
Experimental Animal Models of Phenylketonuria: Pros and Cons

 , , , and
, , , and
Abstract
1. Introduction
2. Phenylketonuria
2.1. Epidemiology of PKU
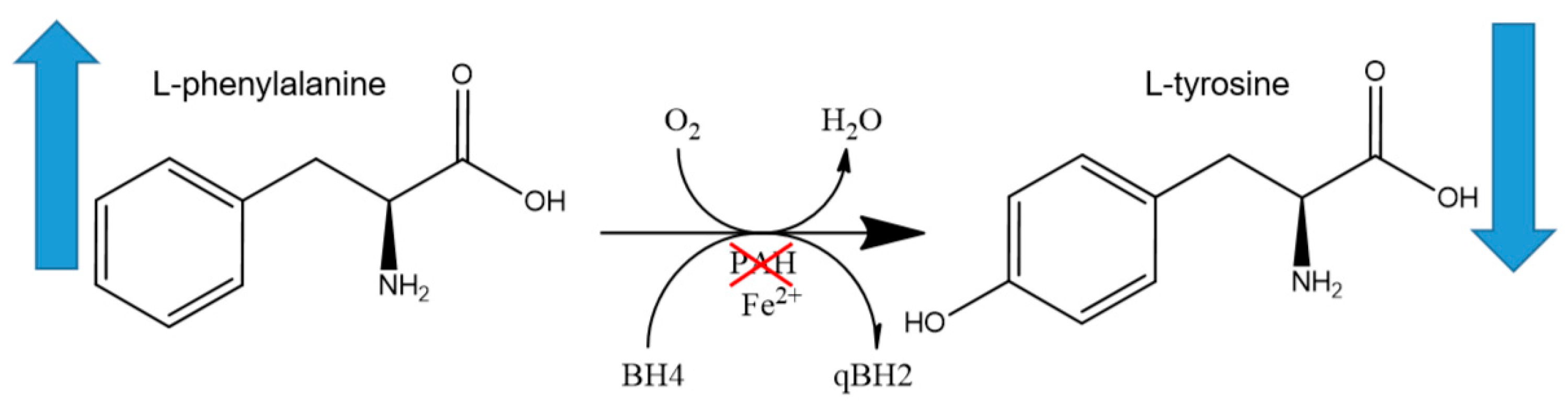
2.2. Genetics and Pathophysiology
3. Animal Models
3.1. Mouse Models
3.1.1. BTBR-Pahenu2/J
3.1.2. B6.BTBR-Pahenu2/MalnJ
3.1.3. BTBR-Pahenu1/J
3.1.4. B6(Cg)-Pahtm1.1(PAH*R408W)Xiwan/J
3.1.5. Mouse Models C57BL/6J-Pahem1Xiwan/J (PAH P281L)
3.1.6. PAH-KO
3.1.7. Pah-R261Q
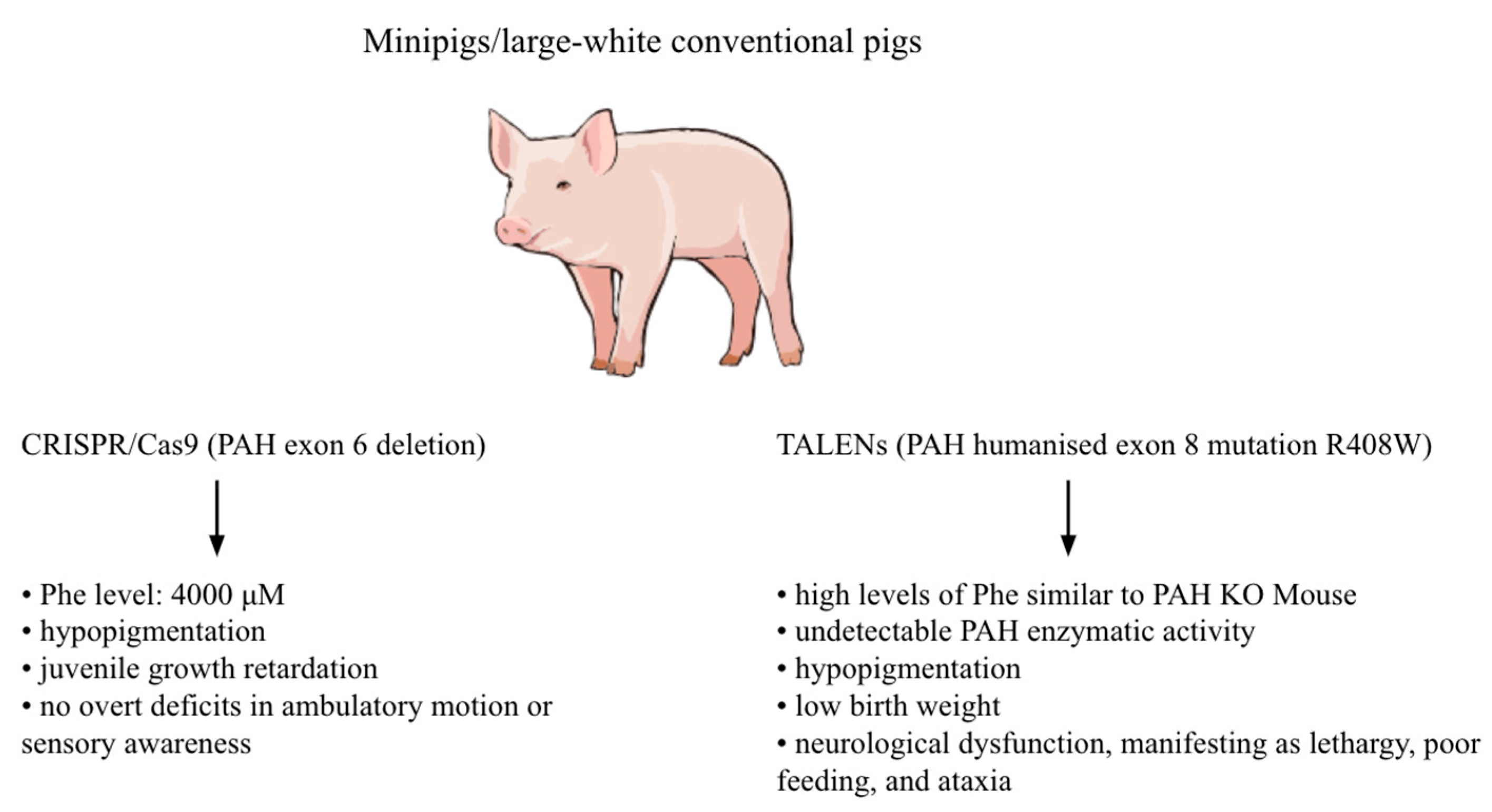
3.2. Pig Models
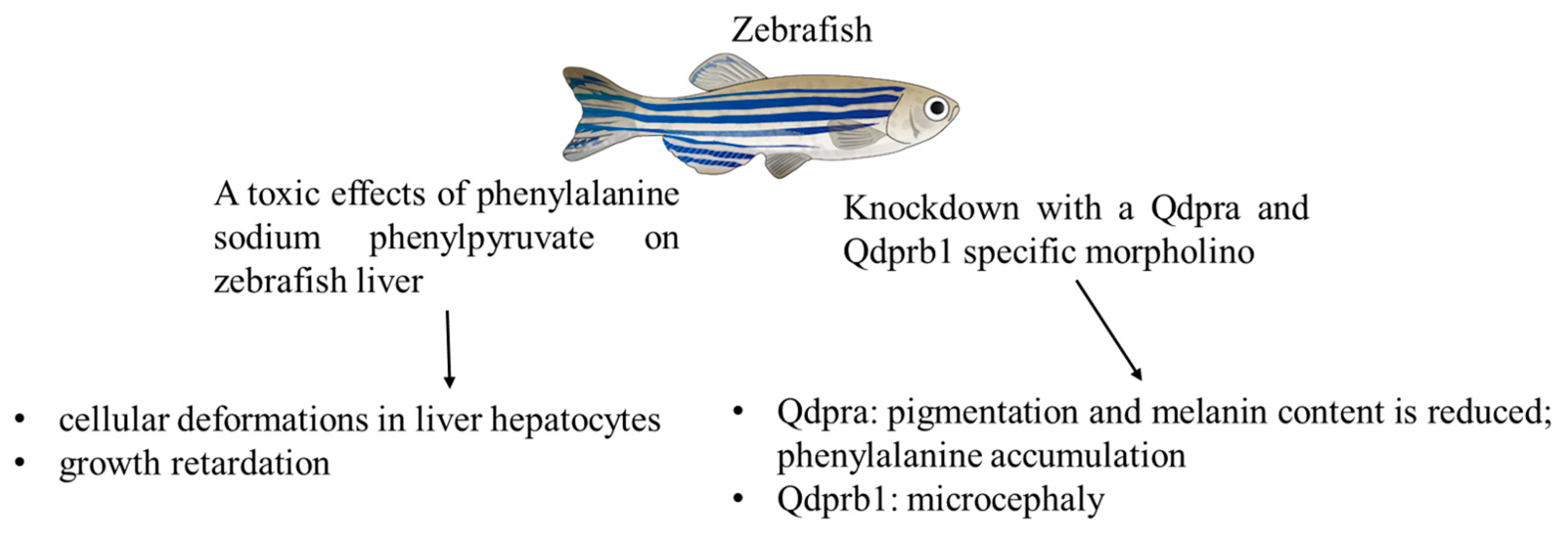
3.3. Zebrafish Models
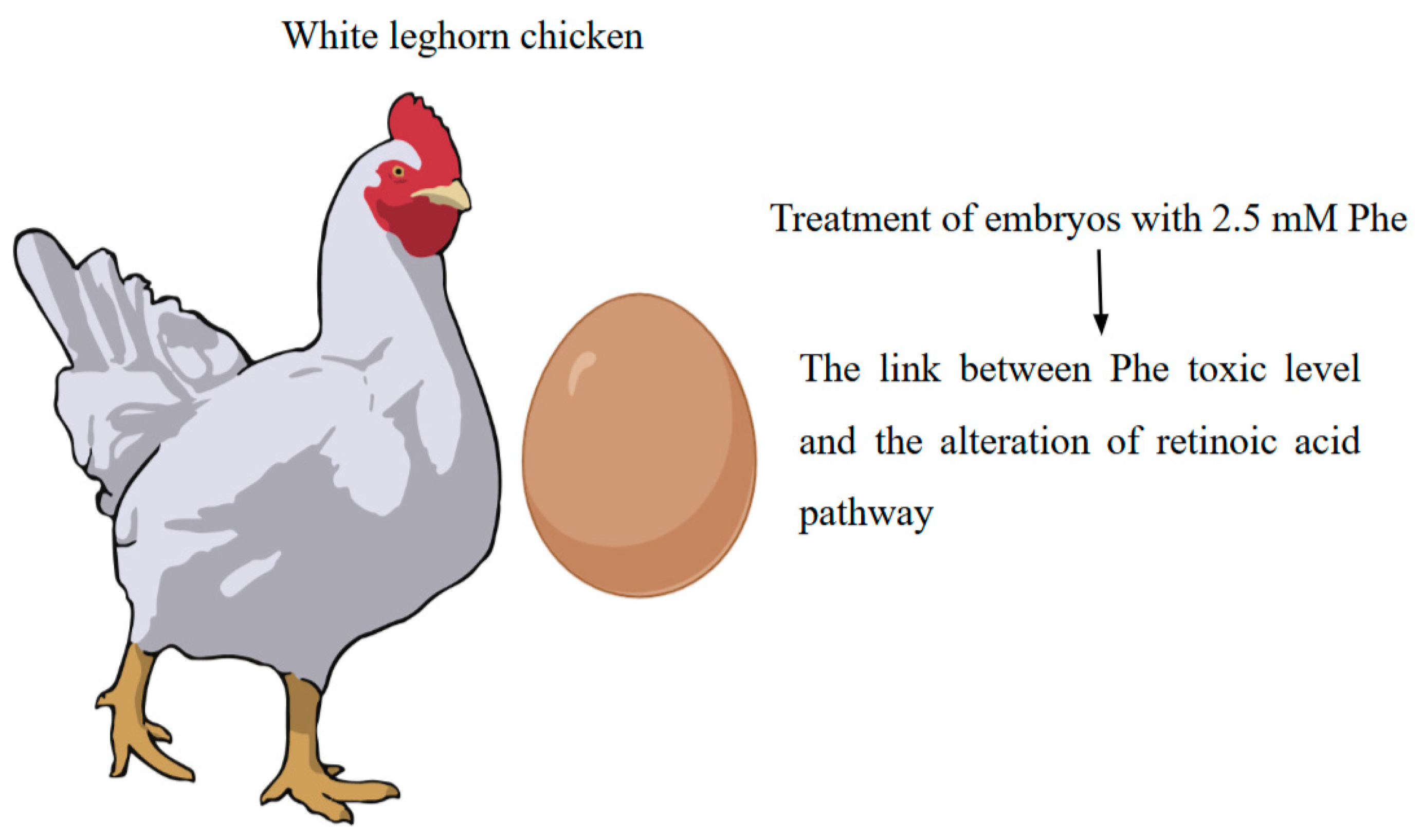
3.4. Avian Models
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| PAH | Phenylalanine hydroxylase |
| PKU | Phenylketonuria |
| Phe | Phenylalanine |
| Tyr | Tyrosine |
| NBS | Newborn bloodspot screening |
| BH4 | Tetrahydrobiopterin |
| HPA | Hyperphenylalaninemia |
| ENU | N-ethyl-N-nitrosourea |
References
- van Spronsen, F.J.; Blau, N.; Harding, C.; Burlina, A.; Longo, N.; Bosch, A.M. Phenylketonuria. Nat. Rev. Dis. Primers 2021, 7, 36. [Google Scholar] [CrossRef]
- Blau, N.; van Spronsen, F.J.; Levy, H.L. Phenylketonuria. Lancet 2010, 376, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Elhawary, N.A.; AlJahdali, I.A.; Abumansour, I.S.; Elhawary, E.N.; Gaboon, N.; Dandini, M.; Madkhali, A.; Alosaimi, W.; Alzahrani, A.; Aljohani, F.; et al. Genetic etiology and clinical challenges of phenylketonuria. Hum. Genom. 2022, 16, 22. [Google Scholar] [CrossRef]
- Centerwall, S.A.; Centerwall, W.R. The Discovery of Phenylketonuria: The Story of a Young Couple, Two Retarded Children, and a Scientist. Pediatrics 2000, 105, 89–103. [Google Scholar] [CrossRef]
- Koppes, E.A.; Redel, B.K.; Johnson, M.A.; Skvorak, K.J.; Ghaloul-Gonzalez, L.; Yates, M.E.; Lewis, D.W.; Gollin, S.M.; Wu, Y.L.; Christ, S.E.; et al. A porcine model of phenylketonuria generated by CRISPR/Cas9 genome editing. JCI Insight. 2020, 5, e141523. [Google Scholar] [CrossRef]
- Shedlovsky, A.; McDonald, J.D.; Symula, D.; Dove, W.F. Mouse models of human phenylketonuria. Genetics 1993, 134, 1205–1210. [Google Scholar] [CrossRef]
- Festing, M.F.W. Design and Statistical Methods in Studies Using Animal Models of Development. ILAR J. 2006, 47, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Følling, I. The discovery of phenylketonuria. Acta Paediatr. 1994, 83, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Irwin, H.R.; Notrica, S.; Fleming, W.; Diego, S. Blood Phenylalanine Levels of Newborn Infants A Routine Screening Program for the Hospital Newborn Nursery. Calif. Med. 1964, 101, 331–333. [Google Scholar]
- Guthrie, R. The origin of newborn screening. Screening 1992, 1, 5–15. [Google Scholar] [CrossRef]
- Hillert, A.; Anikster, Y.; Belanger-Quintana, A.; Burlina, A.; Burton, B.K.; Carducci, C.; Chiesa, A.E.; Christodoulou, J.; Đorđević, M.; Desviat, L.R.; et al. The Genetic Landscape and Epidemiology of Phenylketonuria. Am. J. Hum. Genet. 2020, 107, 234–250. [Google Scholar] [CrossRef]
- Flydal, M.I.; Alcorlo-Pagés, M.; Johannessen, F.G.; Martínez-Caballero, S.; Skjærven, L.; Fernandez-Leiro, R.; Martinez, A.; Hermoso, J.A. Structure of full-length human phenylalanine hydroxylase in complex with tetrahydrobiopterin. Proc. Natl. Acad. Sci. USA 2019, 166, 11229–11234. [Google Scholar] [CrossRef]
- Bailey, N.A.; Mackay, L. Phenylketonuria—Past, Present, and Future Directions. OBM Genet. 2024, 8, 1–21. [Google Scholar] [CrossRef]
- Rovelli, V.; Longo, N. Phenylketonuria and the brain. Mol. Genet. Metab. 2023, 139, 107583. [Google Scholar] [CrossRef]
- de Groot, M.J.; Hoeksma, M.; Blau, N.; Reijngoud, D.J.; van Spronsen, F.J. Pathogenesis of cognitive dysfunction in phenylketonuria: Review of hypotheses. Mol. Genet. Metab. 2010, 99, S86–S89. [Google Scholar] [CrossRef]
- Anderson, P.J.; Leuzzi, V. White matter pathology in phenylketonuria. Mol. Genet. Metab. 2010, 99, S3–S9. [Google Scholar] [CrossRef]
- Adler-Abramovich, L.; Vaks, L.; Carny, O.; Trudler, D.; Magno, A.; Caflisch, A.; Frenkel, D.; Gazit, E. Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nat. Chem. Biol. 2012, 8, 701–706. [Google Scholar] [CrossRef]
- Burgess, N.M.; Kelso, W.; Malpas, C.B.; Winton-Brown, T.; Fazio, T.; Panetta, J.; De Jong, G.; Neath, J.; Atherton, S.; Velakoulis, D.; et al. The effect of improved dietary control on cognitive and psychiatric functioning in adults with phenylketonuria: The ReDAPT study. Orphanet J. Rare Dis. 2021, 16, 35. [Google Scholar] [CrossRef]
- De Giorgi, A.; Nardecchia, F.; Manti, F.; Campistol, J.; Leuzzi, V. Neuroimaging in early-treated phenylketonuria patients and clinical outcome: A systematic review. Mol. Genet. Metab. 2023, 139, 107588. [Google Scholar] [CrossRef] [PubMed]
- Christ, S.E.; Price, M.H.; Bodner, K.E.; Saville, C.; Moffitt, A.J.; Peck, D. Morphometric analysis of gray matter integrity in individuals with early-treated phenylketonuria. Mol. Genet. Metab. 2016, 118, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Bryda, E.C. The Mighty Mouse: The impact of rodents on advances in biomedical research. Mo. Med. 2013, 110, 207–211. [Google Scholar] [PubMed]
- van der Goot, E.; Bruinenberg, V.M.; Hormann, F.M.; Eisel, U.L.M.; van Spronsen, F.J.; Van der Zee, E.A. Hippocampal microglia modifications in C57Bl/6 Pahenu2 and BTBR Pahenu2 phenylketonuria (PKU) mice depend on the genetic background, irrespective of disturbed sleep patterns. Neurobiol. Learn. Mem. 2019, 160, 139–143. [Google Scholar] [CrossRef]
- McDonald, J.D.; Bode, V.C.; Dove, W.F.; Shedlovsky, A. Pahhph-5: A Mouse Mutant Deficient in Phenylalanine Hydroxylase. Proc. Natl. Acad. Sci. USA 1990, 87, 1965–1967. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.L.; Whittaker, M.N.; Qu, P.; Musunuru, K.; Ahrens-Nicklas, R.C.; Wang, X. Efficient in vivo prime editing corrects the most frequent phenylketonuria variant, associated with high unmet medical need. Am. J. Hum. Genet. 2023, 110, 2003–2014. [Google Scholar] [CrossRef]
- Brooks, D.L.; Carrasco, M.J.; Qu, P.; Peranteau, W.H.; Ahrens-Nicklas, R.C.; Musunuru, K.; Alameh, M.-G.; Wang, X. Rapid and definitive treatment of phenylketonuria in variant-humanized mice with corrective editing. Nat. Commun. 2023, 14, 3451. [Google Scholar] [CrossRef]
- Singh, K.; Cornell, C.S.; Jackson, R.; Kabiri, M.; Phipps, M.; Desai, M.; Fogle, R.; Ying, X.; Anarat-Cappillino, G.; Geller, S.; et al. CRISPR/Cas9 generated knockout mice lacking phenylalanine hydroxylase protein as a novel preclinical model for human phenylketonuria. Sci. Rep. 2021, 11, 7254. [Google Scholar] [CrossRef] [PubMed]
- Aubi, O.; Prestegård, K.S.; Jung-Kc, K.; Shi, T.J.S.; Ying, M.; Grindheim, A.K.; Scherer, T.; Ulvik, A.; McCann, A.; Spriet, E.; et al. The Pah-R261Q mouse reveals oxidative stress associated with amyloid-like hepatic aggregation of mutant phenylalanine hydroxylase. Nat. Commun. 2021, 12, 2073. [Google Scholar] [CrossRef]
- Ding, Z.; Georgiev, P.; Thöny, B. Administration-route and gender-independent long-term therapeutic correction of phenylketonuria (PKU) in a mouse model by recombinant adeno-associated virus 8 pseudotyped vector-mediated gene transfer. Gene. Ther. 2006, 13, 587–593. [Google Scholar] [CrossRef]
- Prather, R.S.; Lorson, M.; Ross, J.W.; Whyte, J.J.; Walters, E. Genetically engineered pig models for human diseases. Annu. Rev. Anim. Biosci. 2013, 1, 203–219. [Google Scholar] [CrossRef]
- Lunney, J.K. Advances in Swine Biomedical Model Genomics. Int. J. Biol. Sci. 2007, 3, 179–184. [Google Scholar] [CrossRef]
- Kaiser, R.A.; Carlson, D.F.; Allen, K.L.; Webster, D.A.; VanLith, C.J.; Nicolas, C.T.; Hillin, L.G.; Yu, Y.; Kaiser, C.W.; Wahoff, W.R.; et al. Development of a porcine model of phenylketonuria with a humanized R408W mutation for gene editing. PLoS ONE 2021, 16, e0245831. [Google Scholar] [CrossRef] [PubMed]
- Hou, N.; Du, X.; Wu, S. Advances in pig models of human diseases. Anim. Model. Exp. Med. 2022, 5, 141–152. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef]
- Oka, T.; Nishimura, Y.; Zang, L.; Hirano, M.; Shimada, Y.; Wang, Z.; Umemoto, N.; Kuroyanagi, J.; Nishimura, N.; Tanaka, T. Diet-induced obesity in zebrafish shares common pathophysiological pathways with mammalian obesity. BMC Physiol. 2010, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Sudo, Y.; Sanechika, S.; Yamashita, J.; Shimaguchi, S.; Honda, S.-I.; Sumi-Ichinose, C.; Mori-Kojima, M.; Nakata, R.; Furuta, T.; et al. Disturbed biopterin and folate metabolism in the Qdpr-deficient mouse. FEBS Lett. 2014, 588, 3924–3931. [Google Scholar] [CrossRef]
- Ponzone, A.; Spada, M.; Ferraris, S.; Dianzani, I.; de Sanctis, L. Dihydropteridine reductase deficiency in man: From biology to treatment. Med. Res. Rev. 2004, 24, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Breuer, M.; Patten, S.A. A great catch for investigating inborn errors of metabolism—Insights obtained from zebrafish. Biomolecules. 2020, 10, 1352. [Google Scholar] [CrossRef]
- Bose, S.; Goswami, A.; Mandal, S.; Khan, R.; Maji, H.S. Assessment of Toxicity And Safety Margin of Phenylalanine And Its Metabolites Through In-Vivo And InVitro Model. J. Pharm. Negat. Results 2023, 14, 833. [Google Scholar] [CrossRef]
- Breuer, M.; Guglielmi, L.; Zielonka, M.; Hemberger, V.; Kölker, S.; Okun, J.G.; Hoffmann, G.F.; Carl, M.; Sauer, S.W.; Opladen, T. QDPR homologues in Danio rerio regulate melanin synthesis, early gliogenesis, and glutamine homeostasis. PLoS ONE 2019, 14, e0215162. [Google Scholar] [CrossRef]
- Schartl, M. Beyond the zebrafish: Diverse fish species for modeling human disease. Dis. Model. Mech. 2014, 7, 181–192. [Google Scholar] [CrossRef]
- Braasch, I.; Postlethwait, J.H. Polyploidy in Fish and the Teleost Genome Duplication. In Polyploidy and Genome Evolution; Soltis, P.S., Soltis, D.E., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 341–383. [Google Scholar] [CrossRef]
- Postlethwait, J.H.; Woods, I.G.; Ngo-Hazelett, P.; Yan, Y.-L.; Kelly, P.D.; Chu, F.; Huang, H.; Hill-Force, A.; Talbot, W.S. Zebrafish Comparative Genomics and the Origins of Vertebrate Chromosomes. Genome Res. 2000, 10, 1890–1902. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.N.; Seagraves, N.J. RNA-Seq analysis in an avian model of maternal phenylketonuria. Mol. Genet. Metab. 2019, 126, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Flores-Santin, J.; Burggren, W.W. Beyond the Chicken: Alternative Avian Models for Developmental Physiological Research. Front. Physiol. 2021, 12, 712633. [Google Scholar] [CrossRef] [PubMed]
- Lawal, R.A.; Hanotte, O. Domestic chicken diversity: Origin, distribution, and adaptation. Anim. Genet. 2021, 52, 385–394. [Google Scholar] [CrossRef]
- He, W.; Li, P.; Wu, G. Amino Acid Nutrition and Metabolism in Chickens. In Amino Acids in Nutrition and Health: Amino Acids in the Nutrition of Companion, Zoo and Farm Animals; Wu, G., Ed.; Springer International Publishing: Cham, Switzerland, 2021; pp. 109–131. [Google Scholar] [CrossRef]
- Winn, S.R.; Dudley, S.; Scherer, T.; Rimann, N.; Thöny, B.; Boutros, S.; Krenik, D.; Raber, J.; Harding, C.O. Modeling the cognitive effects of diet discontinuation in adults with phenylketonuria (PKU) using pegvaliase therapy in PAH-deficient mice. Mol. Genet. Metab. 2022, 136, 46–64. [Google Scholar] [CrossRef]
- Veleva, D.; Ay, M.; Ovchinnikov, D.A.; Prowse, A.B.J.; Menezes, M.J.; Nafisinia, M. Generation of fibroblast-derived induced pluripotent stem cell (iPSC) lines from two paediatric patients with phenylketonuria. Stem Cell Res. 2024, 77, 103405. [Google Scholar] [CrossRef]
- Borges, A.C.; Broersen, K.; Leandro, P.; Fernandes, T.G. Engineering Organoids for in vitro Modeling of Phenylketonuria. Front. Mol. Neurosci. 2022, 14, 787242. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Model | Creation Method | Main Features | Application | References |
|---|---|---|---|---|
| BTBR-Pahenu2/J (PAHenu2) | Chemically Induced Mutation (ENU) | Severe hyperphenylalanemia with serum phenylalanine levels elevated 10–20-fold and urinary ketone concentrations significantly increased in adults. Hypopigmented unless maintained on a low phenylalanine diet. | Behaviour, neurological, growth, metabolism, nervous system, pigmentation, urinary system, reproductive system phenotype. | [22] |
| B6.BTBR-Pahenu2/MalnJ (B6 Pahenu2) | Chemically Induced Mutation (ENU) | Hyperphenylalaninemia and other hallmarks of phenylketonuria (PKU) when not maintained on a phenylalanine-free diet. Hyperphenylalaninemia leads to progressive neurological symptoms and seizures. Mice are hypopigmented on a standard diet. | Behaviour/neurological, growth, metabolism, nervous system, pigmentation, urinary system, reproductive system phenotype. | [22] |
| BTBR-Pahenu1/J | Chemically Induced Mutation (ENU) | Consistent with the milder phenotype, homozygotes do not display the pronounced hypopigmentation, or learning and memory deficits. Females do not display a severe maternal effect of smaller and fewer litters and failed survival of pups that are homozygous. Homozygotes have near normal serum phenylalanine levels and elevated brain phenylalanine levels less than twice normal levels. | Homeostasis, metabolism, reproductive system phenotype. | [23] |
| B6(Cg)-Pahtm1.1(PAH*R408W)Xiwan/J (PAH R408W) | Targeted mutation 1.1, Xiao Wan Homologous recombination in C57BL/6 mouse embryonic stem cells was used to replace exon 12 with the orthologous human PAH exon 12 sequence including 500 bp of flanking genomic sequence both 5′ and 3′ of exon 12. | Homozygotes have elevated blood phenylalanine levels, exhibit hypopigmentation due to reduced melanin synthesis, and are smaller in size. Blood phenylalanine levels range from approximately 1000 to 1500 μmol/L. | Testing humanised therapeutics. | [24] |
| C57BL/6J-Pahem1Xiwan/J (PAH P281L) | Endonuclease-mediated mutation. CRISPR/cas9 endonuclease-mediated homology-directed repair was used to replace a portion of exon 7. | Homozygotes have elevated blood phenylalanine levels, exhibit hypopigmentation due to reduced melanin synthesis, and are smaller in size. Blood phenylalanine levels range from 1455 to 2242 μmol/L. | Testing humanised therapeutics. | [25] |
| C57BL/6Smoc-Pahem1Smoc (PAH-KO) | CRISPR/Cas9: The exons 1–13 of mouse PAH gene that encode the full-length protein were knocked out in B-Pah KO mice. | The PAH-KO mice showed lower body weights, Summary of blood Phe at various timepoints is shown (n = 20/group), summary of blood Tyr at various timepoints is shown (n = 20/group). | Behaviour/neurological, growth, metabolism, nervous system, pigmentation phenotype. | [26] |
| C57BL/6Smoc-Pahem(R261Q)Smoc (Pah-R261Q) | R261Q-PAH mutation was generated by CRISPR/Cas9 genome editing technology | The present study detected a mild form of hyperphenylalaninemia in mice with this mutation. Phenylalanine levels have been found to be elevated to a level twice that which is considered normal. It is noteworthy that the patient does not exhibit other phenotypic abnormalities that are characteristic of classical phenylketonuria, such as hypopigmentation. Males exhibited higher body weight. | Model for studying the tissue-specific effects of phenylalanine metabolism disorders. The model is capable of reproducing a key feature of BH4-responsive forms of HFAs. In addition, it facilitates the study of mechanisms of variability in response to therapy and can be used for preclinical evaluation of new therapies. | [27] |
| Animal Model | Pros | Cons |
|---|---|---|
| Mouse models | small size | evolutionary differences between mice and humans |
| high reproductive rate | disparities in size | |
| ease of genetic manipulation | metabolic rate | |
| lifespan | ||
| immune system | ||
| Pig models | anatomy and physiology similar to humans | ethical issues of using large animals |
| high sequence and chromosome structure homology with humans | costs and difficulties in keeping | |
| similar disease progression | small sample size | |
| Zebrafish models | genetic and developmental insights | species differences |
| high-throughput screening | liver metabolism differences | |
| cost-effective | behavioural studies limitations | |
| disease modelling | ||
| Avian models | accessibility | metabolism differences |
| high fecundity | species differences | |
| well-characterised developmental stages | extensive artificial selection | |
| genetic and phenotypic diversity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bobrova, N.A.; Lyubimova, D.I.; Mishina, D.M.; Lobanova, V.S.; Valieva, S.I.; Mityaeva, O.N.; Feoktistova, S.G.; Volchkov, P.Y. Experimental Animal Models of Phenylketonuria: Pros and Cons. Int. J. Mol. Sci. 2025, 26, 5262. https://doi.org/10.3390/ijms26115262
Bobrova NA, Lyubimova DI, Mishina DM, Lobanova VS, Valieva SI, Mityaeva ON, Feoktistova SG, Volchkov PY. Experimental Animal Models of Phenylketonuria: Pros and Cons. International Journal of Molecular Sciences. 2025; 26(11):5262. https://doi.org/10.3390/ijms26115262
Chicago/Turabian StyleBobrova, N. A., D. I. Lyubimova, D. M. Mishina, V. S. Lobanova, S. I. Valieva, O. N. Mityaeva, S. G. Feoktistova, and P. Yu. Volchkov. 2025. "Experimental Animal Models of Phenylketonuria: Pros and Cons" International Journal of Molecular Sciences 26, no. 11: 5262. https://doi.org/10.3390/ijms26115262
APA StyleBobrova, N. A., Lyubimova, D. I., Mishina, D. M., Lobanova, V. S., Valieva, S. I., Mityaeva, O. N., Feoktistova, S. G., & Volchkov, P. Y. (2025). Experimental Animal Models of Phenylketonuria: Pros and Cons. International Journal of Molecular Sciences, 26(11), 5262. https://doi.org/10.3390/ijms26115262