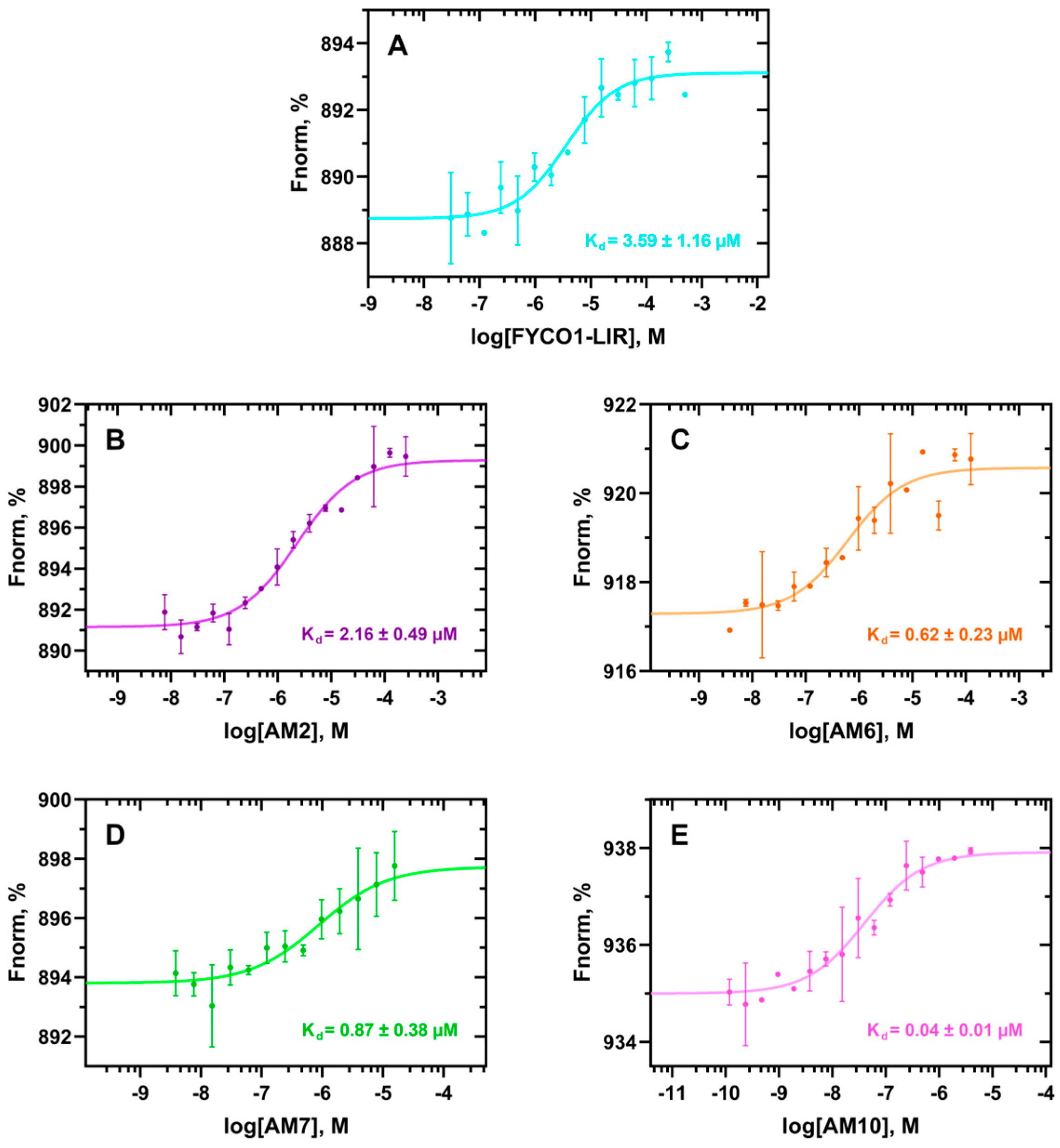
2.5. Biophysical Assays
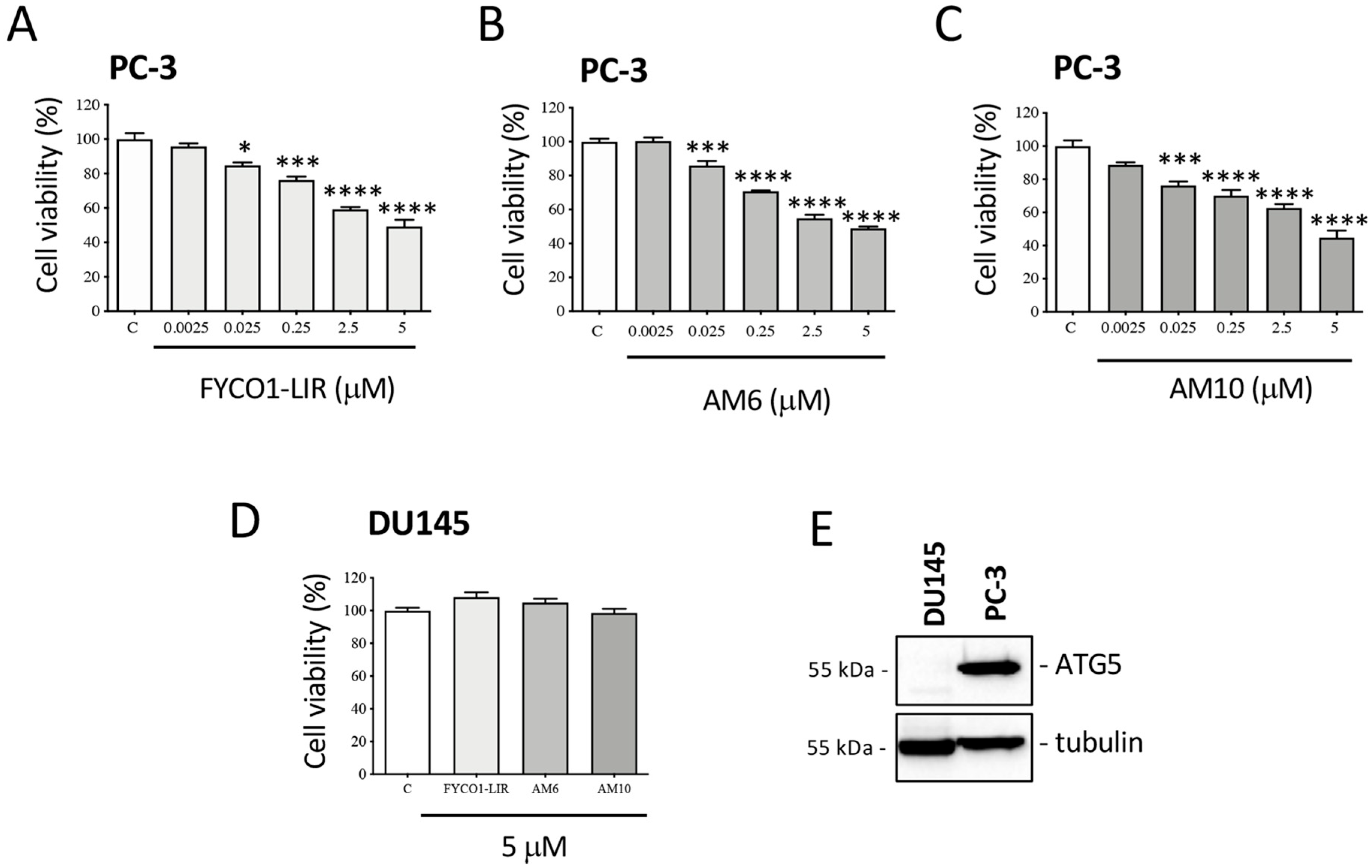
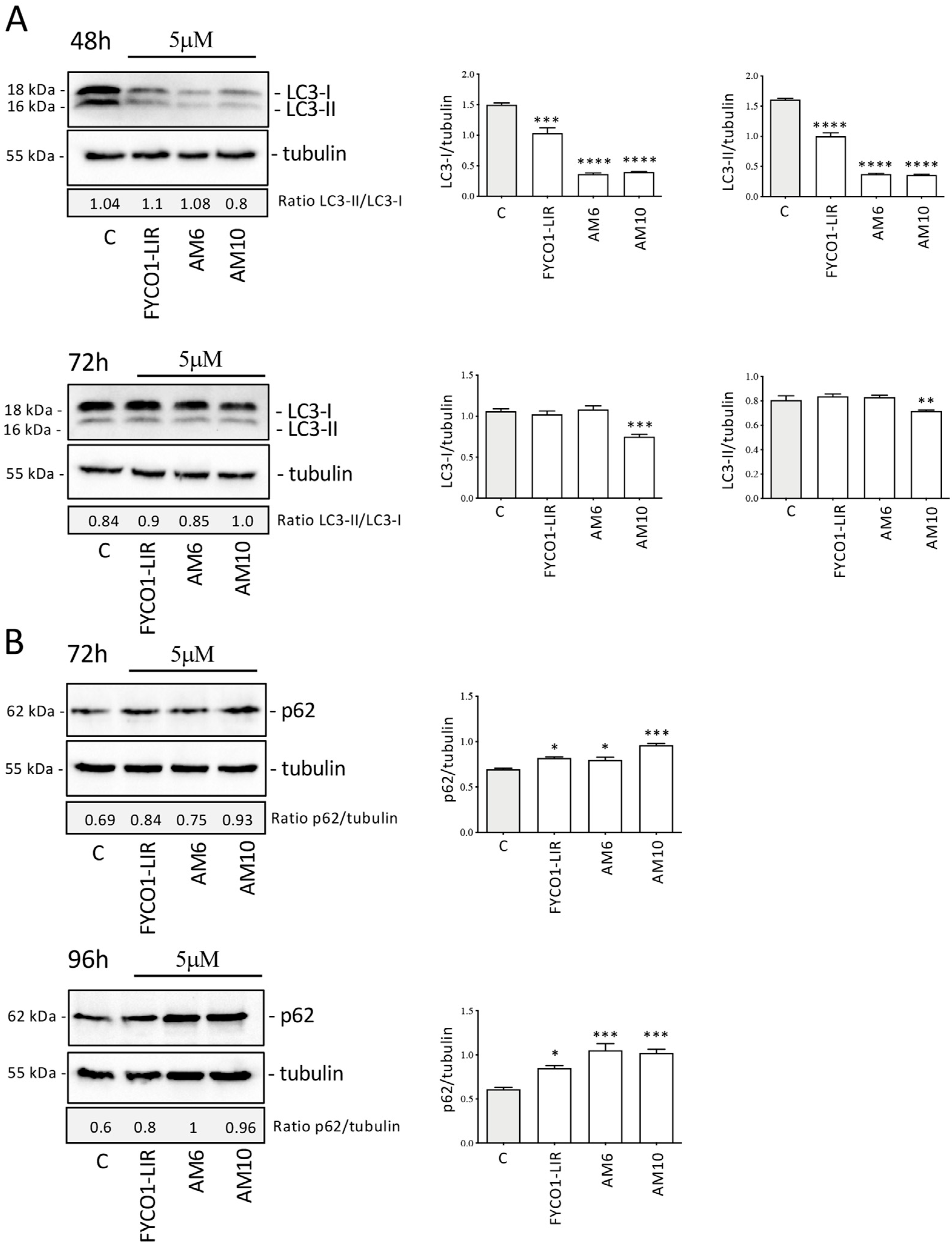
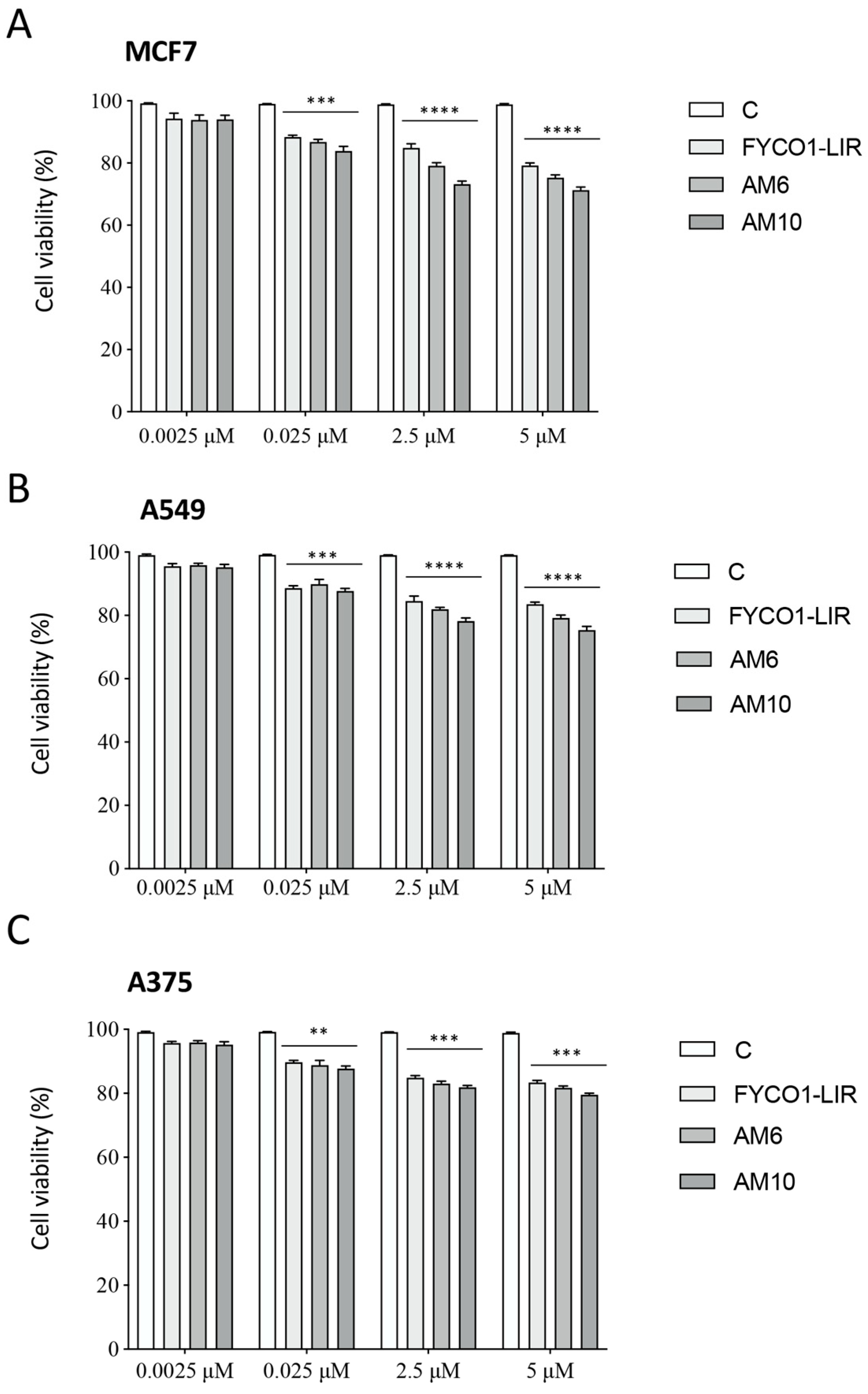
2.6. Biological Assays on PC-3 Cells
3. Materials and Methods
3.1. Computational Design of FYCO1 Analogs
- Peptide Transfer and Adaptation: The DDAVFDIITDEELCQIQESG peptide from the LC3A/FYCO1 complex was transferred onto the LC3B surface. This transfer was feasible because the FYCO1 segment (DAVFDIITDEEL) in the LC3B complex aligned perfectly with its counterpart in the LC3A structure. The sequence was then manually adjusted to match Comb1.
- Refinement: The resulting LC3B/Comb1 model was optimized using energy minimization and MD simulations, following the protocol previously described.
3.2. Synthesis of Peptides
3.3. Biophysical Assays
3.4. Cell Lines
3.5. Cell Viability Studies
3.6. Western Blot (WB) Assay
3.7. Cell Proliferation Studies
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network Organization of the Human Autophagy System. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Klionsky, D.J. Autophagosome Formation: Core Machinery and Adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Brumell, J.H. Bacteria-Autophagy Interplay: A Battle for Survival. Nat. Rev. Microbiol. 2014, 12, 101–114. [Google Scholar] [CrossRef]
- Jacquet, M.; Guittaut, M.; Fraichard, A.; Despouy, G. The Functions of Atg8-Family Proteins in Autophagy and Cancer: Linked or Unrelated? Autophagy 2021, 17, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Hain, A.U.; Miller, A.S.; Levitskaya, J.; Bosch, J. Virtual Screening and Experimental Validation Identify Novel Inhibitors of the Plasmodium Falciparum Atg8-Atg3 Protein-Protein Interaction. ChemMedChem 2016, 11, 900–910. [Google Scholar] [CrossRef]
- Villa, S.; Legnani, L.; Colombo, D.; Gelain, A.; Lammi, C.; Bongiorno, D.; Ilboudo, D.P.; Mcgee, K.E.; Bosch, J.; Grazioso, G. Structure-Based Drug Design, Synthesis and Biological Assays of P. Falciparum Atg3-Atg8 Protein-Protein Interaction Inhibitors. J. Comput. Aided Mol. Des. 2018, 32, 473–486. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin Is an Autophagy Receptor for Damaged Mitochondria in Parkin-Mediated Mitophagy That Is Disrupted by an ALS-Linked Mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef]
- McLendon, P.M.; Ferguson, B.S.; Osinska, H.; Shenuarin Bhuiyan, M.; James, J.; McKinsey, T.A.; Robbins, J. Tubulin Hyperacetylation Is Adaptive in Cardiac Proteotoxicity by Promoting Autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, E5178–E5186. [Google Scholar] [CrossRef]
- Chen, M.; Hong, M.J.; Sun, H.; Wang, L.; Shi, X.; Gilbert, B.E.; Corry, D.B.; Kheradmand, F.; Wang, J. Essential Role for Autophagy in the Maintenance of Immunological Memory against Influenza Infection. Nat. Med. 2014, 20, 503–510. [Google Scholar] [CrossRef]
- Quan, W.; Lim, Y.M.; Lee, M.S. Role of Autophagy in Diabetes and Endoplasmic Reticulum Stress of Pancreatic β-Cells. Exp. Mol. Med. 2012, 44, 81–88. [Google Scholar] [CrossRef]
- Sarparanta, J.; García-Macia, M.; Singh, R. Autophagy and Mitochondria in Obesity and Type 2 Diabetes. Curr. Diabetes Rev. 2016, 13, 352–369. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Yu, F.; Wang, J.; Guo, C.; Fan, X. Autophagy: A New Target for Nonalcoholic Fatty Liver Disease Therapy. Hepat. Med. 2016, ume 8, 27–37. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and Current Strategies for Targeting Autophagy for Cancer Treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Hernández, M.; Arias, A.; Martínez-García, D.; Pérez-Tomás, R.; Quesada, R.; Soto-Cerrato, V. Targeting Autophagy for Cancer Treatment and Tumor Chemosensitization. Cancers 2019, 11, 1599. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, S.; Bozkus, C.C.; Zografos, E.; Athanasiou, A.; Bongiovanni, A.M.; Doulaveris, G.; Bakoyiannis, C.N.; Theodoropoulos, G.E.; Zografos, G.C.; Witkin, S.S.; et al. Targeting Both Autophagy and Immunotherapy in Breast Cancer Treatment. Metabolites 2022, 12, 966. [Google Scholar] [CrossRef]
- Pimentel, J.M.; Zhou, J.Y.; Wu, G.S. Autophagy and Cancer Therapy. Cancer Lett. 2024, 605, 217285. [Google Scholar] [CrossRef]
- Niu, X.; You, Q.; Hou, K.; Tian, Y.; Wei, P.; Zhu, Y.; Gao, B.; Ashrafizadeh, M.; Aref, A.R.; Kalbasi, A.; et al. Autophagy in Cancer Development, Immune Evasion, and Drug Resistance. Drug Resist. Updates 2025, 78, 101170. [Google Scholar] [CrossRef]
- Mortezavi, A.; Salemi, S.; Rupp, N.J.; Rüschoff, J.H.; Hermanns, T.; Poyet, C.; Randazzo, M.; Simon, H.U.; Moch, H.; Sulser, T.; et al. Negative LC3b Immunoreactivity in Cancer Cells Is an Independent Prognostic Predictor of Prostate Cancer Specific Death. Oncotarget 2017, 8, 31765–31774. [Google Scholar] [CrossRef]
- Holah, N.S.; El-Dien, M.M.S.; Mahmoud, S.F. Expression of Autophagy Markers Beclin1 and LC3B in Prostatic Carcinoma: An Immunohistochemical Case-Control Study. Iran. J. Pathol. 2022, 17, 75–84. [Google Scholar] [CrossRef]
- Lamprou, I.; Tsolou, A.; Kakouratos, C.; Mitrakas, A.G.; Xanthopoulou, E.T.; Kassela, K.; Karakasiliotis, I.; Zois, C.E.; Giatromanolaki, A.; Koukourakis, M.I. Suppressed PLIN3 Frequently Occurs in Prostate Cancer, Promoting Docetaxel Resistance via Intensified Autophagy, an Event Reversed by Chloroquine. Med. Oncol. 2021, 38, 116. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Paskeh, M.D.A.; Mirzaei, S.; Gholami, M.H.; Zarrabi, A.; Hashemi, F.; Hushmandi, K.; Hashemi, M.; Nabavi, N.; Crea, F.; et al. Targeting Autophagy in Prostate Cancer: Preclinical and Clinical Evidence for Therapeutic Response. J. Exp. Clin. Cancer Res. 2022, 41, 105. [Google Scholar] [CrossRef] [PubMed]
- Lemos, G.; Fernandes, C.M.A.d.S.; Silva, F.H.; Calmasini, F.B. The Role of Autophagy in Prostate Cancer and Prostatic Diseases: A New Therapeutic Strategy. Prostate Cancer Prostatic Dis. 2024, 27, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wang, R. Small-Molecule Regulators of Autophagy and Their Potential Therapeutic Applications. ChemMedChem 2013, 8, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Fassi, E.M.A.; Garofalo, M.; Sgrignani, J.; Dei Cas, M.; Mori, M.; Roda, G.; Cavalli, A.; Grazioso, G. Focused Design of Novel Cyclic Peptides Endowed with GABARAP-Inhibiting Activity. Int. J. Mol. Sci. 2022, 23, 5070. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Olsvik, H.L.; Lamark, T.; Takagi, K.; Larsen, K.B.; Evjen, G.; Øvervatn, A.; Mizushima, T.; Johansen, X.T. FYCO1 Contains a C-Terminally Extended, LC3A/B-Preferring LC3-Interacting Region (LIR) Motif Required for Efficient Maturation of Autophagosomes during Basal Autophagy. J. Biol. Chem. 2015, 290, 29361–29374. [Google Scholar] [CrossRef]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.-A.; Lamark, T.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 Is a Rab7 Effector That Binds to LC3 and PI3P to Mediate Microtubule plus End–Directed Vesicle Transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef]
- Ma, J.; Becker, C.; Reyes, C.; Underhill, D.M. Cutting Edge: FYCO1 Recruitment to Dectin-1 Phagosomes Is Accelerated by Light Chain 3 Protein and Regulates Phagosome Maturation and Reactive Oxygen Production. J. Immunol. 2014, 192, 1356–1360. [Google Scholar] [CrossRef]
- Raiborg, C.; Wenzel, E.M.; Pedersen, N.M.; Olsvik, H.; Schink, K.O.; Schultz, S.W.; Vietri, M.; Nisi, V.; Bucci, C.; Brech, A.; et al. Repeated ER–Endosome Contacts Promote Endosome Translocation and Neurite Outgrowth. Nature 2015, 520, 234–238. [Google Scholar] [CrossRef]
- Cerulli, R.A.; Shehaj, L.; Brown, H.; Pace, J.; Mei, Y.; Kritzer, J.A. Stapled Peptide Inhibitors of Autophagy Adapter LC3B. ChemBioChem 2020, 21, 2777–2785. [Google Scholar] [CrossRef]
- Albani, M.; Fassi, E.M.A.; Moretti, R.M.; Garofalo, M.; Montagnani Marelli, M.; Roda, G.; Sgrignani, J.; Cavalli, A.; Grazioso, G. Computational Design of Novel Cyclic Peptides Endowed with Autophagy-Inhibiting Activity on Cancer Cell Lines. Int. J. Mol. Sci. 2024, 25, 4622. [Google Scholar] [CrossRef] [PubMed]
- Baliou, S.; Goulielmaki, M.; Ioannou, P.; Cheimonidi, C.; Trougakos, I.P.; Nagl, M.; Kyriakopoulos, A.M.; Zoumpourlis, V. Article Bromamine T (BAT) Exerts Stronger Anti-Cancer Properties than Taurine (Tau). Cancers 2021, 13, 182. [Google Scholar] [CrossRef]
- Potapskyi, E.; Kustrzyńska, K.; Łażewski, D.; Skupin-Mrugalska, P.; Lesyk, R.; Wierzchowski, M. Introducing Bromine to the Molecular Structure as a Strategy for Drug Design. J. Med. Sci. 2024, 93, e1128. [Google Scholar] [CrossRef]
- Xu, L.; Fan, X.; He, Y.; Xia, X.; Zhang, J. Design, Synthesis, and Biological Evaluation of Lysine-Stapled Peptide Inhibitors of P53-MDM2/MDMX Interactions with Potent Antitumor Activity In Vivo. J. Med. Chem. 2024, 67, 17893–17904. [Google Scholar] [CrossRef]
- Walensky, L.D.; Bird, G.H. Hydrocarbon-Stapled Peptides: Principles, Practice, and Progress. J. Med. Chem. 2014, 57, 6275–6288. [Google Scholar] [CrossRef]
- Tombling, B.; Lammi, C.; Lawrence, N.; Gilding, E.; Grazioso, G.; Craik, D.; Wang, C. Bioactive Cyclization Optimizes the Affinity of a Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Peptide Inhibitor. J. Med. Chem. 2021, 64, 2523–2533. [Google Scholar] [CrossRef]
- Ouyang, D.Y.; Xu, L.H.; He, X.H.; Zhang, Y.T.; Zeng, L.H.; Cai, J.Y.; Ren, S. Autophagy Is Differentially Induced in Prostate Cancer LNCaP, DU145 and PC-3 Cells via Distinct Splicing Profiles of ATG5. Autophagy 2013, 9, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Wade, C.A.; Kyprianou, N. Profiling Prostate Cancer Therapeutic Resistance. Int. J. Mol. Sci. 2018, 19, 904. [Google Scholar] [CrossRef]
- Loizzo, D.; Pandolfo, S.D.; Rogers, D.; Cerrato, C.; Di Meo, N.A.; Autorino, R.; Mirone, V.; Ferro, M.; Porta, C.; Stella, A.; et al. Novel Insights into Autophagy and Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 3826. [Google Scholar] [CrossRef]
- Bumbaca, B.; Li, W. Taxane Resistance in Castration-Resistant Prostate Cancer: Mechanisms and Therapeutic Strategies. Acta Pharm. Sin. B 2018, 8, 518–529. [Google Scholar] [CrossRef]
- Giacinti, S.; Poti, G.; Roberto, M.; Macrini, S.; Bassanelli, M.; Di Pietro, F.; Aschelter, A.M.; Ceribelli, A.; Ruggeri, E.M.; Marchetti, P. Molecular Basis of Drug Resistance and Insights for New Treatment Approaches in MCRPC. Anticancer. Res. 2018, 38, 6029–6039. [Google Scholar] [CrossRef] [PubMed]
- Kurganovs, N.J.; Engedal, N. To Eat or Not to Eat: A Critical Review on the Role of Autophagy in Prostate Carcinogenesis and Prostate Cancer Therapeutics. Front. Pharmacol. 2024, 15, 1419806. [Google Scholar] [CrossRef] [PubMed]
- Pickard, R.D.; Spencer, B.H.; McFarland, A.J.; Bernaitis, N.; Davey, A.K.; Perkins, A.V.; Chess-Williams, R.; McDermott, C.M.; Forbes, A.; Christie, D.; et al. Paradoxical Effects of the Autophagy Inhibitor 3-Methyladenine on Docetaxel-Induced Toxicity in PC-3 and LNCaP Prostate Cancer Cells. Naunyn Schmiedebergs Arch. Pharmacol. 2015, 388, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; He, W.Y.; Zeng, Y.Z.; Hossain, A.; Gou, X. Inhibiting Autophagy Overcomes Docetaxel Resistance in Castration-Resistant Prostate Cancer Cells. Int. Urol. Nephrol. 2018, 50, 675–686. [Google Scholar] [CrossRef]
- Cristofani, R.; Montagnani Marelli, M.; Cicardi, M.E.; Fontana, F.; Marzagalli, M.; Limonta, P.; Poletti, A.; Moretti, R.M. Dual Role of Autophagy on Docetaxel-Sensitivity in Prostate Cancer Cells. Cell Death Dis. 2018, 9, 889. [Google Scholar] [CrossRef]
- Hu, F.; Zhao, Y.; Yu, Y.; Fang, J.M.; Cui, R.; Liu, Z.Q.; Guo, X.L.; Xu, Q. Docetaxel-Mediated Autophagy Promotes Chemoresistance in Castration-Resistant Prostate Cancer Cells by Inhibiting STAT3. Cancer Lett. 2018, 416, 24–30. [Google Scholar] [CrossRef]
- Zeng, J.; Liu, W.; Fan, Y.Z.; He, D.L.; Li, L. PrLZ Increases Prostate Cancer Docetaxel Resistance by Inhibiting LKB1/AMPK-Mediated Autophagy. Theranostics 2018, 8, 109–123. [Google Scholar] [CrossRef]
- Peng, K.; Sun, A.; Zhu, J.; Gao, J.; Li, Y.; Shao, G.; Yang, W.; Lin, Q. Restoration of the ATG5-Dependent Autophagy Sensitizes DU145 Prostate Cancer Cells to Chemotherapeutic Drugs. Oncol. Lett. 2021, 22, 638. [Google Scholar] [CrossRef]
- Liu, B.; Miyake, H.; Nishikawa, M.; Tei, H.; Fujisawa, M. Expression Profile of Autophagy-Related Markers in Localized Prostate Cancer: Correlation with Biochemical Recurrence after Radical Prostatectomy. Urology 2015, 85, 1424–1430. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, FL, USA, 11–17 November 2006. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-Chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, Y.; Gong, Y.; Li, F.; Guo, Y.; Hu, S.; Liu, J.; Pan, L. Structural Basis of FYCO1 and MAP1LC3A Interaction Reveals a Novel Binding Mode for Atg8-Family Proteins. Autophagy 2016, 12, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.J.; Ha, H.; Lee, B.S.; Kim, B.H.; Song, H.K.; Kim, Y.K. LC3B Is an RNA-Binding Protein to Trigger Rapid MRNA Degradation during Autophagy. Nat. Commun. 2022, 13, 1436. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 ). The curly bracket (
). The curly bracket ( ) of AM7 joins two cysteines bis-alkylated by a para-dibromomethylbenzene (see the Materials and Methods section for details).
). The curly bracket () of AM7 joins two cysteines bis-alkylated by a para-dibromomethylbenzene (see the Materials and Methods section for details).
) of AM7 joins two cysteines bis-alkylated by a para-dibromomethylbenzene (see the Materials and Methods section for details).
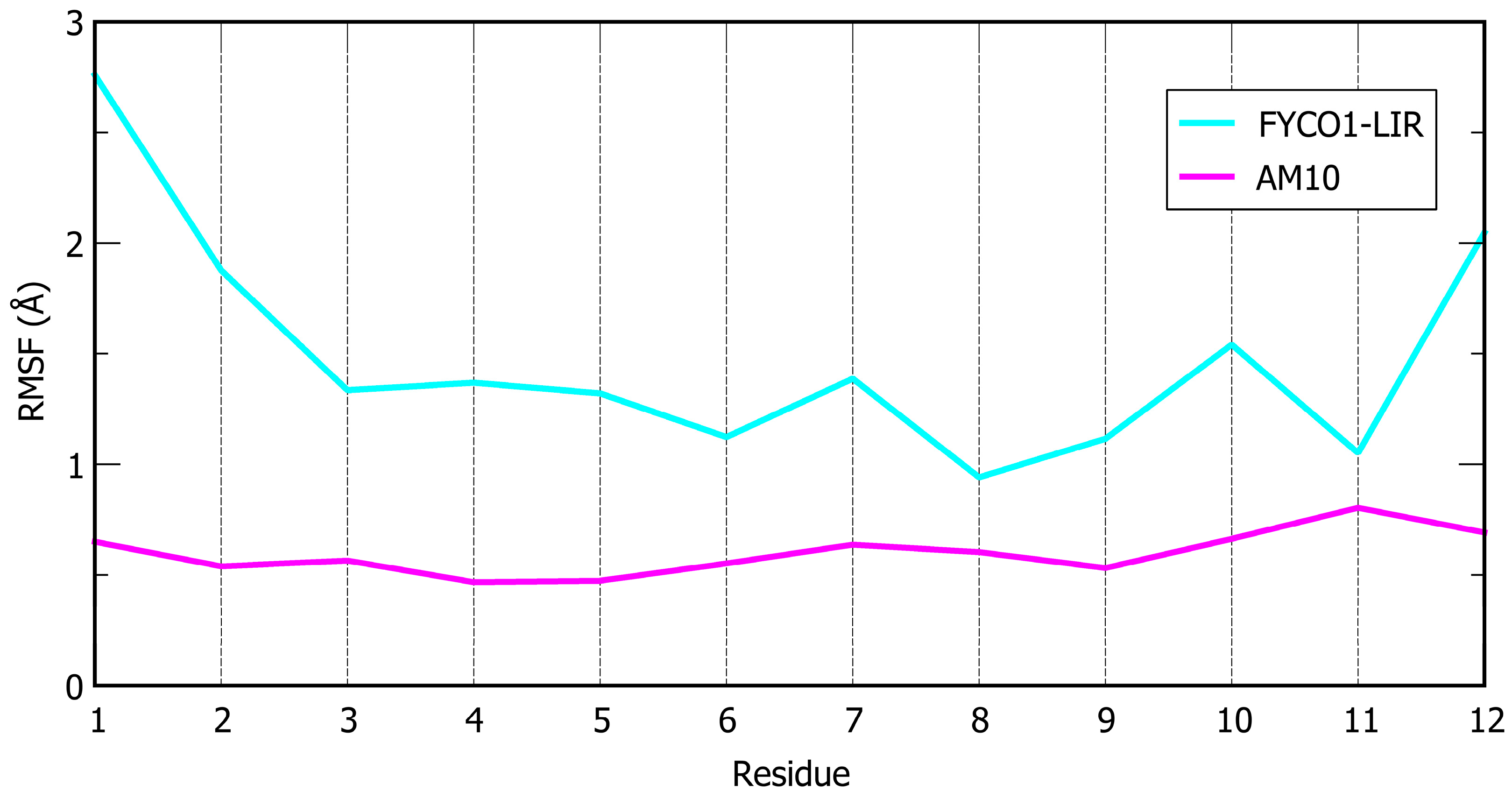
). The curly bracket () of AM7 joins two cysteines bis-alkylated by a para-dibromomethylbenzene (see the Materials and Methods section for details).| Peptide | Sequence | ΔG* (kcal/mol) | SD (kcal/mol) | Avg. Cα RMSF (Å) |
|---|---|---|---|---|
| FYCO1-LIR | DAVFDIITDEEL | −110.6 | 7.0 | 1.49 |
| Sequence mutation | ||||
| AM1 | DAVFDIMTDEEL | −113.0 | 7.3 | 1.52 |
| AM2 | DAVFIDIITDEEL | −119.2 | 10.1 | 1.29 |
| AM3 | DAVFBrDIITDEEL | −111.6 | 7.7 | 1.44 |
| AM4 | DAVFIDIMTDEEL | −123.8 | 5.8 | 1.13 |
| Backbone rigidification | ||||
| AM5 | DAVFDIITCEEC | −109.5 | 6.2 | 1.26 |
| AM6 | DAVFDIMTCEEC | −126.6 | 8.4 | 1.38 |
| AM7 | DAVFDIITCEEC | −118.5 | 8.3 | 1.00 |
| AM8 | DAVFIDIITCEEC | −109.2 | 5.3 | 1.03 |
| AM9 | DAVFBrDIITCEEC | −110.4 | 7.3 | 1.17 |
| AM10 | DAVFIDIMTCEEC | −137.1 | 6.1 | 0.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fassi, E.M.A.; Moretti, R.M.; Montagnani Marelli, M.; Garofalo, M.; Gori, A.; Pesce, C.; Albani, M.; Milano, E.G.; Sgrignani, J.; Cavalli, A.; et al. FYCO1 Peptide Analogs: Design and Characterization of Autophagy Inhibitors as Co-Adjuvants in Taxane Chemotherapy of Prostate Cancer. Int. J. Mol. Sci. 2025, 26, 5365. https://doi.org/10.3390/ijms26115365
Fassi EMA, Moretti RM, Montagnani Marelli M, Garofalo M, Gori A, Pesce C, Albani M, Milano EG, Sgrignani J, Cavalli A, et al. FYCO1 Peptide Analogs: Design and Characterization of Autophagy Inhibitors as Co-Adjuvants in Taxane Chemotherapy of Prostate Cancer. International Journal of Molecular Sciences. 2025; 26(11):5365. https://doi.org/10.3390/ijms26115365
Chicago/Turabian StyleFassi, Enrico Mario Alessandro, Roberta Manuela Moretti, Marina Montagnani Marelli, Mariangela Garofalo, Alessandro Gori, Cristiano Pesce, Marco Albani, Erica Ginevra Milano, Jacopo Sgrignani, Andrea Cavalli, and et al. 2025. "FYCO1 Peptide Analogs: Design and Characterization of Autophagy Inhibitors as Co-Adjuvants in Taxane Chemotherapy of Prostate Cancer" International Journal of Molecular Sciences 26, no. 11: 5365. https://doi.org/10.3390/ijms26115365
APA StyleFassi, E. M. A., Moretti, R. M., Montagnani Marelli, M., Garofalo, M., Gori, A., Pesce, C., Albani, M., Milano, E. G., Sgrignani, J., Cavalli, A., & Grazioso, G. (2025). FYCO1 Peptide Analogs: Design and Characterization of Autophagy Inhibitors as Co-Adjuvants in Taxane Chemotherapy of Prostate Cancer. International Journal of Molecular Sciences, 26(11), 5365. https://doi.org/10.3390/ijms26115365