
Amyotrophic Lateral Sclerosis: Pathophysiological Mechanisms and Treatment Strategies (Part 2)


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
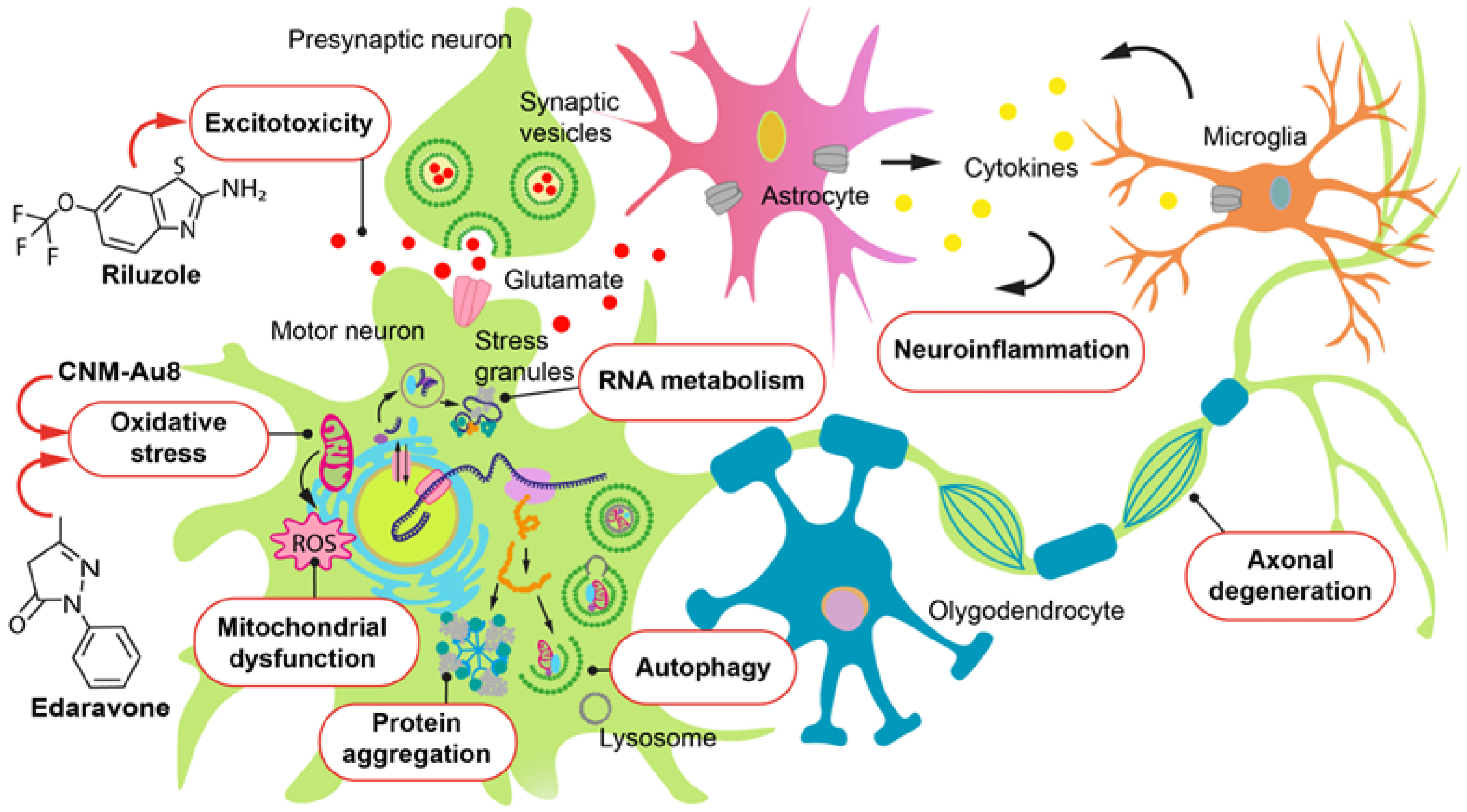
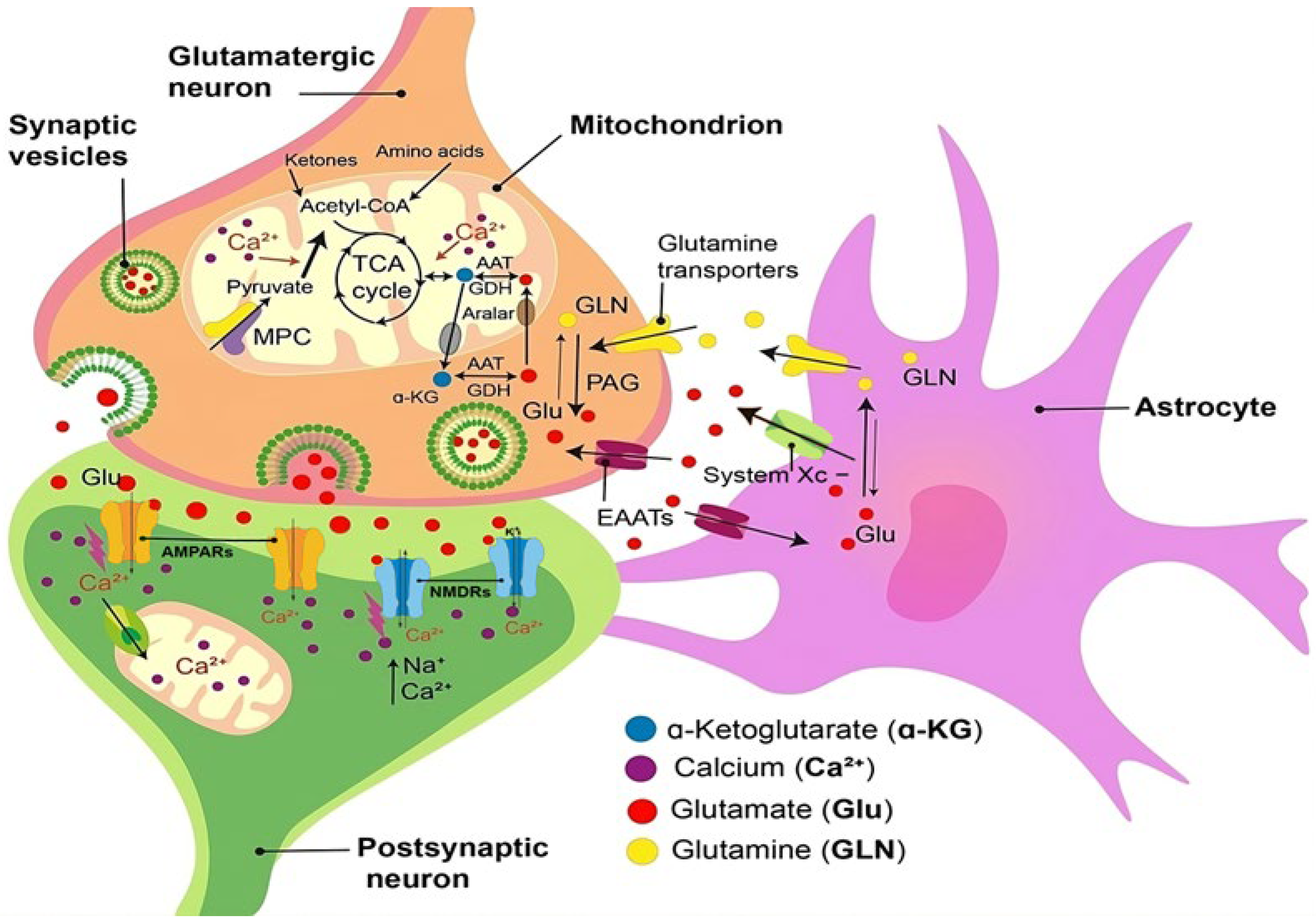
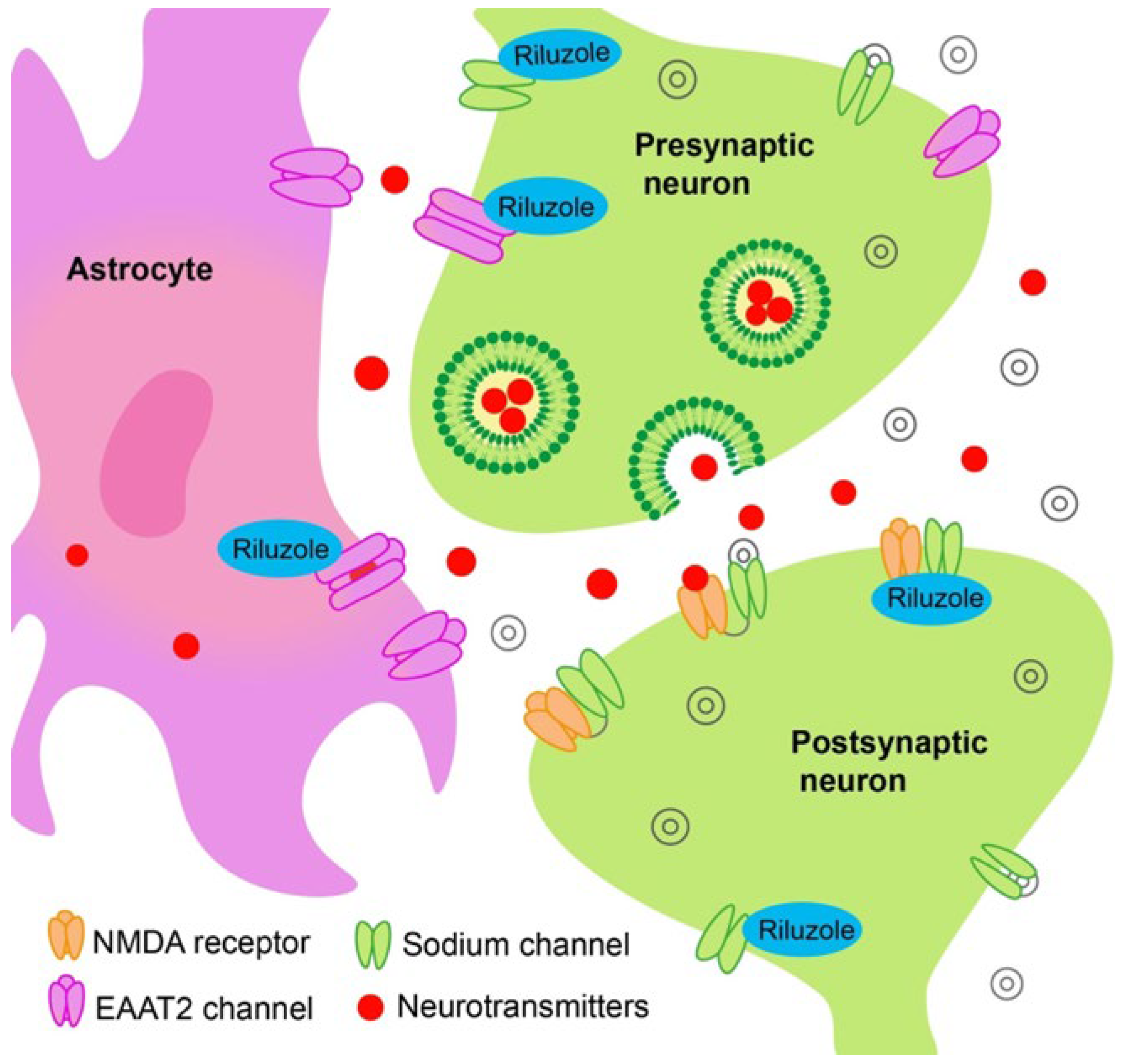
2. Glutamate Excitotoxicity and Antiglutamatergic Drugs
Other Drugs with Effect on Glutamate-Mediated Neurotransmission
3. Oxidative Stress and Antioxidant Drugs
4. Edaravone
5. Gold Nanocrystals, the Drug CNM-Au8
6. Verdiperstat
7. Mitochondrial Dysfunction and Cytoprotective and Antiapoptotic Drugs AMX0035
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C. Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, T.; Ura, M.; Nittono, H. Intrapersonal and interpersonal processes of social exclusion. Front. Neurosci. 2015, 9, 62. [Google Scholar] [CrossRef]
- Turner, M.R.; Barnwell, J.; Al-Chalabi, A.; Eisen, A. Young-onset amyotrophic lateral sclerosis: Historical and other observations. Brain 2012, 135 Pt 9, 2883–2891. [Google Scholar] [CrossRef]
- Liu, Z.J.; Lin, H.X.; Liu, G.L.; Tao, Q.Q.; Ni, W.; Xiao, B.G.; Wu, Z.Y. The investigation of genetic and clinical features in Chinese patients with juvenile amyotrophic lateral sclerosis. Clin. Genet. 2017, 92, 267–273. [Google Scholar] [CrossRef]
- Rudenskaya, G.E.; Nikitin, S.S.; Shatokhina, O.L.; Shchagina, O.A. Juvenile amyotrophic lateral sclerosis type 4: Case report and review. Neuromuscul. Dis. 2022, 12, 52–58. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.; Heverin, M.; McLaughlin, R.L.; Hardiman, O. Lifetime Risk and Heritability of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2019, 76, 1367–1374. [Google Scholar] [CrossRef]
- Gosset, P.; Camu, W.; Raoul, C.; Mezghrani, A. Prionoids in amyotrophic lateral sclerosis. Brain Commun. 2022, 4, fcac145. [Google Scholar] [CrossRef]
- Xu, L.; Liu, T.; Liu, L.; Yao, X.; Chen, L.; Fan, D.; Zhan, S.; Wang, S. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. 2020, 267, 944–953. [Google Scholar] [CrossRef]
- Pateri, M.I.; Pilotto, S.; Borghero, G.; Pili, F.; Pierri, V.; Ercoli, T.; Gigante, A.F.; Muroni, A.; Defazio, G. Increasing prevalence 2015–2019 of amyotrophic lateral sclerosis in Sardinia, Italy. Neurol. Sci. 2023, 44, 2781–2786. [Google Scholar] [CrossRef] [PubMed]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef]
- Gowland, A.; Opie-Martin, S.; Scott, K.M.; Jones, A.R.; Mehta, P.R.; Batts, C.J.; Ellis, C.M.; Leigh, N.; Shaw, C.E.; Sreedharan, J.; et al. Predicting the future of ALS: The impact of demographic change and potential new treatments on the prevalence of ALS in the United Kingdom, 2020–2116. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 264–274. [Google Scholar] [CrossRef]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’h, P. Causative Genes in Amyotrophic Lateral Sclerosis and Protein Degradation Pathways: A Link to Neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Lee, V.M.; Trojanowski, J.Q. TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 211–220. [Google Scholar] [CrossRef]
- Tan, R.H.; Ke, Y.D.; Ittner, L.M.; Halliday, G.M. ALS/FTLD: Experimental models and reality. Acta Neuropathol. 2017, 133, 177–196. [Google Scholar] [CrossRef]
- Appleby-Mallinder, C.; Schaber, E.; Kirby, J.; Shaw, P.J.; Cooper-Knock, J.; Heath, P.R.; Highley, J.R. TDP43 proteinopathy is associated with aberrant DNA methylation in human amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2021, 47, 61–72. [Google Scholar] [CrossRef]
- Koike, Y.; Sugai, A.; Hara, N.; Ito, J.; Yokoseki, A.; Ishihara, T.; Yamagishi, T.; Tsuboguchi, S.; Tada, M.; Ikeuchi, T.; et al. Age-related demethylation of the TDP-43 autoregulatory region in the human motor cortex. Commun. Biol. 2021, 4, 1107. [Google Scholar] [CrossRef] [PubMed]
- Zufiría, M.; Gil-Bea, F.J.; Fernández-Torrón, R.; Poza, J.J.; Muñoz-Blanco, J.L.; Rojas-García, R.; Riancho, J.; López de Munain, A. ALS: A bucket of genes, environment, metabolism and unknown ingredients. Prog. Neurobiol. 2016, 142, 104–129. [Google Scholar] [CrossRef]
- Andrew, A.; Zhou, J.; Gui, J.; Harrison, A.; Shi, X.; Li, M.; Guetti, B.; Nathan, R.; Tischbein, M.; Pioro, E.P.; et al. Pesticides applied to crops and amyotrophic lateral sclerosis risk in the U.S. Neurotoxicology 2021, 87, 128–135. [Google Scholar] [CrossRef]
- Xue, Y.C.; Feuer, R.; Cashman, N.; Luo, H. Enteroviral Infection: The Forgotten Link to Amyotrophic Lateral Sclerosis? Front. Mol. Neurosci. 2018, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Recent advances in the diagnosis and prognosis of amyotrophic lateral sclerosis. Lancet Neurol. 2022, 21, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Arnold, F.J.; Putka, A.F.; Raychaudhuri, U.; Hsu, S.; Bedlack, R.S.; Bennett, C.L.; La Spada, A.R. Revisiting Glutamate Excitotoxicity in Amyotrophic Lateral Sclerosis and Age-Related Neurodegeneration. Int. J. Mol. Sci. 2024, 25, 5587. [Google Scholar] [CrossRef] [PubMed]
- Hemerková, P.; Vališ, M. Role of Oxidative Stress in the Pathogenesis of Amyotrophic Lateral Sclerosis: Antioxidant Metalloenzymes and Therapeutic Strategies. Biomolecules 2021, 11, 437. [Google Scholar] [CrossRef]
- Genin, E.C.; Abou-Ali, M.; Paquis-Flucklinger, V. Mitochondria, A Key Target in Amyotrophic Lateral Sclerosis Pathogenesis. Genes 2023, 14, 1981. [Google Scholar] [CrossRef]
- De Marchi, F.; Franjkic, T.; Schito, P.; Russo, T.; Nimac, J.; Chami, A.A.; Mele, A.; Vidatic, L.; Kriz, J.; Julien, J.-P.; et al. Emerging Trends in the Field of Inflammation and Proteinopathy in ALS/FTD Spectrum Disorder. Biomedicines 2023, 11, 1599. [Google Scholar] [CrossRef]
- Chua, J.P.; De Calbiac, H.; Kabashi, E.; Barmada, S.J. Autophagy and ALS: Mechanistic insights and therapeutic implications. Autophagy 2022, 18, 254–282. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef]
- King, A.E.; Woodhouse, A.; Kirkcaldie, M.T.; Vickers, J.C. Excitotoxicity in ALS: Overstimulation, or overreaction? Exp. Neurol. 2016, 275, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.S.; Bjork, R.A.; Handal, A. Retrieval-induced forgetting in an eyewitness-memory paradigm. Psychon. Bull. Rev. 1995, 2, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Wuolikainen, A.; Moritz, T.; Marklund, S.L.; Antti, H.; Andersen, P.M. Disease-related changes in the cerebrospinal fluid metabolome in amyotrophic lateral sclerosis detected by GC/TOFMS. PLoS ONE 2011, 6, e17947. [Google Scholar] [CrossRef] [PubMed]
- Sonay, A.Y.; Mc Larney, B.E.; Apfelbaum, E.; Grimm, J. In vivo imaging of ferroptosis through nanodynamic changes in lipid membranes. Nat. Nanotechnol. 2025, 20, 123–130. [Google Scholar]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef]
- Rosenblum, L.T.; Trotti, D. EAAT2 and the Molecular Signature of Amyotrophic Lateral Sclerosis. Adv. Neurobiol. 2017, 16, 117–136. [Google Scholar]
- Khodorov, B. Glutamate-induced deregulation of calcium homeostasis and mitochondrial dysfunction in mammalian central neurones. Prog. Biophys. Mol. Biol. 2004, 86, 279–351. [Google Scholar] [CrossRef]
- Plotegher, N.; Filadi, R.; Pizzo, P.; Duchen, M.R. Excitotoxicity Revisited: Mitochondria on the Verge of a Nervous Breakdown. Trends Neurosci. 2021, 44, 342–351. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.P.; Dietrich, W.D.; Keane, R.W. Activation and regulation of cellular inflammasomes: Gaps in our knowledge for central nervous system injury. J. Cereb. Blood Flow. Metab. 2014, 34, 369–375. [Google Scholar] [CrossRef]
- Olmos, G.; Llado, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Schmidt, T.; Golla, M.; Lehmann, L.; Weber, J.J.; Hübener-Schmid, J.; Riess, O. In vivo assessment of riluzole as a potential therapeutic drug for spinocerebellar ataxia type 3. J. Neurochem. 2016, 138, 150–162. [Google Scholar] [CrossRef]
- Sala, G.; Arosio, A.; Conti, E.; Beretta, S.; Lunetta, C.; Riva, N.; Ferrarese, C.; Tremolizzo, L. Riluzole Selective Antioxidant Effects in Cell Models Expressing Amyotrophic Lateral Sclerosis Endophenotypes. Clin. Psychopharmacol. Neurosci. 2019, 17, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Chio, A.; Mazzini, L.; Mora, G. Disease-modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology 2020, 167, 107986. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Beltran, S.; Bakkouche, S.E.; Couratier, P. Therapeutic news in ALS. Rev. Neurol. 2021, 177, 544–549. [Google Scholar] [CrossRef]
- Johnson, S.A.; Fang, T.; De Marchi, F.; Neel, D.; Van Weehaeghe, D.; Berry, J.D.; Paganoni, S. Pharmacotherapy for Amyotrophic Lateral Sclerosis: A Review of Approved and Upcoming Agents. Drugs 2022, 82, 1367–1388. [Google Scholar] [CrossRef]
- Deflorio, C.; Onesti, E.; Lauro, C.; Tartaglia, G.; Giovannelli, A.; Limatola, C.; Inghilleri, M.; Grassi, F. Partial block by riluzole of muscle sodium channels in myotubes from amyotrophic lateral sclerosis patients. Neurol. Res. Int. 2014, 2014, 946073. [Google Scholar] [CrossRef]
- Doble, A. The pharmacology and mechanism of action of riluzole. Neurology 1996, 47, 233–241. [Google Scholar] [CrossRef]
- Martin, D.; Thompson, M.A.; Nadler, J.V. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur. J. Pharmacol. 1993, 250, 473–476. [Google Scholar] [CrossRef]
- Lazarevic, V.; Yang, Y.; Ivanova, D.; Fejtova, A.; Svenningsson, P. Riluzole attenuates the efficacy of glutamatergic transmission by interfering with the size of the readily releasable neurotransmitter pool. Neuropharmacology 2018, 143, 38–48. [Google Scholar] [CrossRef]
- Frizzo, M.E.D.S.; Dall’Onder, L.P.; Dalcin, K.B.; Souza, D.O. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell Mol. Neurobiol. 2004, 24, 123–128. [Google Scholar] [CrossRef]
- Mignani, S.; Majoral, J.P.; Desaphy, J.F.; Lentini, G. From riluzole to dexpramipexole via substituted-benzothiazole derivatives for amyotrophic lateral sclerosis disease treatment: Case studies. Molecules 2020, 25, 3320. [Google Scholar] [CrossRef] [PubMed]
- Bissaro, B.; Várnai, A.; Røhr, Å.K.; Eijsink, V.G. Oxidoreductases and Reactive Oxygen Species in Conversion of Lignocellulosic Biomass. Microbiol. Mol. Biol. Rev. 2018, 82, e00029-18. [Google Scholar] [CrossRef] [PubMed]
- Ko, V.I.; Ong, K.; Cleveland, D.W.; Yu, H.; Ravits, J.M. CK1δ/ε kinases regulate TDP-43 phosphorylation and are therapeutic targets for ALS-related TDP-43 hyperphosphorylation. Neurobiol. Dis. 2024, 196, 106516. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Župunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.L.; Della Gatta, P.A.; Le, S.; Berning, B.A.; Mehta, P.; Jacobs, K.R.; Gul, H.; Gil, R.S.; Hedl, T.J.; Riddell, W.R.; et al. Riluzole does not ameliorate disease caused by cytoplasmic TDP-43 in a mouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 2021, 54, 6237–6255. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Lacomblez, L.; Bensimon, G.; Leigh, P.N.; Guillet, P.; Meininger, V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Lancet 1996, 347, 1425–1431. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Delumeau, J.C.; Bejuit, R.; Truffinet, P.; Meininger, V.; Riluzole/ALS Study Group II. A study of riluzole in the treatment of advanced stage or elderly patients with amyotrophic lateral sclerosis. J. Neurol. 2002, 249, 609–615. [Google Scholar] [CrossRef]
- Meininger, V.; Lacomblez, L.; Salachas, F. What has changed with riluzole? J. Neurol. 2000, 247 (Suppl. S6), VI19–VI22. [Google Scholar] [CrossRef]
- Brooks, B.R.; Sanjak, M. Disease-modifying drug therapies. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2004, 5 (Suppl. S1), 68–75. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Bakker, M.; Sham, P.; Shaw, C.E.; Leigh, P.N.; Al-Chalabi, A. Prognostic modelling of therapeutic interventions in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2002, 3, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.D.; O’Brien, M.R.; Joshi, M. Audit of outcomes in motor neuron disease (MND) patients treated with riluzole. Amyotroph. Lateral Scler. 2006, 7, 67–71. [Google Scholar] [CrossRef]
- Georgoulopoulou, E.; Fini, N.; Vinceti, M.; Monelli, M.; Vacondio, P.; Bianconi, G.; Sola, P.; Nichelli, P.; Mandrioli, J. The impact of clinical factors, riluzole and therapeutic interventions on ALS survival: A population-based study in Modena, Italy. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Stevic, Z.; Kostic-Dedic, S.; Peric, S.; Dedic, V.; Basta, I.; Rakocevic-Stojanovic, V.; Lavrnic, D. Prognostic factors and survival of ALS patients from Belgrade, Serbia. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 508–514. [Google Scholar] [CrossRef]
- Favero, F.M.; Voos, M.C.; Castro, I.; Caromano, F.A.; Oliveira, A.S.B. Epidemiological and clinical factors impact on the benefit of riluzole in the survival rates of patients with ALS. Arq. Neuropsiquiatr. 2017, 75, 515–522. [Google Scholar] [CrossRef]
- Andrews, J.A.; Jackson, C.E.; Heiman-Patterson, T.D.; Bettica, P.; Brooks, B.R.; Pioro, E.P. Real-world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 509–518. [Google Scholar] [CrossRef]
- Fang, T.; Al Khleifat, A.; Meurgey, J.H.; Jones, A.; Leigh, P.N.; Bensimon, G.; Al-Chalabi, A. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: A retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018, 17, 416–422. [Google Scholar] [CrossRef]
- Atassi, N.; Berry, J.; Shui, A.; Zach, N.; Sherman, A.; Sinani, E.; Walker, J.; Katsovskiy, I.; Schoenfeld, D.; Cudkowicz, M.; et al. The PRO-ACT database: Design, initial analyses and predictive features. Neurology 2014, 83, 1719–1725. [Google Scholar] [CrossRef]
- Thakore, N.J.; Lapin, B.R.; Pioro, E.P. Stage-specific riluzole effect in amyotrophic lateral sclerosis: A retrospective study. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 140–143. [Google Scholar] [CrossRef]
- De Jongh, A.D.; van Eijk, R.P.A.; van den Berg, L.H. Evidence for a multimodal effect of riluzole in patients with ALS? J. Neurol. Neurosurg. Psychiatry 2019, 90, 1183–1184. [Google Scholar] [CrossRef] [PubMed]
- Traynor, B.J.; Alexander, M.; Corr, B.; Frost, E.; Hardiman, O. An outcome study of riluzole in amyotrophic lateral sclerosis—A population-based study in Ireland, 1996–2000. J. Neurol. 2003, 250, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Zoccolella, S.; Beghi, E.; Palagano, G.; Fraddosio, A.; Guerra, V.; Samarelli, V.; Lepore, V.; Simone, I.L.; Lamberti, P.; Serlenga, L.; et al. Riluzole and amyotrophic lateral sclerosis survival: A population-based study in southern Italy. Eur. J. Neurol. 2007, 14, 262–268. [Google Scholar] [CrossRef]
- Chen, L.; Liu, X.; Tang, L.; Zhang, N.; Fan, D. Long-Term Use of Riluzole Could Improve the Prognosis of Sporadic Amyotrophic Lateral Sclerosis Patients: A Real-World Cohort Study in China. Front. Aging Neurosci. 2016, 8, 246. [Google Scholar] [CrossRef] [PubMed]
- Cetin, H.; Rath, J.; Füzi, J.; Reichardt, B.; Fülöp, G.; Koppi, S.; Erdler, M.; Ransmayr, G.; Weber, J.; Neumann, K.; et al. Epidemiology of amyotrophic lateral sclerosis and effect of riluzole on disease course. Neuroepidemiology 2015, 44, 6–15. [Google Scholar] [CrossRef]
- Mandrioli, J.; Malerba, S.A.; Beghi, E.; Fini, N.; Fasano, A.; Zucchi, E.; De Pasqua, S.; Guidi, C.; Terlizzi, E.; Sette, E.; et al. Riluzole and other prognostic factors in ALS: A population-based registry study in Italy. J. Neurol. 2018, 265, 817–827. [Google Scholar] [CrossRef]
- Debove, C.; Zeisser, P.; Salzman, P.M.; Powe, L.K.; Truffinet, P. The Rilutek (riluzole) Global Early Access Programme: An open-label safety evaluation in the treatment of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2001, 2, 153–158. [Google Scholar] [CrossRef]
- Geronimo, A.; Albertson, R.M.; Noto, J.; Simmons, Z. Ten years of riluzole use in a tertiary ALS clinic. Muscle Nerve 2022, 65, 659–666. [Google Scholar] [CrossRef]
- Wagner, M.L.; Landis, B.E. Riluzole: A new agent for amyotrophic lateral sclerosis. Ann. Pharmacother. 1997, 31, 738–744. [Google Scholar] [CrossRef]
- Weber, G.; Bitterman, H. Riluzole-induced neutropenia. Neurology 2004, 62, 1648. [Google Scholar] [CrossRef]
- Yanagisawa, N.; Tashiro, K.; Tohgi, H.; Mizuno, Y.; Kowa, H.; Kimuma, J. Efficacy and safety of riluzole in patients with amyotrophic lateral sclerosis: Double-blind placebo-controlled study in Japan. Igakuno Ayumi 1997, 182, 851–866. [Google Scholar]
- Ianiro, G.; Cammarota, G.; Milani, A.; Mettimano, M.; Gasbarrini, A. Moderately severe acute pancreatitis associated with riluzole. J. Clin. Gastroenterol. 2014, 48, 563. [Google Scholar] [CrossRef]
- Rodrigo, L.; Moreno, M.; Calleja, S.; Mateos, V.; Andrade, R.J.; Lucena, I.M. Riluzole-induced acute pancreatitis. Am. J. Gastroenterol. 2001, 96, 2268–2269. [Google Scholar] [CrossRef] [PubMed]
- Drory, V.E.; Sidi, I.; Korczyn, A.D. Riluzole-induced pancreatitis. Neurology 1999, 52, 892–893. [Google Scholar] [CrossRef]
- Falcao de Campos, C.; de Carvalho, M. Riluzole-induced recurrent pancreatitis. J. Clin. Neurosci. 2017, 45, 153–154. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Bravver, E.; Desai, U.; Jalali, N.; Dawson, W.B.; Bockenek, W.L.; Lindblom, S.S.; Paling, F.A.; Hawkins, S.; Antunez, P.; et al. Early Treatment Effects of Riluzole in ALS: Isometric Strength Improvement in Sentinel Muscles Correlates with Improved Survival (4121). Neurology 2020, 94 (Suppl. S15), 4121. [Google Scholar] [CrossRef]
- Onesti, E.; Schettino, I.; Gori, M.C.; Frasca, V.; Ceccanti, M.; Cambieri, C.; Ruoppolo, G.; Inghilleri, M. Dysphagia in Amyotrophic Lateral Sclerosis: Impact on Patient Behavior, Diet Adaptation, and Riluzole Management. Front. Neurol. 2017, 8, 94. [Google Scholar] [CrossRef]
- Hinchcliffe, M.; Smith, A. Riluzole: Real-world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degener. Neurol. Neuromuscul. Dis. 2017, 7, 61–70. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bettica, P.; Cazzaniga, S. Riluzole Oral Suspension: Bioavailability Following Percutaneous Gastrostomy Tube-modeled Administration Versus Direct Oral Administration. Clin. Ther. 2019, 41, 2490–2499. [Google Scholar] [CrossRef]
- Pascuzzi, R.M.; Shefner, J.; Chappell, A.S.; Bjerke, J.S.; Tamura, R.; Chaudhry, V.; Clawson, L.; Haas, L.; Rothstein, J.D. A phase II trial of talampanel in subjects with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2010, 11, 266–271. [Google Scholar] [CrossRef]
- Akamatsu, M.; Yamashita, T.; Hirose, N.; Teramoto, S.; Kwak, S. The AMPA receptor antagonist perampanel robustly rescues amyotrophic lateral sclerosis (ALS) pathology in sporadic ALS model mice. Sci. Rep. 2016, 6, 28649. [Google Scholar] [CrossRef] [PubMed]
- Turalde, C.W.R.; Moalong, K.M.C.; Espiritu, A.I.; Prado, M., Jr. Perampanel for amyotrophic lateral sclerosis: A systematic review and meta-analysis. Neurol. Sci. 2022, 43, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Cudkowicz, M.E.; Titus, S.; Kearney, M.; Yu, H.; Sherman, A.; Schoenfeld, D.; Hayden, D.; Shui, A.; Benjamin Brooks, B.; Conwit, R.; et al. Safety and Efficacy of Ceftriaxone for Amyotrophic Lateral Sclerosis: A Multi-Stage, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 2014, 13, 1083–1091. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S. Oxidative stress in amyotrophic lateral sclerosis: Pathophysiology and opportunities for pharmacological intervention. Oxidative Med. Cell. Longev. 2020, 2020, 5021694. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Carrì, M.T.; Valle, C.; Bozzo, F.; Cozzolino, M. Oxidative stress and mitochondrial damage: Importance in non-SOD1 ALS. Front. Cell Neurosci. 2015, 9, 41. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.K.; Singh, R.L.; Kalita, J.; Misra, U.K. Oxidant-antioxidant imbalance in the erythrocytes of sporadic amyotrophic lateral sclerosis patients correlates with the progression of disease. Neurochem. Int. 2008, 52, 1284–1289. [Google Scholar] [CrossRef]
- Kim, K. Glutathione in the nervous system as a potential therapeutic target to control the development and progression of amyotrophic lateral sclerosis. Antioxidants 2021, 10, 1011. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Jimenez-Villegas, J.; Ferraiuolo, L.; Mead, R.J.; Shaw, P.J.; Cuadrado, A.; Rojo, A.I. NRF2 as a therapeutic opportunity to impact in the molecular roadmap of ALS. Free Radic. Biol. Med. 2021, 173, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Pan, L.H.; Watanabe, M.; Kato, T.; Itoyama, Y. Induction of nitrotyrosine-like immunoreactivity in the lower motor neuron of amyotrophic lateral sclerosis. Neurosci. Lett. 1995, 199, 152–154. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Edaravone Acute Infarction Study Group. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction. Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc. Dis. 2003, 15, 222–229. [Google Scholar] [CrossRef]
- Miyaji, Y.; Yoshimura, S.; Sakai, N.; Yamagami, H.; Egashira, Y.; Shirakawa, M.; Uchida, K.; Kageyama, H.; Tomogane, Y. Effect of edaravone on favorable outcome in patients with acute cerebral large vessel occlusion: Subanalysis of RESCUE-Japan registry. Neurol. Med. Chir. 2015, 55, 241–247. [Google Scholar] [CrossRef]
- Cruz, M.P. Edaravone (Radicava): A Novel Neuroprotective Agent for the Treatment of Amyotrophic Lateral Sclerosis. Pharm. Ther. 2018, 43, 25–28. [Google Scholar]
- Writing Group, Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well-defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Lunetta, C.; Moglia, C.; Lizio, A.; Caponnetto, C.; Dubbioso, R.; Giannini, F.; Matà, S.; Mazzini, L.; Sabatelli, M.; Siciliano, G.; et al. The Italian multicenter experience with edaravone in amyotrophic lateral sclerosis. J. Neurol. 2020, 267, 3258–3267. [Google Scholar] [CrossRef]
- Witzel, S.; Maier, A.; Steinbach, R.; Grosskreutz, J.; Koch, J.C.; Sarikidi, A.; Petri, S.; Günther, R.; Wolf, J.; Hermann, A.; et al. Safety and effectiveness of long-term intravenous administration of edaravone for treatment of patients with amyotrophic lateral sclerosis. JAMA Neurol. 2022, 79, 121–130. [Google Scholar] [CrossRef]
- Brooks, B.R.; Berry, J.D.; Ciepielewska, M.; Liu, Y.; Zambrano, G.S.; Zhang, J.; Hagan, M. Intravenous edaravone treatment in ALS and survival: An exploratory, retrospective, administrative claims analysis. eClinicalMedicine 2022, 52, 101590. [Google Scholar] [CrossRef] [PubMed]
- Vu, M.; Tortorice, K.; Zacher, J.; Dong, D.; Hur, K.; Zhang, R.; Good, C.B.; Glassman, P.A.; Cunningham, F.E. Assessment of Use and Safety of Edaravone for Amyotrophic Lateral Sclerosis in the Veterans Affairs Health Care System. JAMA Netw. Open 2020, 3, e2014645. [Google Scholar] [CrossRef]
- Jayasinghe, M.; Prathiraja, O.; Perera, P.B.; Jena, R.; Silva, M.S.; Weerawarna, P.S.H.; Singhal, M.; Kayani, A.M.A.; Karnakoti, S.; Jain, S. The Role of Mesenchymal Stem Cells in the Treatment of Type 1 Diabetes. Cureus 2022, 14, e27337. [Google Scholar] [CrossRef] [PubMed]
- Nourelden, A.Z.; Kamal, I.; Hagrass, A.I.; Tawfik, A.G.; Elhady, M.M.; Fathallah, A.H.; Eshag, M.M.E.; Zaazouee, M.S. Safety and efficacy of edaravone in patients with amyotrophic lateral sclerosis: A systematic review and meta-analysis. Neurol. Sci. 2023, 44, 3429–3442. [Google Scholar] [CrossRef]
- Shang, Q.; Zhou, J.; Ye, J.; Chen, M. Adverse events reporting of edaravone: A real-world analysis from FAERS database. Sci. Rep. 2025, 15, 8148. [Google Scholar] [CrossRef]
- Shimizu, H.; Nishimura, Y.; Shiide, Y.; Yoshida, K.; Hirai, M.; Matsuda, M.; Nakamaru, Y.; Kato, Y.; Kondo, K. Bioequivalence study of oral suspension and intravenous formulation of edaravone in healthy adult subjects. Clin. Pharmacol. Drug Dev. 2021, 10, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Ferrer. Reports Top-Line Results from Phase 3 ADORE Study in ALS. News Release. Ferrer. Published 10 January 2024. Available online: https://www.ferrer.com/en/results-study-ADORE-ALS (accessed on 15 January 2024).
- Robinson, A.P.; Zhang, J.Z.; Titus, H.E.; Karl, M.; Merzliakov, M.; Dorfman, A.R.; Karlik, S.; Stewart, M.G.; Watt, R.K.; Facer, B.D.; et al. Nanocatalytic activity of clean-surfaced, faceted nanocrystalline gold enhances remyelination in animal models of multiple sclerosis. Sci. Rep. 2020, 10, 1936. [Google Scholar] [CrossRef]
- Vucic, S.; Menon, P.; Huynh, W.; Mahoney, C.; Ho, K.S.; Hartford, A.; Rynders, A.; Evan, J.; Evan, J.; Ligozio, S.; et al. Efficacy and safety of CNM-Au8 in amyotrophic lateral sclerosis (RESCUE-ALS study): A phase 2, randomised, double-blind, placebo-controlled trial and open label extension. eClinicalMedicine 2023, 60, 102036. [Google Scholar] [CrossRef]
- Cudkowicz, M.E. NCT04297683: HEALEY ALS Platform Trial—Master Protocol; ClinicalTrials.gov; U.S. National Library of Medicine: Bethesda, MD, USA, 2021.
- Cudkowicz, M.E. NCT04414345: HEALEY ALS Platform Trial—Regimen C CNM-Au8; ClinicalTrials.gov; U.S. National Library of Medicine: Bethesda, MD, USA, 2021.
- Miller, R.; Bradley, W.; Cudkowicz, M.; Hubble, J.; Meininger, V.; Mitsumoto, H.; Moore, D.; Pohlmann, H.; Sauer, D.; Silani, V.; et al. Phase II/III randomized trial of TCH346 in patients with ALS. Neurology 2007, 69, 776–784. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Hong, S.H.; Lee, Y.J.; Chung, S.S.; Jung, H.S.; Park, S.G.; Park, K.S. Tauroursodeoxycholate (TUDCA), chemical chaperone, enhances function of islets by reducing ER stress. Biochem. Biophys. Res. Commun. 2010, 397, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Ghaffar, A.; Elhossary, G.G.; Mahmoud, A.M.; Elshazly, A.H.M.; Hassanin, O.A.; Saleh, A.; Mansour, S.M.; Metwally, F.G.; Hanafy, L.K.; Karam, S.H.; et al. Potential prophylactic effect of chemical chaperones for alleviation of endoplasmic reticulum stress in experimental diabetic cataract. Bull. Natl. Res. Cent. 2019, 43, 71. [Google Scholar] [CrossRef]
- Khalaf, K.; Tornese, P.; Cocco, A.; Albanese, A. Tauroursodeoxycholic acid: A potential therapeutic tool in neurodegenerative diseases. Transl. Neurodegener. 2022, 11, 33. [Google Scholar] [CrossRef]
- Rodrigues, C.M.P.; Fan, G.; Wong, P.Y.; Kren, B.T.; Steer, C.J. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol. Med. 1998, 4, 165–178. [Google Scholar] [CrossRef]
- Rodrigues, C.M.P.; Fan, G.; Ma, X.; Kren, B.T.; Steer, C.J. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J. Clin. Investig. 1998, 101, 2790–2799. [Google Scholar] [CrossRef]
- Rodrigues, C.M.P.; Ma, X.; Linehan-Stieers, C.; Fan, G.; Kren, B.T.; Steer, C.J. Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ. 1999, 6, 842–854. [Google Scholar] [CrossRef]
- Castro, R.E.; Amaral, J.D.; Solá, S.; Kren, B.T.; Steer, C.J.; Rodrigues, C.M. Differential regulation of cyclin D1 and cell death by bile acids in primary rat hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G327–G334. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.E.; Lalli, S.; Monsurrò, M.R.; Sagnelli, A.; Taiello, A.C.; Reggiori, B.; La Bella, V.; Tedeschi, G.; Albanese, A. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2016, 23, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, E.; Musazzi, U.M.; Fedele, G.; Martinelli, I.; Gianferrari, G.; Simonini, C.; Fini, N.; Ghezzi, A.; Caputo, M.; Sette, E.; et al. Effect of tauroursodeoxycholic acid on survival and safety in amyotrophic lateral sclerosis: A retrospective population-based cohort study. eClinicalMedicine 2023, 65, 102256. [Google Scholar] [CrossRef]
- Cortez, D.; Marin, R.; Toledo-Flores, D.; Froidevaux, L.; Liechti, A.; Waters, P.D.; Grützner, F.; Kaessmann, H. Origins and functional evolution of Y chromosomes across mammals. Nature 2014, 508, 488–493. [Google Scholar] [CrossRef]
- Ryu, H.; Smith, K.; Camelo, S.I.; Carreras, I.; Lee, J.; Iglesias, A.H.; Dangond, F.; Cormier, K.A.; Cudkowicz, M.E.; Brown, R.H., Jr.; et al. phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 2005, 93, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Margaret, A.; Owegi, M.A.; et al. Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Macklin, E.A.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve 2021, 63, 31–39. [Google Scholar] [CrossRef]
- Lin, T.J.; Cheng, G.C.; Wu, L.Y.; Lai, W.Y.; Ling, T.Y.; Kuo, Y.C.; Huang, Y.H. Potential of cellular therapy for ALS: Current strategies and future prospects. Front. Cell Dev. Biol. 2022, 10, 851613, Erratum in Front. Cell Dev. Biol. 2023, 11, 1292681. https://doi.org/10.3389/fcell.2023.1292681. [Google Scholar] [CrossRef] [PubMed]
- Shiryaeva, O.; Tolochko, C.; Alekseeva, T.; Dyachuk, V. Targets and gene therapy of ALS (Part 1). Int. J. Mol. Sci. 2025, 26, 4063. [Google Scholar] [CrossRef]
- Richardson, P.J.; Smith, D.P.; de Giorgio, A.; Snetkov, X.; Almond-Thynne, J.; Cronin, S.; Mead, R.J.; McDermott, C.J.; Shaw, P.J. Janus kinase inhibitors are potential therapeutics for amyotrophic lateral sclerosis. Transl. Neurodegener. 2023, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.L.; Ma, S.; Tai, W.; Zhong, X.; Ni, H.; Zou, Y.; Wang, J.; Zhang, C.-L. Chemical screens in aging-relevant human motor neurons identify MAP4Ks as therapeutic targets for amyotrophic lateral sclerosis. bioRxiv 2023. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, C.L. MAP4K inhibition as a potential therapy for amyotrophic lateral sclerosis. Neural. Regen. Res. 2024, 19, 1639–1640. [Google Scholar] [CrossRef]
- Liu, M.L.; Ma, S.; Tai, W.; Zhong, X.; Ni, H.; Zou, Y.; Wang, J.; Zhang, C.-L. Screens in aging-relevant human ALS-motor neurons identify MAP4Ks as therapeutic targets for the disease. Cell Death Dis. 2024, 15, 4. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolochko, C.; Shiryaeva, O.; Alekseeva, T.; Dyachuk, V. Amyotrophic Lateral Sclerosis: Pathophysiological Mechanisms and Treatment Strategies (Part 2). Int. J. Mol. Sci. 2025, 26, 5240. https://doi.org/10.3390/ijms26115240
Tolochko C, Shiryaeva O, Alekseeva T, Dyachuk V. Amyotrophic Lateral Sclerosis: Pathophysiological Mechanisms and Treatment Strategies (Part 2). International Journal of Molecular Sciences. 2025; 26(11):5240. https://doi.org/10.3390/ijms26115240
Chicago/Turabian StyleTolochko, Christina, Olga Shiryaeva, Tatiana Alekseeva, and Vyacheslav Dyachuk. 2025. "Amyotrophic Lateral Sclerosis: Pathophysiological Mechanisms and Treatment Strategies (Part 2)" International Journal of Molecular Sciences 26, no. 11: 5240. https://doi.org/10.3390/ijms26115240
APA StyleTolochko, C., Shiryaeva, O., Alekseeva, T., & Dyachuk, V. (2025). Amyotrophic Lateral Sclerosis: Pathophysiological Mechanisms and Treatment Strategies (Part 2). International Journal of Molecular Sciences, 26(11), 5240. https://doi.org/10.3390/ijms26115240