
Stromal Hedgehog Signaling Is Associated with Favorable Outcomes in Pancreatic Cancer

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
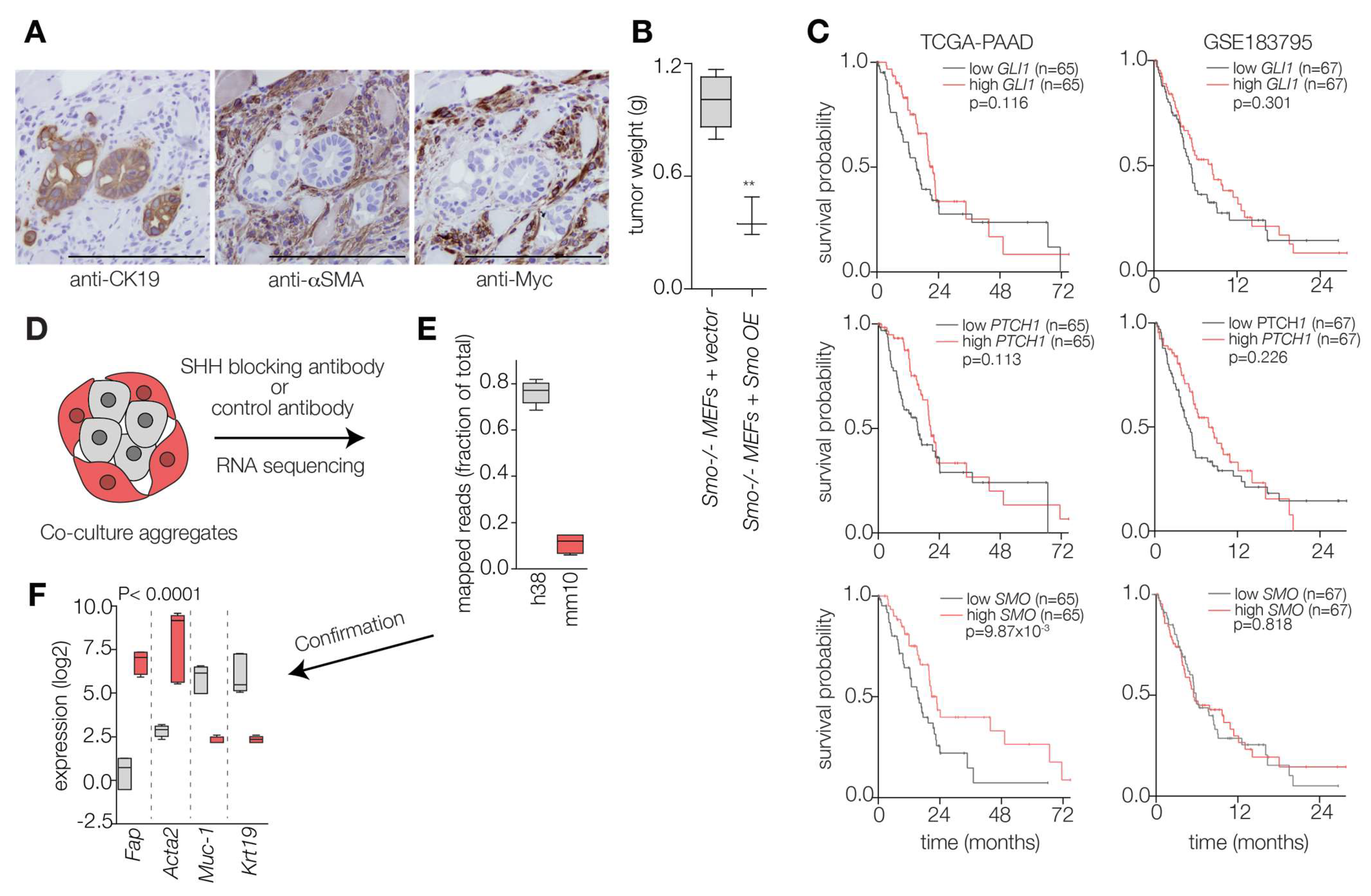
2.1. Hedgehog Ligand Abundance Is Not Prognostic in PDAC
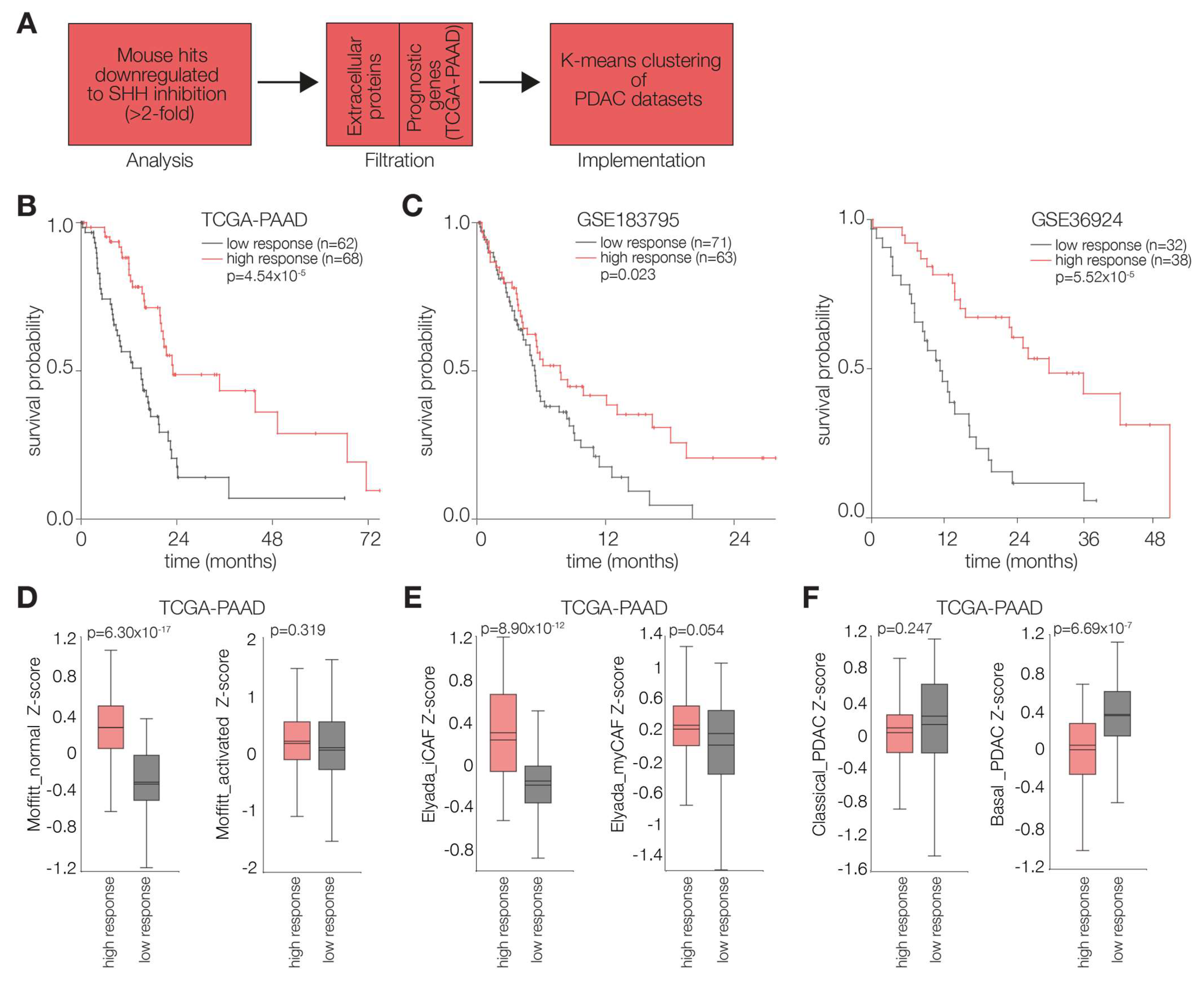
2.2. Hedgehog Response in the PDAC Microenvironment Impacts Tumor Progression
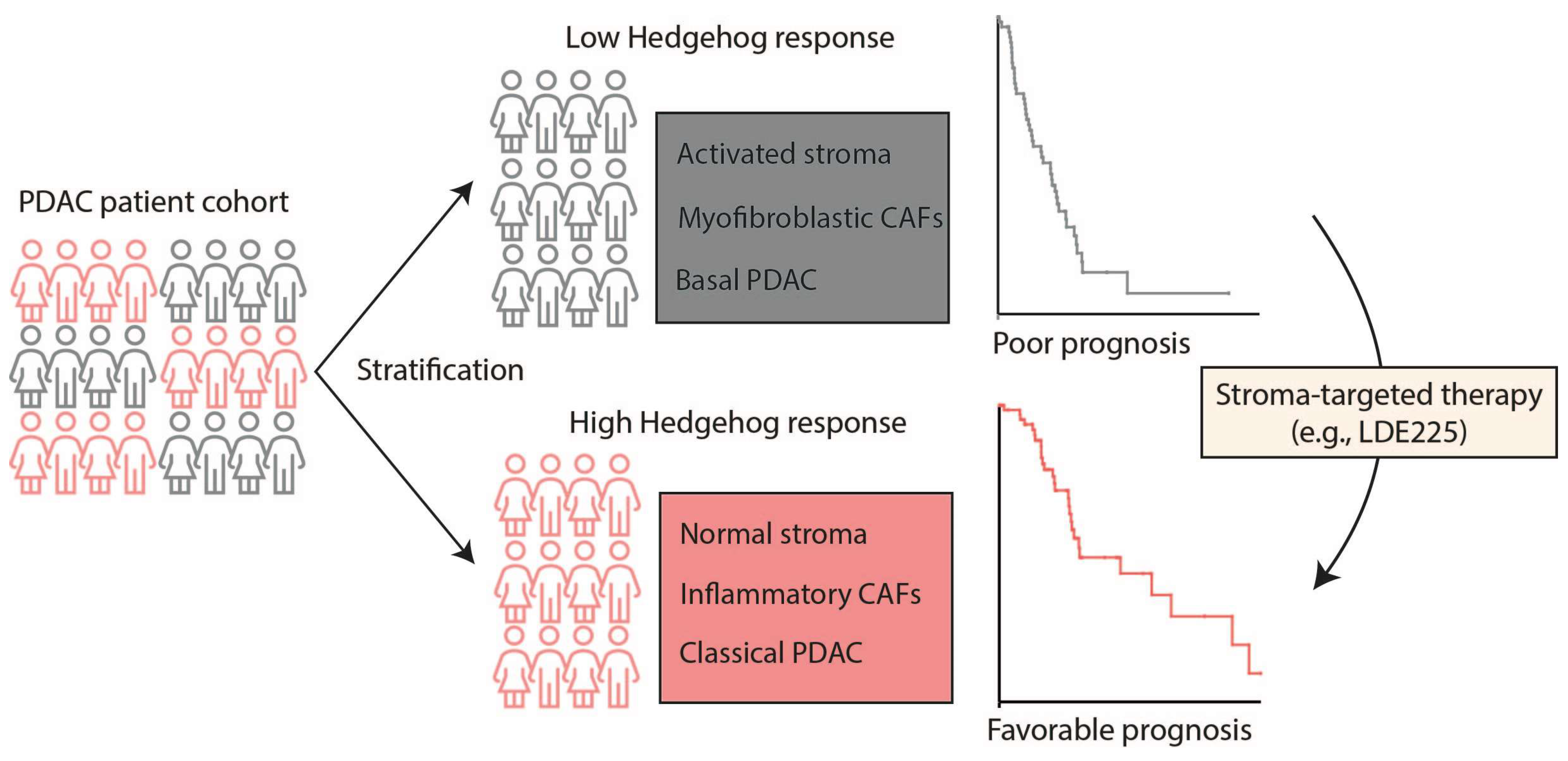
2.3. PDAC Tumors Characterized by High Stromal Hh Response Are Favorably Prognostic
3. Discussion and Conclusions
4. Limitations and Perspectives
5. Materials and Methods
5.1. Cell Culture and Generation of Antibodies
5.2. Co-Culture Model
5.3. Gene Silencing
5.4. Lentivirus Generation and Transduction
5.5. Quantitative RT-PCR
- hGAPDH Fw: GAAGGTGAAGGTCGGAGTC; Rv: TGGAAGATGGTGATGGGATT
- hSHH Fw: GCTCGGTGAAAGCAGAGAAC; Rv: CCAGGAAAGTGAGGAAGTCG
- hDISP1 Fw: ACAGCTTTTTCTGCGACGTT; Rv: TCCATAATGTCTCCCCTCCA
- mGapdh Fw: CTCATGACCACAGTCCATGC; Rv: CACATTGGGGGTAGGAACAC
- mGli1 Fw: ATAGGGTCTCGGGGTCTCA; Rv: CGGCTGACTGTGTAAGCAGA
- mPtch1 Fw: CTCAGGCAATACGAAGCACA; Rv: GACAAGGAGCCAGAGTCCAG
5.6. Preparation of RNA-Seq Libraries
5.7. Animals and PDAC PDX Model
5.8. Reporter Assay
5.9. Slide Preparation and Immunohistochemical Staining
5.10. Statistics and Bioinformatic Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Halbrook, C.J.; Lyssiotis, C.A.; Pasca di Magliano, M.; Maitra, A. Pancreatic cancer: Advances and challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Deng, G.; Ma, Y.; Si, H.; Wang, Z.; Dai, G. Survival outcome of different treatment sequences in patients with locally advanced and metastatic pancreatic cancer. BMC Cancer 2024, 24, 67. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Manoukian, P.; Bijlsma, M.; Laarhoven, H. The Cellular Origins of Cancer-Associated Fibroblasts and Their Opposing Contributions to Pancreatic Cancer Growth. Front. Cell Dev. Biol. 2021, 9, 743907. [Google Scholar] [CrossRef] [PubMed]
- Hutton, C.; Heider, F.; Blanco-Gomez, A.; Banyard, A.; Kononov, A.; Zhang, X.; Karim, S.; Paulus-Hock, V.; Watt, D.; Steele, N.; et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 2021, 39, 1227–1244.e20. [Google Scholar] [CrossRef]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-Positive Cancer-Associated Fibroblasts Inhibit Pancreatic Carcinogenesis. Cancer Res. 2019, 79, 5367–5381. [Google Scholar] [CrossRef] [PubMed]
- Dings, M.P.G.; Manoukian, P.; Waasdorp, C.; Quik, J.S.E.; Strijker, M.; Lodestijn, S.C.; van Neerven, S.M.; Moreno, L.F.; de Oliveira, R.L.; Bonsing, B.A.; et al. Serum levels of iCAF-derived osteoglycin predict favorable outcome in pancreatic cancer. Int. J. Cancer 2023, 152, 511–523. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Hwang, W.L.; Jagadeesh, K.A.; Guo, J.A.; Hoffman, H.I.; Yadollahpour, P.; Reeves, J.W.; Mohan, R.; Drokhlyansky, E.; Van Wittenberghe, N.; Ashenberg, O.; et al. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat. Genet. 2022, 54, 1178–1191. [Google Scholar] [CrossRef]
- Javadrashid, D.; Baghbanzadeh, A.; Derakhshani, A.; Leone, P.; Silvestris, N.; Racanelli, V.; Solimando, A.G.; Baradaran, B. Pancreatic Cancer Signaling Pathways, Genetic Alterations, and Tumor Microenvironment: The Barriers Affecting the Method of Treatment. Biomedicines 2021, 9, 373. [Google Scholar] [CrossRef]
- Grauel, A.L.; Nguyen, B.; Ruddy, D.; Laszewski, T.; Schwartz, S.; Chang, J.; Chen, J.; Piquet, M.; Pelletier, M.; Yan, Z.; et al. TGFβ-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat. Commun. 2020, 11, 6315. [Google Scholar] [CrossRef] [PubMed]
- Ferrantella, A.R.; Garg, B.; Giri, B.; Modi, S.; Sethi, V.; Kurtom, S.; Ramakrishnan, S.; Gilboa, E.; Saluja, A.; Dudeja, V. Secretion of SDF1/CXCL12 by Stromal Fibroblasts Shields Pancreatic Cancer from Immune Response. J. Am. Coll. Surg. 2018, 227 (Suppl. 1), S248. [Google Scholar] [CrossRef]
- Blaauboer, A.; Sideras, K.; van Eijck, C.H.J.; Hofland, L.J. Type I interferons in pancreatic cancer and development of new therapeutic approaches. Crit. Rev. Oncol. Hematol. 2021, 159, 103204. [Google Scholar] [CrossRef]
- Qian, L.; Yu, S.; Yin, C.; Zhu, B.; Chen, Z.; Meng, Z.; Wang, P. Plasma IFN-γ-inducible chemokines CXCL9 and CXCL10 correlate with survival and chemotherapeutic efficacy in advanced pancreatic ductal adenocarcinoma. Pancreatology 2019, 19, 340–345. [Google Scholar] [CrossRef] [PubMed]
- House, I.G.; Savas, P.; Lai, J.; Chen, A.X.Y.; Oliver, A.J.; Teo, Z.L.; Todd, K.L.; Henderson, M.A.; Giuffrida, L.; Petley, E.V.; et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin. Cancer Res. 2020, 26, 487–504. [Google Scholar] [CrossRef] [PubMed]
- Armas-López, L.; Zúñiga, J.; Arrieta, O.; Ávila-Moreno, F. The Hedgehog-GLI pathway in embryonic development and cancer: Implications for pulmonary oncology therapy. Oncotarget 2017, 8, 60684–60703. [Google Scholar] [CrossRef]
- Chamberlain, C.E.; Jeong, J.; Guo, C.; Allen, B.L.; McMahon, A.P. Notochord-derived Shh concentrates in close association with the apically positioned basal body in neural target cells and forms a dynamic gradient during neural patterning. Development 2008, 135, 1097–1106. [Google Scholar] [CrossRef]
- Kim, S.K.; Melton, D.A. Pancreas development is promoted by cyclopamine, a Hedgehog signaling inhibitor. Proc. Natl. Acad. Sci. USA 1998, 95, 13036–13041. [Google Scholar] [CrossRef]
- Xuan, S.; Sussel, L. GATA4 and GATA6 regulate pancreatic endoderm identity through inhibition of hedgehog signaling. Development 2016, 143, 780–786. [Google Scholar] [CrossRef]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.-C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef]
- Suchors, C.; Kim, J. Canonical Hedgehog Pathway and Noncanonical GLI Transcription Factor Activation in Cancer. Cells 2022, 11, 2523. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, Y.; Li, X. Structure of human Dispatched-1 provides insights into Hedgehog ligand biogenesis. Life Sci. Alliance 2020, 3, e202000776. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Schlotman, K.E.; Xie, J. Deciphering the role of hedgehog signaling in pancreatic cancer. J. Biomed. Res. 2016, 30, 353–360. [Google Scholar]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in αSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 39, 548–565.e6. [Google Scholar] [CrossRef] [PubMed]
- Dijk, F.; Veenstra, V.L.; Soer, E.C.; Dings, M.P.G.; Zhao, L.; Halfwerk, J.B.; Hooijer, G.K.; Damhofer, H.; Marzano, M.; Steins, A.; et al. Unsupervised class discovery in pancreatic ductal adenocarcinoma reveals cell-intrinsic mesenchymal features and high concordance between existing classification systems. Sci. Rep. 2020, 10, 337. [Google Scholar] [CrossRef]
- Adams, C.R.; Htwe, H.H.; Marsh, T.; Wang, A.L.; Montoya, M.L.; Subbaraj, L.; Tward, A.D.; Bardeesy, N.; Perera, R.M. Transcriptional control of subtype switching ensures adaptation and growth of pancreatic cancer. eLife 2019, 8, e45313. [Google Scholar] [CrossRef]
- Damhofer, H.; Veenstra, V.L.; Tol, J.A.M.G.; van Laarhoven, H.W.M.; Medema, J.P.; Bijlsma, M.F. Blocking Hedgehog release from pancreatic cancer cells increases paracrine signaling potency. J. Cell Sci. 2015, 128, 129–139. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Damhofer, H.; Medema, J.P.; Veenstra, V.L.; Badea, L.; Popescu, I.; Roelink, H.; Bijlsma, M.F. Assessment of the stromal contribution to Sonic Hedgehog-dependent pancreatic adenocarcinoma. Mol. Oncol. 2013, 7, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Yang, S.; Tang, W.; Azizian, A.; Gaedcke, J.; Ströbel, P.; Wang, L.; Cawley, H.; Ohara, Y.; Valenzuela, P.; Zhang, L.; et al. Dysregulation of HNF1B/Clusterin axis enhances disease progression in a highly aggressive subset of pancreatic cancer patients. Carcinogenesis 2022, 43, 1198–1210. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef]
- Liang, C.; Shi, S.; Meng, Q.; Liang, D.; Ji, S.; Zhang, B.; Qin, Y.; Xu, J.; Ni, Q.; Yu, X. Complex roles of the stroma in the intrinsic resistance to gemcitabine in pancreatic cancer: Where we are and where we are going. Exp. Mol. Med. 2017, 49, e406. [Google Scholar] [CrossRef]
- Walter, K.; Omura, N.; Hong, S.M.; Griffith, M.; Vincent, A.; Borges, M.; Goggins, M. Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clin. Cancer Res. 2010, 16, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Francescone, R.; Crawford, H.C.; Vendramini-Costa, D.B. Rethinking the Roles of Cancer-Associated Fibroblasts in Pancreatic Cancer. Cell. Mol. Gastroenterol. Hepatol. 2024, 17, 737–743. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa with Nab-Paclitaxel Plus Gemcitabine for Patients with Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Kocher, H.M.; Sasieni, P.; Corrie, P.; McNamara, M.G.; Sarker, D.; Froeling, F.E.M.; Christie, A.; Gillmore, R.; Khan, K.; Propper, D. Study protocol: Multi-centre, randomised controlled clinical trial exploring stromal targeting in locally advanced pancreatic cancer; STARPAC2. BMC Cancer 2025, 25, 106. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manoukian, P.; Damhofer, H.; Zhao, L.; van Laarhoven, H.W.M.; Bijlsma, M.F. Stromal Hedgehog Signaling Is Associated with Favorable Outcomes in Pancreatic Cancer. Int. J. Mol. Sci. 2025, 26, 5200. https://doi.org/10.3390/ijms26115200
Manoukian P, Damhofer H, Zhao L, van Laarhoven HWM, Bijlsma MF. Stromal Hedgehog Signaling Is Associated with Favorable Outcomes in Pancreatic Cancer. International Journal of Molecular Sciences. 2025; 26(11):5200. https://doi.org/10.3390/ijms26115200
Chicago/Turabian StyleManoukian, Paul, Helene Damhofer, Lan Zhao, Hanneke W. M. van Laarhoven, and Maarten F. Bijlsma. 2025. "Stromal Hedgehog Signaling Is Associated with Favorable Outcomes in Pancreatic Cancer" International Journal of Molecular Sciences 26, no. 11: 5200. https://doi.org/10.3390/ijms26115200
APA StyleManoukian, P., Damhofer, H., Zhao, L., van Laarhoven, H. W. M., & Bijlsma, M. F. (2025). Stromal Hedgehog Signaling Is Associated with Favorable Outcomes in Pancreatic Cancer. International Journal of Molecular Sciences, 26(11), 5200. https://doi.org/10.3390/ijms26115200