

Are You a Friend or an Enemy? The Dual Action of Methylglyoxal on Brain Microvascular Endothelial Cells
 , , and
, , and 
Abstract

1. Introduction
2. Results
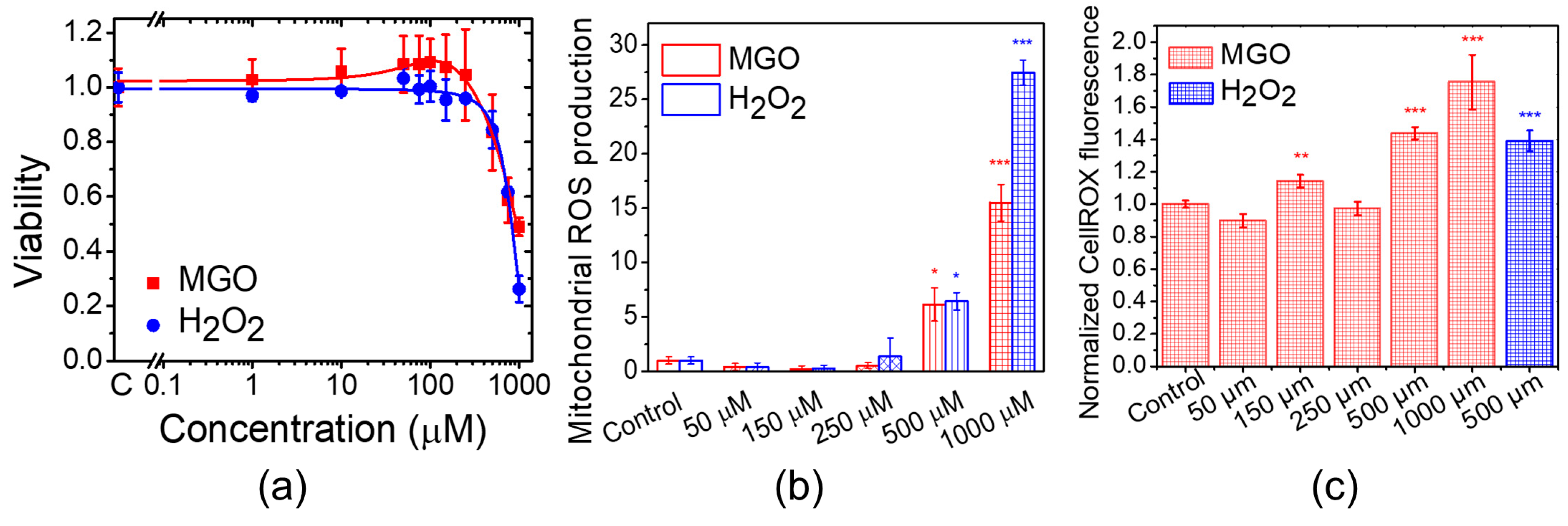
2.1. Methylglyoxal Inhibits Cell Proliferation and Stimulates Mitochondrial ROS Production in Brain Microvascular Endothelial Cells
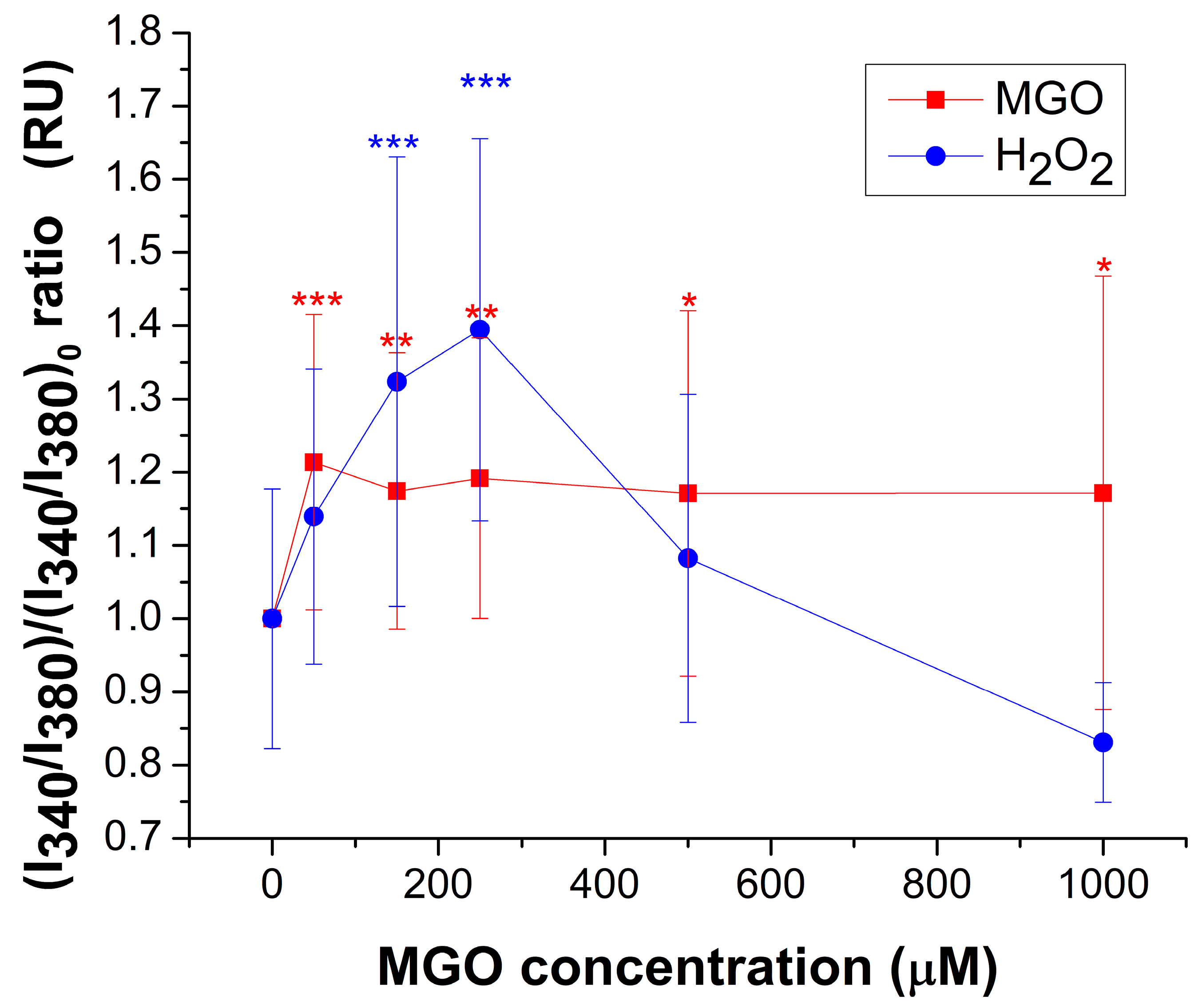
2.2. Methylglyoxal Modulates the Equilibrium of Free Cytosolic Calcium Ions in Brain Endothelial Cells
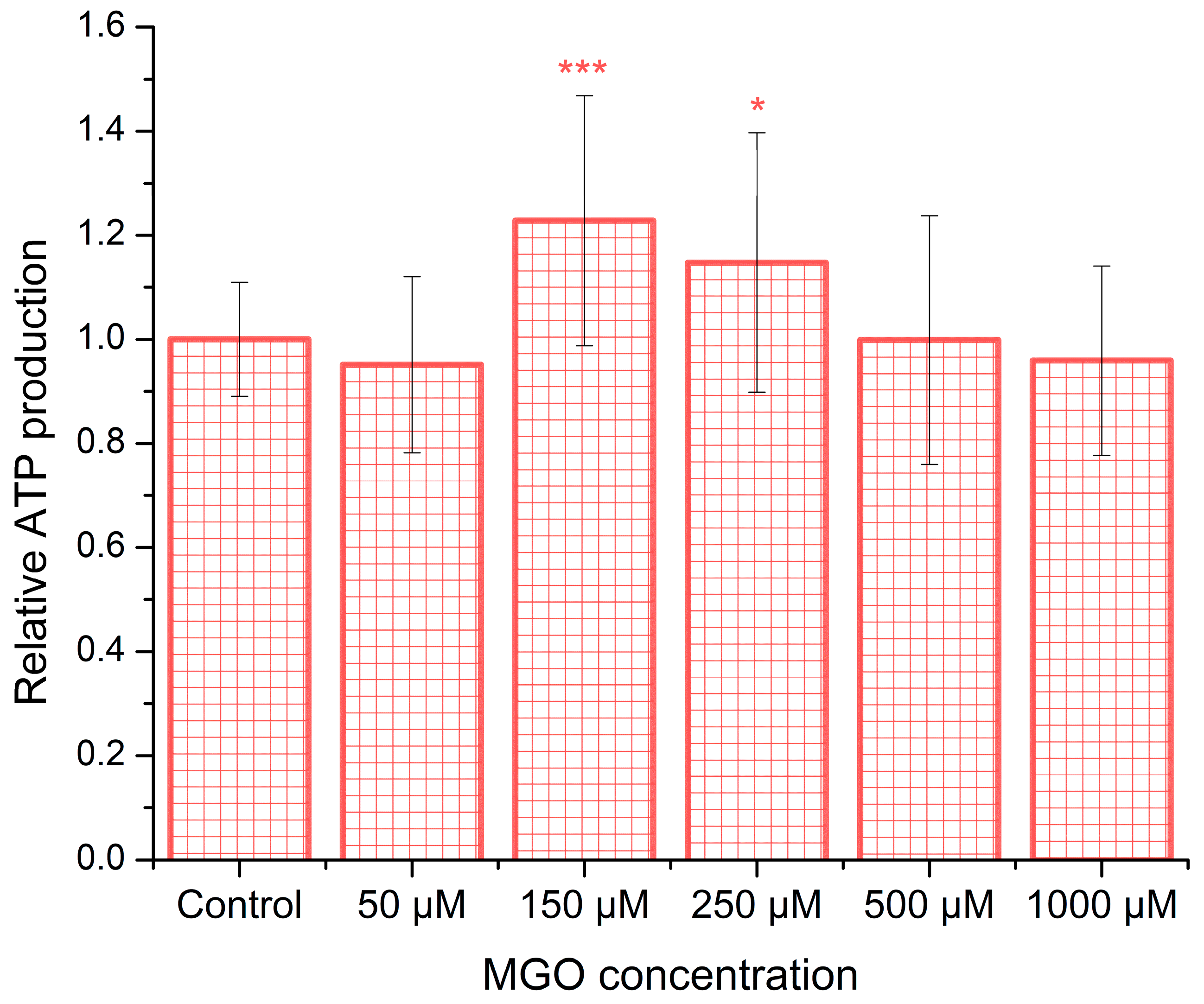
2.3. MGO Upregulates ATP Release, Exhibiting a Window-like Dose–Response Pattern
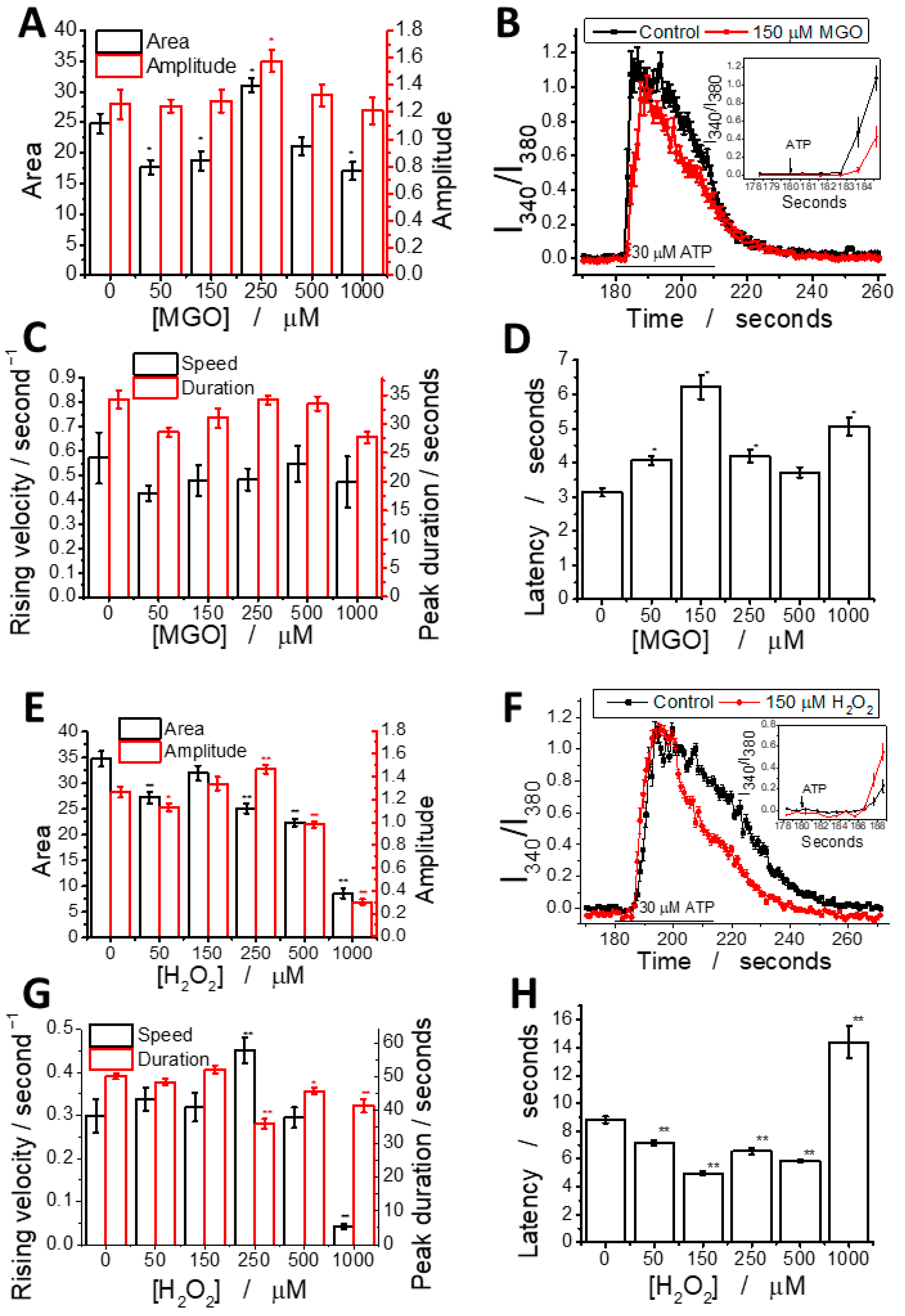
2.4. Methylglyoxal Modulates ATP-Induced Cytosolic Calcium Transients in Brain Endothelial Cells
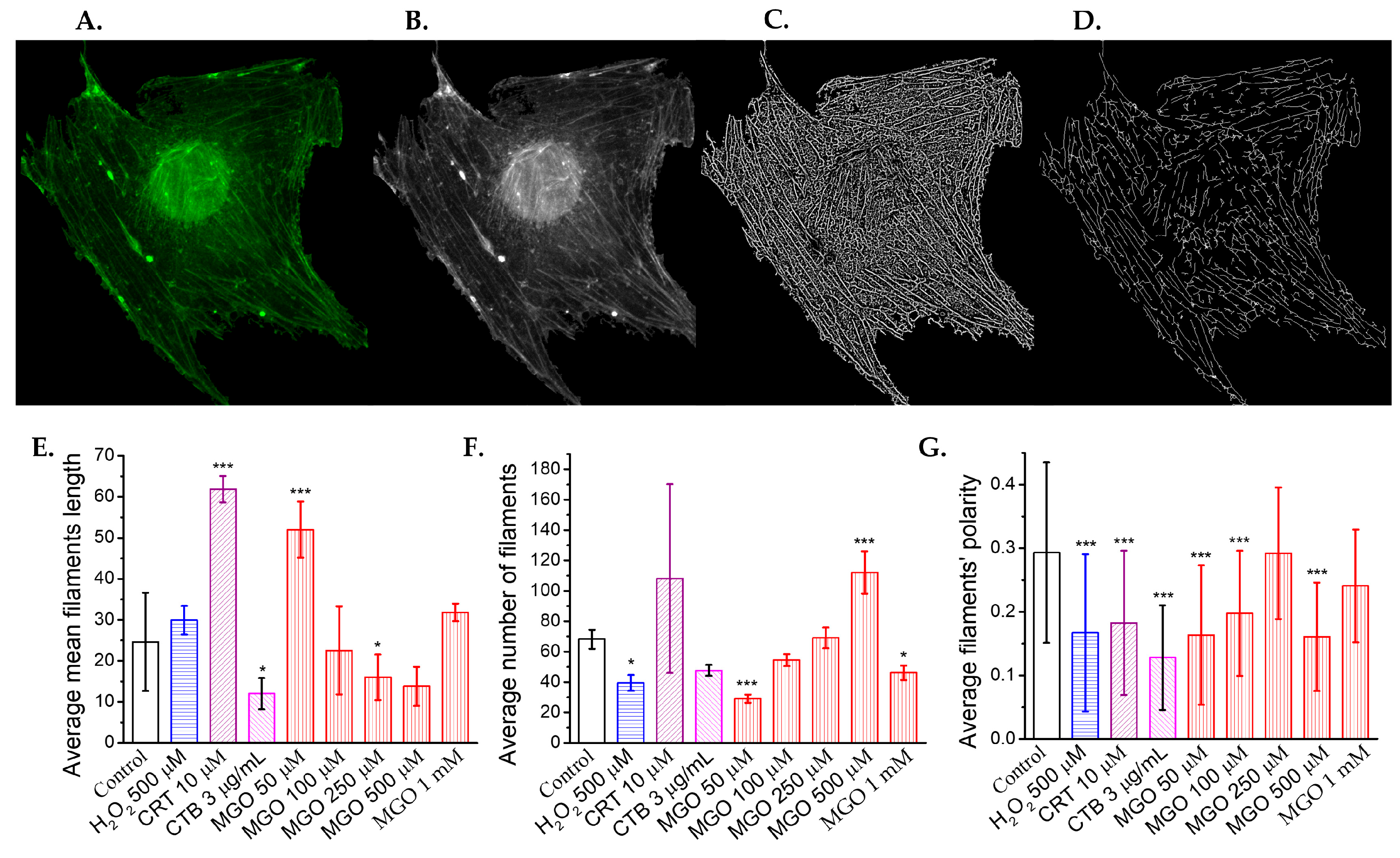
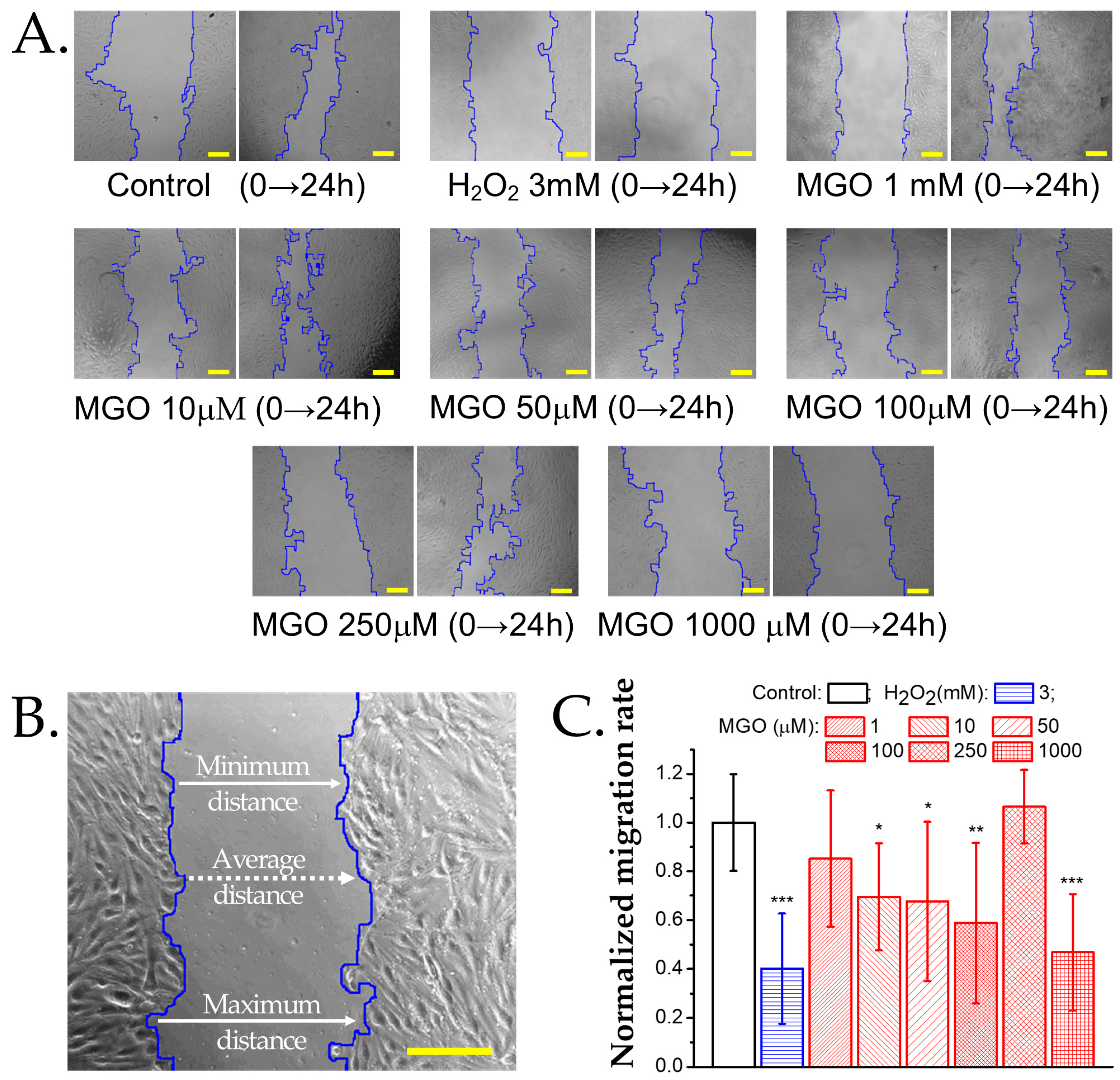
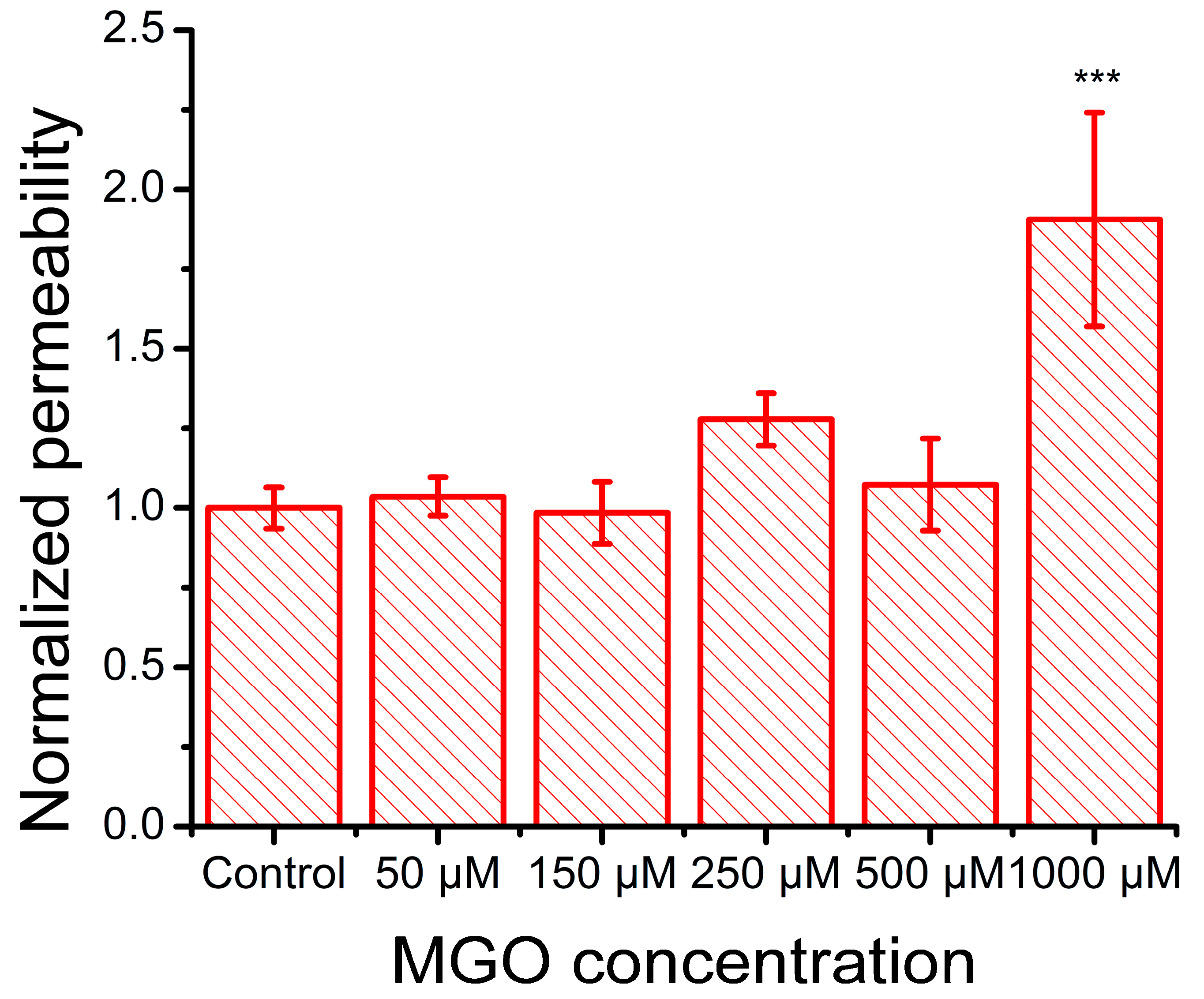
2.5. Methylglyoxal Induces Cytoskeletal Reorganization in Brain Microvascular Endothelial Cells, Leading to Impaired Migration and Increased Monolayer Permeability
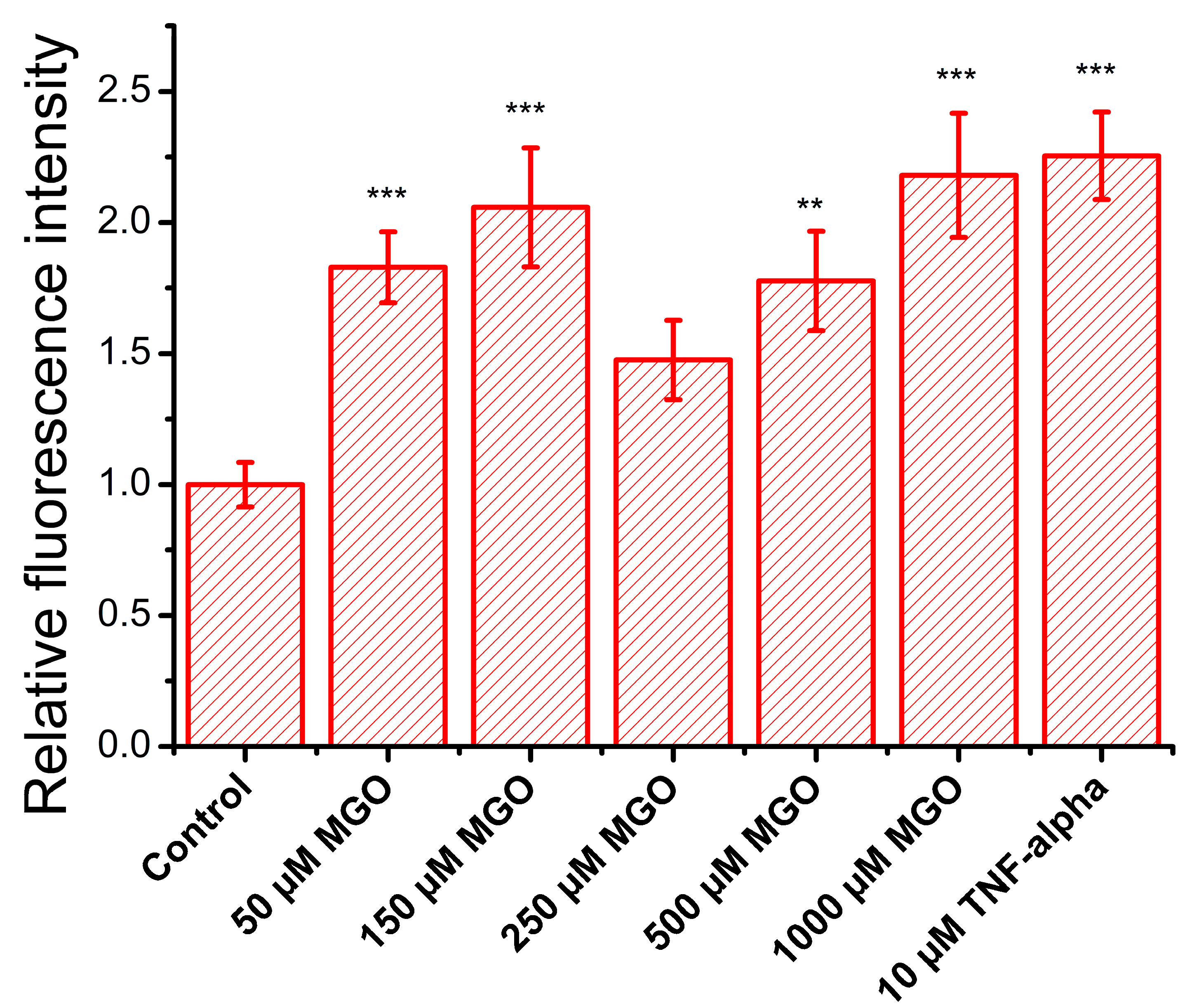
2.6. MGO Enhances Leukocyte Adhesion to Brain Microvascular Endothelial Cells
3. Discussion
4. Materials and Methods
4.1. Brain Endothelial Cells
4.2. Cell Viability Assay
4.3. Mitochondrial and Total ROS Production
4.4. ATP Production
4.5. Calcium Imaging and Calcium-Signal Parameter Analysis
4.6. Adhesion Assay
4.7. Permeabilization Assay
4.8. Wound Healing Assay
4.9. Fluorescent Staining of Actin Filaments and Confocal Image Acquisition
4.10. Confocal Image Processing for Cytoskeletal Changes
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schalkwijk, C.G.; Van Bezu, J.; Van Der Schors, R.C.; Uchida, K.; Stehouwer, C.D.A.; Van Hinsbergh, V.W.M. Heat-shock Protein 27 Is a Major Methylglyoxal-modified Protein in Endothelial Cells. FEBS Lett. 2006, 580, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Wang, H.; Song, H.; Yang, R.; Wang, L.; Xue, X.; Sun, W.; Hu, J. Lactate Is Answerable for Brain Function and Treating Brain Diseases: Energy Substrates and Signal Molecule. Front. Nutr. 2022, 9, 800901. [Google Scholar] [CrossRef] [PubMed]
- De Arriba, S.G.; Stuchbury, G.; Yarin, J.; Burnell, J.; Loske, C.; Münch, G. Methylglyoxal Impairs Glucose Metabolism and Leads to Energy Depletion in Neuronal Cells—Protection by Carbonyl Scavengers. Neurobiol. Aging 2007, 28, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.L.; Guillausseau, P.J. Advanced Glycation End Products, Their Receptors and Diabetic Angiopathy. Diabetes Metab. 2001, 27, 535–542. [Google Scholar] [CrossRef]
- Münch, G.; Westcott, B.; Menini, T.; Gugliucci, A. Advanced Glycation Endproducts and Their Pathogenic Roles in Neurological Disorders. Amino Acids 2012, 42, 1221–1236. [Google Scholar] [CrossRef]
- Maessen, D.E.M.; Stehouwer, C.D.A.; Schalkwijk, C.G. The Role of Methylglyoxal and the Glyoxalase System in Diabetes and Other Age-Related Diseases. Clin. Sci. 2015, 128, 839–861. [Google Scholar] [CrossRef]
- Ramachandra Bhat, L.; Vedantham, S.; Krishnan, U.M.; Rayappan, J.B.B. Methylglyoxal—An Emerging Biomarker for Diabetes Mellitus Diagnosis and Its Detection Methods. Biosens. Bioelectron. 2019, 133, 107–124. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Micali, L.R.; Wouters, K. Advanced Glycation Endproducts in Diabetes-Related Macrovascular Complications: Focus on Methylglyoxal. Trends Endocrinol. Metab. 2023, 34, 49–60. [Google Scholar] [CrossRef]
- Lai, S.W.T.; Lopez Gonzalez, E.D.J.; Zoukari, T.; Ki, P.; Shuck, S.C. Methylglyoxal and Its Adducts: Induction, Repair, and Association with Disease. Chem. Res. Toxicol. 2022, 35, 1720–1746. [Google Scholar] [CrossRef]
- Bellier, J.; Nokin, M.-J.; Lardé, E.; Karoyan, P.; Peulen, O.; Castronovo, V.; Bellahcène, A. Methylglyoxal, a Potent Inducer of AGEs, Connects between Diabetes and Cancer. Diabetes Res. Clin. Pract. 2019, 148, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Kuhla, B.; Lüth, H.; Haferburg, D.; Boeck, K.; Arendt, T.; Münch, G. Methylglyoxal, Glyoxal, and Their Detoxification in Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2005, 1043, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Zambonin, L.; Hrelia, S. Role of Methylglyoxal in Alzheimer’s Disease. BioMed. Res. Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R. Diabetes as a Risk Factor for Alzheimer’s Disease in the Middle East and Its Shared Pathological Mediators. Saudi J. Biol. Sci. 2020, 27, 736–750. [Google Scholar] [CrossRef]
- Hipkiss, A.R. On the Relationship between Energy Metabolism, Proteostasis, Aging and Parkinson’s Disease: Possible Causative Role of Methylglyoxal and Alleviative Potential of Carnosine. Aging Dis. 2017, 8, 334. [Google Scholar] [CrossRef]
- Distler, M.G.; Gorfinkle, N.; Papale, L.A.; Wuenschell, G.E.; Termini, J.; Escayg, A.; Winawer, M.R.; Palmer, A.A. Glyoxalase 1 and Its Substrate Methylglyoxal Are Novel Regulators of Seizure Susceptibility. Epilepsia 2013, 54, 649–657. [Google Scholar] [CrossRef]
- Fang, L.; Li, X.; Zhong, Y.; Yu, J.; Yu, L.; Dai, H.; Yan, M. Autophagy Protects Human Brain Microvascular Endothelial Cells against Methylglyoxal-induced Injuries, Reproducible in a Cerebral Ischemic Model in Diabetic Rats. J. Neurochem. 2015, 135, 431–440. [Google Scholar] [CrossRef]
- Li, W.; Chen, Z.; Yan, M.; He, P.; Chen, Z.; Dai, H. The Protective Role of Isorhamnetin on Human Brain Microvascular Endothelial Cells from Cytotoxicity Induced by Methylglyoxal and Oxygen–Glucose Deprivation. J. Neurochem. 2016, 136, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Fabene, P.F.; Mora, G.N.; Martinello, M.; Rossi, B.; Merigo, F.; Ottoboni, L.; Bach, S.; Angiari, S.; Benati, D.; Chakir, A.; et al. A Role for Leukocyte-Endothelial Adhesion Mechanisms in Epilepsy. Nat. Med. 2008, 14, 1377–1383. [Google Scholar] [CrossRef]
- Bertini, G.; Bramanti, P.; Constantin, G.; Pellitteri, M.; Radu, B.M.; Radu, M.; Fabene, P.F. New Players in the Neurovascular Unit: Insights from Experimental and Clinical Epilepsy. Neurochem. Int. 2013, 63, 652–659. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–Brain Barrier Breakdown in Alzheimer Disease and Other Neurodegenerative Disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Karakatsani, M.E.; Blesa, J.; Konofagou, E.E. Blood–Brain Barrier Opening with Focused Ultrasound in Experimental Models of Parkinson’s Disease. Mov. Disord. 2019, 34, 1252–1261. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, W.; Lu, H.; Cai, S. Methylglyoxal in the Brain: From Glycolytic Metabolite to Signalling Molecule. Molecules 2022, 27, 7905. [Google Scholar] [CrossRef]
- Itoh, Y.; Esaki, T.; Shimoji, K.; Cook, M.; Law, M.J.; Kaufman, E.; Sokoloff, L. Dichloroacetate Effects on Glucose and Lactate Oxidation by Neurons and Astroglia in Vitro and on Glucose Utilization by Brain in Vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4879–4884. [Google Scholar] [CrossRef]
- Salmina, A.B.; Kuvacheva, N.V.; Morgun, A.V.; Komleva, Y.K.; Pozhilenkova, E.A.; Lopatina, O.L.; Gorina, Y.V.; Taranushenko, T.E.; Petrova, L.L. Glycolysis-Mediated Control of Blood-Brain Barrier Development and Function. Int. J. Biochem. Cell Biol. 2015, 64, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; De Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef] [PubMed]
- Tóth, A.E.; Walter, F.R.; Bocsik, A.; Sántha, P.; Veszelka, S.; Nagy, L.; Puskás, L.G.; Couraud, P.-O.; Takata, F.; Dohgu, S.; et al. Edaravone Protects against Methylglyoxal-Induced Barrier Damage in Human Brain Endothelial Cells. PLoS ONE 2014, 9, e100152. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, K.-A.; Kim, J.-H.; Kim, E.-H.; Bae, O.-N. Methylglyoxal-Induced Dysfunction in Brain Endothelial Cells via the Suppression of Akt/HIF-1α Pathway and Activation of Mitophagy Associated with Increased Reactive Oxygen Species. Antioxidants 2020, 9, 820. [Google Scholar] [CrossRef]
- Kim, J.; Kim, C.-S.; Lee, Y.M.; Jo, K.; Shin, S.D.; Kim, J.S. Methylglyoxal Induces Hyperpermeability of the Blood–Retinal Barrier via the Loss of Tight Junction Proteins and the Activation of Matrix Metalloproteinases. Graefes Arch. Clin. Exp. Ophthalmol. 2012, 250, 691–697. [Google Scholar] [CrossRef]
- Li, W.; Maloney, R.E.; Circu, M.L.; Alexander, J.S.; Aw, T.Y. Acute Carbonyl Stress Induces Occludin Glycation and Brain Microvascular Endothelial Barrier Dysfunction: Role for Glutathione-Dependent Metabolism of Methylglyoxal. Free Radic. Biol. Med. 2013, 54, 51–61. [Google Scholar] [CrossRef]
- Hussain, M.; Bork, K.; Gnanapragassam, V.S.; Bennmann, D.; Jacobs, K.; Navarette-Santos, A.; Hofmann, B.; Simm, A.; Danker, K.; Horstkorte, R. Novel Insights in the Dysfunction of Human Blood-Brain Barrier after Glycation. Mech. Ageing Dev. 2016, 155, 48–54. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.M.; Coleman, G.; Bhatwadekar, A.; Gardiner, T.A.; Stitt, A.W. Advanced Glycation of the Arg-Gly-Asp (RGD) Tripeptide Motif Modulates Retinal Microvascular Endothelial Cell Dysfunction. Mol. Vis. 2009, 15, 1509–1520. [Google Scholar]
- Radu, B.M.; Dumitrescu, D.I.; Mustaciosu, C.C.; Radu, M. Dual Effect of Methylglyoxal on the Intracellular Ca2+ Signaling and Neurite Outgrowth in Mouse Sensory Neurons. Cell Mol. Neurobiol. 2012, 32, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, A.A.; Dmitrenko, V.V. Pitfalls of the MTT Assay: Direct and off-Target Effects of Inhibitors Can Result in over/Underestimation of Cell Viability. Gene 2015, 574, 193–203. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am. J. Physiol.-Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Radu, B.M.; Radu, M.; Tognoli, C.; Benati, D.; Merigo, F.; Assfalg, M.; Solani, E.; Stranieri, C.; Ceccon, A.; Fratta Pasini, A.M.; et al. Are They in or out? The Elusive Interaction between Qtracker® 800 Vascular Labels and Brain Endothelial Cells. Nanomedicine 2015, 10, 3329–3342. [Google Scholar] [CrossRef]
- Ohkawara, S.; Tanaka-Kagawa, T.; Furukawa, Y.; Jinno, H. Methylglyoxal Activates the Human Transient Receptor Potential Ankyrin 1 Channel. J. Toxicol. Sci. 2012, 37, 831–835. [Google Scholar] [CrossRef]
- Lichtenstein, N.; Geiger, B.; Kam, Z. Quantitative Analysis of Cytoskeletal Organization by Digital Fluorescent Microscopy. Cytom. Part A J. Int. Soc. Anal. Cytol. 2003, 54, 8–18. [Google Scholar] [CrossRef]
- Shasby, D.M.; Shasby, S.S.; Sullivan, J.M.; Peach, M.J. Role of Endothelial Cell Cytoskeleton in Control of Endothelial Permeability. Circ. Res. 1982, 51, 657–661. [Google Scholar] [CrossRef]
- Mardilovich, K.; Baugh, M.; Crighton, D.; Kowalczyk, D.; Gabrielsen, M.; Munro, J.; Croft, D.R.; Lourenco, F.; James, D.; Kalna, G.; et al. LIM Kinase Inhibitors Disrupt Mitotic Microtubule Organization and Impair Tumor Cell Proliferation. Oncotarget 2015, 6, 38469–38486. [Google Scholar] [CrossRef]
- Shemiakova, T.; Ivanova, E.; Grechko, A.V.; Gerasimova, E.V.; Sobenin, I.A.; Orekhov, A.N. Mitochondrial Dysfunction and DNA Damage in the Context of Pathogenesis of Atherosclerosis. Biomedicines 2020, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Parveen, A.; Do, M.H.; Kang, M.C.; Yumnam, S.; Kim, S.Y. Molecular Mechanisms of Methylglyoxal-Induced Aortic Endothelial Dysfunction in Human Vascular Endothelial Cells. Cell Death Dis. 2020, 11, 403. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Wang, R.; Olson, D.J.H.; Mousseau, D.D.; Ross, A.R.S.; Wu, L. Modification of Akt1 by Methylglyoxal Promotes the Proliferation of Vascular Smooth Muscle Cells. FASEB J. 2011, 25, 1746–1757. [Google Scholar] [CrossRef]
- Paone, S.; Baxter, A.A.; Hulett, M.D.; Poon, I.K.H. Endothelial Cell Apoptosis and the Role of Endothelial Cell-Derived Extracellular Vesicles in the Progression of Atherosclerosis. Cell. Mol. Life Sci. 2019, 76, 1093–1106. [Google Scholar] [CrossRef]
- Dhar, A.; Dhar, I.; Desai, K.M.; Wu, L. Methylglyoxal Scavengers Attenuate Endothelial Dysfunction Induced by Methylglyoxal and High Concentrations of Glucose. Br. J. Pharmacol. 2010, 161, 1843–1856. [Google Scholar] [CrossRef] [PubMed]
- Alomar, F.; Singh, J.; Jang, H.S.; Rozanzki, G.J.; Shao, C.H.; Padanilam, B.J.; Mayhan, W.G.; Bidasee, K.R. Smooth Muscle-Generated Methylglyoxal Impairs Endothelial Cell-Mediated Vasodilatation of Cerebral Microvessels in Type 1 Diabetic Rats. Br. J. Pharmacol. 2016, 173, 3307–3326. [Google Scholar] [CrossRef]
- Avdonin, P.V.; Nadeev, A.D.; Mironova, G.Y.; Zharkikh, I.L.; Avdonin, P.P.; Goncharov, N.V. Enhancement by Hydrogen Peroxide of Calcium Signals in Endothelial Cells Induced by 5-HT1B and 5-HT2B Receptor Agonists. Oxidative Med. Cell. Longev. 2019, 1, 1701478. [Google Scholar] [CrossRef]
- Suresh, K.; Servinsky, L.; Reyes, J.; Baksh, S.; Undem, C.; Caterina, M.; Pearse, D.B.; Shimoda, L.A. Hydrogen Peroxide-Induced Calcium Influx in Lung Microvascular Endothelial Cells Involves TRPV4. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, 1467–1477. [Google Scholar] [CrossRef]
- Quintero, M.; Colombo, S.L.; Godfrey, A.; Moncada, S. Mitochondria as Signaling Organelles in the Vascular Endothelium. Proc. Natl. Acad. Sci. USA 2006, 103, 5379–5384. [Google Scholar] [CrossRef]
- Atawia, R.T.; Batori, R.K.; Jordan, C.R.; Kennard, S.; Antonova, G.; Bruder-Nascimento, T.; Mehta, V.; Saeed, M.I.; Patel, V.S.; Fukai, T.; et al. Type 1 Diabetes Impairs Endothelium-Dependent Relaxation Via Increasing Endothelial Cell Glycolysis Through Advanced Glycation End Products, PFKFB3 and Nox-1-Mediated Mechanisms. Hypertension 2023, 80, 10. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, Y.X.; Bu, W.J.; Li, P.; Chen, J.H.; Cao, M.; Dong, Y.C.; Sun, Z.J.; Dong, D.L. Piezo1 Channel Activation Stimulates ATP Production Through Enhancing Mitochondrial Respiration and Glycolysis in Vascular Endothelial Cells. Br. J. Pharmacol. 2023, 180, 1862–1877. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Endothelial Ca2+ Signaling, Angiogenesis and Vasculogenesis: Just What It Takes to Make a Blood Vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef] [PubMed]
- Thaning, P.; Bune, L.T.; Hellsten, Y.; Pilegaard, H.; Saltin, B.; Rosenmeier, J.B. Attenuated Purinergic Receptor Function in Patients with Type 2 Diabetes. Diabetes 2010, 59, 182–189. [Google Scholar] [CrossRef]
- Bierhaus, A.; Fleming, T.; Stoyanov, S.; Leffler, A.; Babes, A.; Neacsu, C.; Sauer, S.K.; Eberhardt, M.; Schnölzer, M.; Lasitschka, F.; et al. Methylglyoxal Modification of Nav1.8 Facilitates Nociceptive Neuron Firing and Causes Hyperalgesia in Diabetic Neuropathy. Nat. Med. 2012, 18, 926–933. [Google Scholar] [CrossRef]
- Ciobanu, A.C.; Selescu, T.; Gasler, I.; Soltuzu, L.; Babes, A. Glycolytic Metabolite Methylglyoxal Inhibits Cold and Menthol Activation of the Transient Receptor Potential Melastatin Type 8 Channel. J. Neurosci. Res. 2016, 94, 282–294. [Google Scholar] [CrossRef]
- Chan, C.M.; Huang, D.Y.; Huang, Y.P.; Hsu, S.H.; Kang, L.Y.; Shen, C.M.; Lin, W.W. Methylglyoxal Induces Cell Death Through Endoplasmic Reticulum Stress-Associated ROS Production and Mitochondrial Dysfunction. J. Cell. Mol. Med. 2016, 20, 1749–1760. [Google Scholar] [CrossRef]
- Wu, K.; Cheng, K.; Wong, K.; Shiao, L.; Leung, Y.; Chang, L. ARC 118925XX Stimulates Cation Influx in bEND.3 Endothelial Cells. Fundam. Clin. Pharma 2019, 33, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Nunes, P.; Demaurex, N. Redox Regulation of Store-Operated Ca2+ Entry. Antioxid. Redox Signal. 2014, 21, 915–932. [Google Scholar] [CrossRef]
- Rengifo, J.; Gibson, C.J.; Winkler, E.; Collin, T.; Ehrlich, B.E. Regulation of the Inositol 1,4,5- Trisphosphate Receptor Type 1 by O-GlcNAc Glycosylation. J. Neurosci. 2007, 27, 13813–13821. [Google Scholar] [CrossRef]
- Boeynaems, J.M.; Pearson, J.D. P2 Purinoceptors on Vascular Endothelial Cells: Physiological Significance and Transduction Mechanisms. Trends Pharmacol. Sci. 1990, 11, 34–37. [Google Scholar] [CrossRef]
- Motte, S.; Pirotton, S.; Boeynaems, J.M. Heterogeneity of ATP Receptors in Aortic Endothelial Cells. Circ. Res. 1993, 72, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, M.; Pontecorvi, P. Microenvironment Promotes Cytoskeleton Remodeling and Adaptive Phenotypic transition. Biocell 2022, 46, 1357–1362. [Google Scholar] [CrossRef]
- Izadi, M.; Hou, W.; Qualmann, B.; Kessels, M.M. Direct Effects of Ca2+/ Calmodulin on Actin Filament Formation. Biochem. Biophys. Res. Commun. 2018, 506, 355–360. [Google Scholar] [CrossRef]
- Bravo-Cordero, J.J.; Magalhaes, M.A.O.; Eddy, R.J.; Hodgson, L.; Condeelis, J. Functions of Cofilin in Cell Locomotion and Invasion. Nat. Rev. Mol. Cell Biol. 2013, 14, 405–415. [Google Scholar] [CrossRef]
- Xu, Q.; Huff, L.P.; Fujii, M.; Griendling, K.K. Redox Regulation of the Actin Cytoskeleton and its Role in the Vascular System. Free Radic. Biol. Med. 2018, 109, 84–107. [Google Scholar] [CrossRef] [PubMed]
- Huot, J.; Houle, F.; Spitz, D.R.; Landry, J. HSP27 Phosphorylation-Mediated resistance Against Actin Fragmentation and Cell Death Induced by Oxidative Stress. Cancer Res. 1996, 56, 273–279. [Google Scholar]
- Janmey, P.A.; Hvidt, S.; Oster, G.F.; Lamb, J.; Stossel, T.P.; Hartwig, J.H. Effect of ATP on Actin Filament Stiffness. Nature 1990, 347, 95–99. [Google Scholar] [CrossRef]
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin Dynamics, Architecture, and Mechanics in Cell Motility. Physiol. Rev. 2014, 94, 235–263. [Google Scholar] [CrossRef]
- Zanotelli, M.R.; Goldblatt, Z.E.; Miller, J.P.; Bordeleau, F.; Li, J.; VanderBurgh, J.A.; Lampi, M.C.; King, M.R.; Reinhart-King, C.A. Regulation of ATP Utilization during Metastatic Cell Migration by Collagen Architecture. MBoC 2018, 29, 1–9. [Google Scholar] [CrossRef]
- Hu, T.Y.; Liu, C.-L.; Chen, J.-Y.; Hu, M.-L. Curcumin Ameliorates Methylglyoxal-Induced Alterations of Cellular Morphology and Hyperpermeability in Human Umbilical Vein Endothelial Cells. J. Funct. Foods 2013, 5, 745–754. [Google Scholar] [CrossRef]
- Su, Y.; Lei, X.; Wu, L.; Liu, L. The Role of Endothelial Cell Adhesion Molecules P-Selectin, E-Selectin and Intercellular Adhesion Molecule-1 in Leucocyte Recruitment Induced by Exogeneous Methylglyoxal. Immunology 2012, 137, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Qadri, S.M.; Hossain, M.; Wu, L.; Liu, L. Uncoupling of eNos Contributes to Redox-Sensitive Leukocyte Recruitment and Microvascular Leakage Elicited by Methylglyoxal. Biochem. Pharmacol. 2013, 86, 1762–1774. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Qadri, S.M.; Cayabyab, F.S.; Wu, L.; Liu, L. Regulation of Methylglyoxal-Elicited Leukocyte Recruitment by Endothelial SGK1/GSK3 Signaling. Biochem. Biophys. Acta-Mol. Cell Res. 2014, 1843, 2481–2491. [Google Scholar] [CrossRef]
- Rom, S.; Heldt, N.A.; Gajghate, S.; Seliga, A.; Reichenbach, N.L.; Persidsky, Y. Hyperglycemia and Advanced Glycation End Products Disrupt BBB and Promote Occludin and Claudin-5 Protein Secretion on Extracellular Microvesicles. Sci. Rep. 2020, 10, 7274. [Google Scholar] [CrossRef]
- Chen, P.C.; Chang, Y.C.; Tsai, K.L.; Shen, C.H.; Lee, S.D. Vitexin Suppresses High-Glucose-Upregulated Adhesion Molecule Expression in Endothelial Cells through Inhibiting NF-kB Signaling Pathway. Am. Chem. Soc. 2024, 9, 32727–32734. [Google Scholar] [CrossRef]
- Fleming, T.; von Nettelbladt, B.; Morgenstern, J.; Campos, M.; Le Marois, M.; Bartosova, M.; Hausser, I.; Schwab, C.; Fischer, A.; Nawroth, P.P.; et al. Methylglyoxal Induces Endothelial Dysfunction via Stunning-like Phenotype. bioRxiv 2021. [Google Scholar] [CrossRef]
- Radu, B.M.; Osculati, A.N.M.; Suku, E.; Banciu, A.; Tsenov, G.; Merigo, F.; Di Chio, M.; Banciu, D.D.; Tognoli, C.; Kacer, P.; et al. All Muscarinic Acetylcholine Receptors (M1-M5) are Expressed in Murine Brain Microvascular Endothelium. Sci. Rep. 2017, 7, 1. [Google Scholar] [CrossRef]
- Versele, R.; Sevin, E.; Gosselet, F.; Fenart, L.; Candela, P. TNF-α and IL-1β Modulate Blood-Brain Barrier Permeability and Decrease Amyloid-β Peptide Efflux in a Human Blood-Brain Barrier Model. Int. J. Mol. Sci. 2022, 23, 10235. [Google Scholar] [CrossRef]
- Reyes-Aldasoro, C.C.; Wilson, I.; Prise, V.E.; Barber, P.R.; Ameer-Beg, M.; Vojnovic, B.; Cunningham, V.J.; Tozer, G.M. Estimation of Apparent Tumor Vascular Permeability from Multiphoton Fluorescence Microscopic Images of P22 Rat Sarcomas In Vivo. Microcirculation 2008, 15, 65–79. [Google Scholar] [CrossRef]
- Grada, A.; Otero Vinas, M.; Prieto Castrillo, F.; Obagi, Z.; Falanga, V. Research Techniques Made Simple: Analysis of Collective Cell Migration Using the Wound Healing Assay. J. Investig. Dermatol. 2017, 137, e11–e16. [Google Scholar] [CrossRef]
- Rusu, C.M.; Bacalum, M.; Deftu, A.T.; Radu, B.M.; Radu, M. Digital Analysis of Actin Filaments Images Obtained by Fluorescence Microscopy. Rom. J. Biophys. 2018, 4, 28. [Google Scholar]
- Schober, P.; Boer, C.; Schwarte, L.A. Correlation Coefficients: Appropriate Use and Interpretation. Anesth. Analg. 2018, 126, 1763–1768. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Value |
|---|---|
| Kernel matrix size (L) | 6 |
| Angular resolution (K) | 10 |
| Threshold correlation coefficient (TC) | 0.55 |
| Threshold for the normalized standard deviation (M) | 0.3 |
| Threshold for the ratio of the normalized standard deviations (N) | 2.1 |
| Threshold pixel intensity (T) | 0.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moisă, R.; Rusu, C.M.; Deftu, A.T.; Bacalum, M.; Radu, M.; Radu, B.M. Are You a Friend or an Enemy? The Dual Action of Methylglyoxal on Brain Microvascular Endothelial Cells. Int. J. Mol. Sci. 2025, 26, 5104. https://doi.org/10.3390/ijms26115104
Moisă R, Rusu CM, Deftu AT, Bacalum M, Radu M, Radu BM. Are You a Friend or an Enemy? The Dual Action of Methylglyoxal on Brain Microvascular Endothelial Cells. International Journal of Molecular Sciences. 2025; 26(11):5104. https://doi.org/10.3390/ijms26115104
Chicago/Turabian StyleMoisă (Stoica), Roberta, Călin Mircea Rusu, Antonia Teona Deftu, Mihaela Bacalum, Mihai Radu, and Beatrice Mihaela Radu. 2025. "Are You a Friend or an Enemy? The Dual Action of Methylglyoxal on Brain Microvascular Endothelial Cells" International Journal of Molecular Sciences 26, no. 11: 5104. https://doi.org/10.3390/ijms26115104
APA StyleMoisă, R., Rusu, C. M., Deftu, A. T., Bacalum, M., Radu, M., & Radu, B. M. (2025). Are You a Friend or an Enemy? The Dual Action of Methylglyoxal on Brain Microvascular Endothelial Cells. International Journal of Molecular Sciences, 26(11), 5104. https://doi.org/10.3390/ijms26115104