1. Introduction
The ability to avoid the immune response is considered a hallmark of cancer and is recognized as a necessary step for tumor progression [
1,
2]. Cancer cells evade immune detection by a variety of methods, including the downregulation of MHC I, the upregulation of PD-L1, and the release of regulatory cytokines, which lead to the suppression of immune cells in the tumor microenvironment [
3]. The goal of cancer immunotherapy is to augment or reactivate the patient’s immune response and to enhance the ability of the immune system to recognize tumor antigens and eliminate cancer cells [
4].
The adaptive immune response is primarily carried out by cytotoxic T lymphocytes (CTLs) and B cells. Both are antigen-specific effector cells that require antigen presentation and activation by antigen-presenting cells (APCs), such as macrophages and dendritic cells (DCs) [
5]. APCs bridge the innate immune and adaptive immune responses by processing available antigens from their environment and displaying them to T cells and B cells, along with co-stimulatory factors [
5,
6]. These antigens are often obtained through the coating of pathogens with complement and subsequent internalization into APCs mediated by complement receptor 3. APCs process the pathogen and display antigens to effector cells through MHC I and MHC II, while danger-associated molecular patterns from the pathogen lead to APC activation. APCs are often immunosuppressed in cancer patients due to the lower expression of MHC II, CD80/86, and mTORC1 and the increased production of immunosuppressive cytokines such as IL-6 and IL-10 [
7]. APC suppression plays a central role in the inhibition of the adaptive immune response and in the reduced efficacy of applied immunotherapies [
8]. To overcome this suppression, there exists a need for treatments that target immunosuppressed APCs for reactivation while simultaneously delivering antigen.
Our laboratory has developed a liposome-based delivery system that utilizes part of the complement cascade to target APCs and simultaneously deliver antigen and adjuvants [
9]. The liposomes contain a lipid with a linker group, orthopyridyl disulfide (OPSS), on the exterior of the liposomes, where it forms covalent disulfide bonds with other molecules under physiological conditions. In serum, the OPSS linker groups readily bind to complement protein C3 and mediate the delivery of the liposomes to immune cells that express C3b receptors. This set of immune cells includes all three APC subtypes: B cells, macrophages, and DCs [
10]. Prior research in our laboratory using model tumor antigen ovalbumin (OVA) has shown the ability of this system to target APCs, elicit a significant increase in activated antigen-specific T cells, and suppress growth in established A20-OVA tumors [
11]. One drawback of using OVA peptide as a mock antigen is that it is derived from a different species than the mice being vaccinated and can result in robust immune responses that may not necessarily be replicated when tumor antigens are substituted into the system. However, it is a well-characterized antigen and could be a good indicator of how our liposomal delivery system would perform if used to deliver foreign peptides from viruses and bacteria.
Previous iterations of the C3-liposome system utilized a passive method for encapsulating hydrophilic antigens during extrusion [
11,
12,
13]. This process could be challenging to scale up to pharmaceutically relevant quantities, given the costs associated with the low loading efficiency of the antigen and the methods required for separating liposomes from unencapsulated antigens. To improve upon our method, we have explored the possibility of conjugating peptides directly to the exterior of the liposomes through the same OPSS groups used to bind complement C3 after the liposomes have been formed. To evaluate the efficacy of this system, liposomes were covalently linked to an ovalbumin-derived peptide with an additional four amino acids (-GGGC) at the C-terminus (OVA-C), a strategy that has been successful for attaching peptides to nanoparticles using SMPH as an intermediary [
14]. At physiological pH, the OPSS groups bind to the available cysteine residue on the OVA-C peptides, conjugating OVA-C to the exterior of the pre-formed liposomes and leaving sufficient OPSS groups available for the subsequent binding of endogenous complement C3. Following vaccination with OVA-C liposomes, mice were evaluated for humoral and cellular immune responses. We demonstrate that targeted antigen delivery to APCs utilizing OVA-C liposomes enhances the adaptive T cell and antibody response in mice. Our study suggests this peptide + C3-liposome conjugation method may allow for an efficient delivery system that could be utilized in personalized cancer therapies.
3. Discussion
The results of this study demonstrate the ability of C3-liposomes to bind antigens to external OPSS groups, to target APCs via an endogenous complement-dependent mechanism, and to elicit a robust adaptive immune response. An HPLC analysis confirmed the efficient binding and retention of OVA-C peptide on the liposome surface. Importantly, this covalent binding competed with, but did not completely block, subsequent complement C3 binding. This result indicates that OPSS functional groups on C3-liposomes can support the disulfide conjugation of both the antigen and complement, leaving the interior of the liposome free for the delivery of any drug or package (i.e., protein or nucleic acid) of interest. Furthermore, these experiments demonstrate the flexibility of the system for the conjugation of hydrophilic molecules bearing a sulfhydryl group to the exterior of OPSS-liposomes while still retaining targeted uptake by APCs.
In vivo biodistribution experiments demonstrated that the C3-mediated targeting mechanism remains active in both the blood and spleen despite partial displacement of the complement corona by the antigen peptide. Interestingly, uptake into splenic myeloid cells differed from previous findings [
9], in that control liposomes lacking OPSS exhibited higher-than-expected uptake. Several factors could account for this discrepancy. In these reported experiments, liposomes were only incubated for 1 h, compared to 4 h in prior experiments. Extended incubation time could lead to more pronounced differences in uptake between C3-targeted and control liposomes, as previously reported. Additionally, changes in the lipid composition may have increased non-specific uptake through a complement-independent mechanism. Lipid composition is known to strongly influence liposome biodistribution [
15], suggesting that the further optimization of lipid formulation and time points is warranted to enhance uptake specificity.
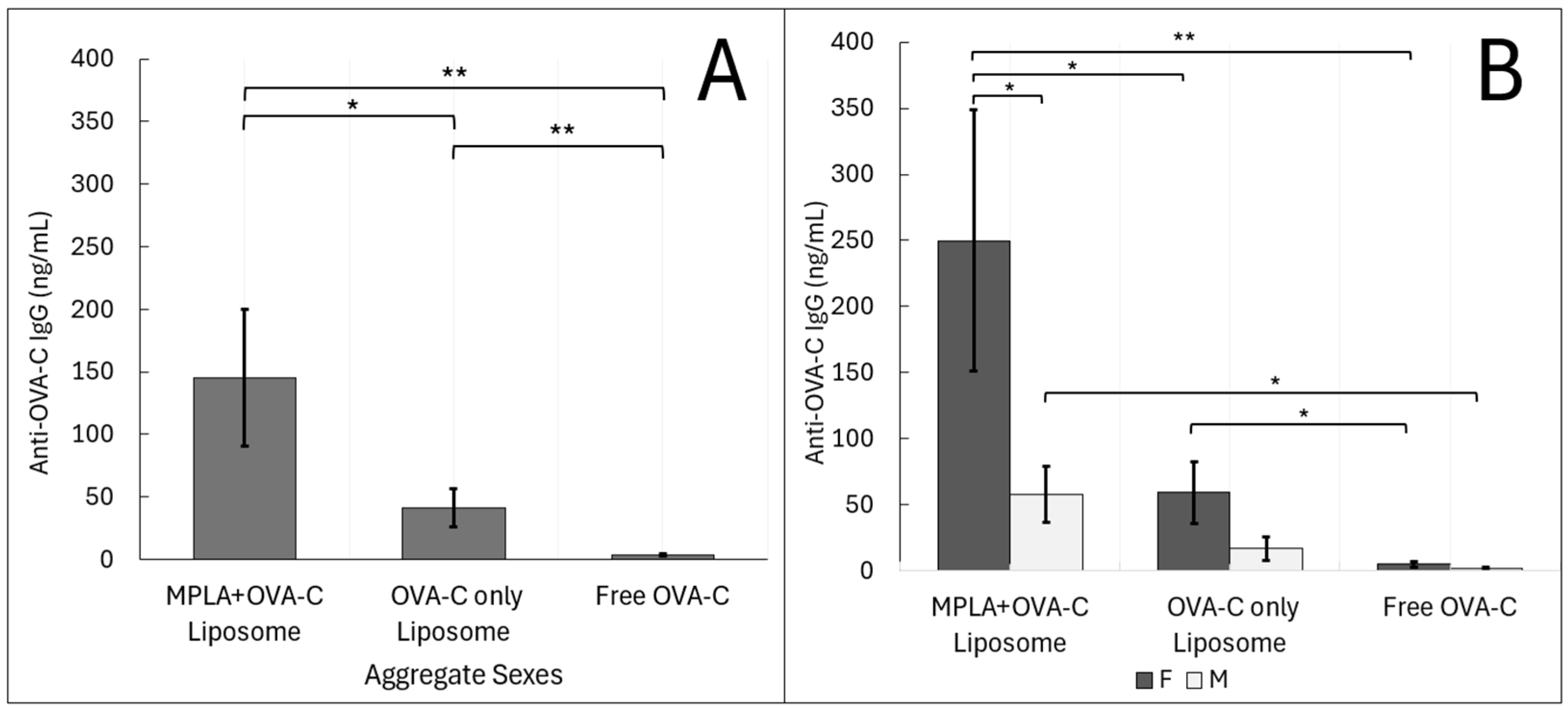
OVA-C-conjugated liposomes generated significantly stronger adaptive immune responses than free OVA-C peptide, which is poorly immunogenic. Both cellular and humoral responses were markedly enhanced in mice vaccinated with OVA-C liposomes. The inclusion of MPLA in the liposomes further enhanced the IgG antibody response, although it had a limited effect on T cell activation. MPLA was selected as an adjuvant due to its hydrophobic nature, allowing for incorporation into liposomal membranes, and for its known ability to activate TLR4 signaling pathways. TLR4 activation plays a crucial role in APC maturation, antigen presentation, and T cell differentiation and maturation [
16]. While MPLA effectively boosted humoral responses, alternative adjuvants targeting TLR9 (e.g., CpG) or TLR3 (e.g., poly I:C) may differentially modulate the adaptive response, potentially enhancing cellular immunity through distinct signaling cascades.
Sex-based differences in immune responses to vaccination were observed, with female mice having stronger adaptive responses overall. Statistically significant differences were observed only in the humoral responses of female mice vaccinated with MPLA + OVA-C liposomes compared to their male counterparts. Among males, the addition of MPLA did not significantly alter antibody concentrations. Interestingly, while the addition of MPLA in the vaccination of female mice increased antibody concentrations, it also increased variability between mice. In females, MPLA more than doubled the standard error in both ELISpot and ELSA results, suggesting heterogeneous responses within this group. In contrast, MPLA reduced variability in T cell responses among male mice. This MPLA-associated increase in immune variability among females may stem from sex-specific differences in TLR4 signaling and hormone regulation. Other studies have shown mixed data on TLR4 expression and antigen presentation in macrophages between male and female mice, with some data suggesting a consistently greater expression of TLR4 in female-derived macrophages [
17] and some studies suggesting no difference, or even greater sensitivity to LPS in male-derived macrophages [
18]. Additional data suggest that when a TLR4 agonist is used in combination with TLR7/8 and TLR9 agonists, sex differences in IgG responses to a vaccine may be reduced compared to vaccination with a TLR7/8 agonist alone [
12]. A recent review by Popotas [
19] showed that estrogen and progesterone levels can modulate TLR4 expression throughout the estrous cycle. Indeed, the activation of estrogen receptor alpha has been demonstrated to inhibit TLR4 signaling [
20] in murine macrophages, and estrogen receptor elements have been found within the promoter for murine TLR4 [
21], inhibiting the expression of TLR4. Given that the female mice in our experiments were collected in multiple, separate cohorts, without the synchronization of estrus stages, hormonal fluctuations may partially explain the increased variance observed in female immune responses. Further study of sex differences in TLR agonist signaling is needed to corroborate these findings and examine the increased sensitivity to MPLA in female mice. The myeloid-targeting ability of our C3-liposomal system may be a useful tool for future in vivo experiments.
Some of the manufacturing challenges of nanoparticle-based vaccines include the stability of the nanoparticle formulation, the cost of peptides and proteins, and the difficulty of purifying the nanoparticles from unconjugated components when scaling up to pharmaceutically relevant quantities. Our present system addresses these difficulties first by greatly increasing the conjugation efficiency from 5% with passive encapsulation to 84.6% with the strategy of disulfide-binding peptide to the exterior of the liposomes [
12]. This level of conjugation will greatly decrease the quantity of peptide needed and could avoid a purification step, as the vast majority of the peptide is bound to the liposomes. In addition, the liposome formulation showed impressive stability over 15 months with no visible aggregation, a modest increase in liposome size from 255 ± 98 nm to 368 ± 124 nm, and a retained ability to internalize in vitro, as demonstrated by uptake into bone marrow myeloid cells (
Figure S1).
In conclusion, this study demonstrates the effectiveness of a novel liposomal delivery platform targeting myeloid cells and eliciting both potent T cell and B cell responses in mice. The incorporation of MPLA adjuvant into the C3-liposomes significantly enhanced the humoral immune response but also revealed sex-based differences in immunogenicity, highlighting the importance of considering sex as a biological variable in future research. The modular design of our liposomal system allows for the flexible, targeted delivery of single or multiple antigens, making it a promising tool for vaccine development in cancer and other disease applications. Furthermore, the ability to rapidly synthesize and conjugate custom peptides to the exterior of our complement C3-targeted liposomes makes this delivery system potentially useful in future scenarios, such as for vaccines in emerging epidemics with rapidly evolving organisms or for personalized cancer immunotherapy targeting identified neoantigens in rapidly mutating tumors. Although the scope of this work did not allow for testing the immune response to tumor antigens, future studies will explore the liposome delivery system using tumor antigens such as TRP2 in B16F10 melanomas to treat mouse models of cancer.
4. Materials and Methods
4.1. Materials
1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[PDP-poly(ethylene glycol)-2000] (DSPE-PEG(2000)-PDP) for liposome preparation were purchased from Avanti Polar Lipids (Alabaster, AL, USA). As with previous publications, we refer to the PDP group as OPSS [
9]. Size exclusion chromatography was performed using CL4B Sepharose gel (Sigma-Aldrich, St. Louis, MO, USA). Fluorescently tagged lipid, lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (Rhodamine-PE), was purchased from Life Technologies (Grand Island, NY, USA). Monophosphoryl Lipid A (MPLA) was purchased from InvivoGen (San Diego, CA, USA). The peptide sequence ISQAVHAAHAEINEAGRGGGC was synthesized by Genscript (Piscataway, NJ, USA). CL4B Sepharose gel used in the size exclusion chromatography of liposomes was purchased from Sigma-Aldrich (St. Louis, MO, USA). Alexafluor700 CD11b antibody was purchased from BioLegend (San Diego, CA, USA). For the ELISA, 96-well flat-bottom Maxisorp plates, streptavidin, and SMPH (succinimidyl 6-((beta-maleimidopropionamido)hexanoate)) were ordered from Thermo Scientific (Pittsburgh, PA, USA). Goat anti-murine IgG-horseradish peroxidase (HRP), used as the secondary antibody, was ordered from Abcam (ab6789) (Waltham, MA, USA). Murine anti-OVA IgG, used as a standard, was ordered from Sigma-Aldrich (St. Louis, MO, USA). Mouse IFN-γ Single-Color ELISpot kits were purchased from ImmunoSpot (Cleveland, OH, USA). Mouse serum with functional complement was ordered from Innovative Research (Novi, MI, USA). All other chemicals and reagents were purchased from Thermo Fisher Scientific (Pittsburgh, PA, USA).
4.2. Liposome Preparation
Liposomes were prepared from lipid stocks suspended in chloroform. DSPC:DPPC:DSPE–rhodamine:MPLA:DSPE-PEG(2000)-PDP (DSPE-PEG(2000)-OPSS) were mixed at a molar ratio of 21:72:1:1:5 for MPLA+ liposomes and without MPLA for liposomes that did not contain the adjuvant. Control liposomes had a similar composition except with DSPE-PEG(2000) substituted for DSPE-PEG(2000)-OPSS. The lipids were mixed and dried at 47 °C under gaseous nitrogen for one hour and then further dried under vacuum at room temperature (RT) for two hours to remove any remaining chloroform. The lipid film was hydrated in 0.7 mL of water at 47 °C and extruded through a 400 nm polycarbonate membrane filter 9 times (Avestin, Ottawa, ON, Canada). After extrusion, liposomes were incubated overnight at RT with a custom peptide ISQAVHAAHAEINEAGRGGGC (OVA-C) synthesized by Genscript. In total, 25 μL of 1.2 mg/mL peptide in 1× PBS (pH 7.4) OVA-C was added to 200 μL of liposomes to obtain a final concentration of 133 μg/mL.
Liposome-bound antigen concentration was quantified using HPLC. Liposomes and free OVA-C vaccinations were normalized to a concentration of 35 μg/mL OVA-C. Liposome conjugation efficiency was calculated by the following equation:
For in vitro experiments, following the CL4B column, liposomes were incubated for 30 min at RT with 10% v/v C3+ C57BL/6 mouse serum. Following antigen and complement C3 incubation, the liposomes were column-purified using a CL4B Sepharose column hydrated in 1× PBS and collected in a 0.7 mL fraction. Resulting liposomes were analyzed by HPLC and normalized to 35 μg/mL OVA-C. In vivo experiments relied on endogenous complement.
4.3. HPLC Analysis
C3-liposomes with conjugated OVA-C were analyzed using a 1200 Infinity series liquid chromatography system with diode array detection controlled by MassHunter v. B.06.00 (Agilent, Santa Clara, CA, USA). First, 100 µL aliquots of liposomes were incubated with 1 µL 1M dithiothreitol for 30 min at room temperature. Then, 10 µL injections were run on a Zorbax 300SB-C8 column (100 × 2.1 mm, 3.5 µm) (Agilent, Santa Clara, CA, USA). The column temperature was maintained at 35 °C for the method. OVA-C was eluted using a gradient method with solvent A consisting of water and solvent B consisting of acetonitrile, both containing 0.1% trifluoroacetic acid, using a flow rate of 0.3 mL/min. The gradient increased the proportion of acetonitrile from 10% to 95% over a 10-minute period, followed by a 3-minute hold before returning to the starting conditions. OVA-C was eluted at 5.48 min, the primary complement C3 peak occurred at approximately 8.39 min, and the total method run time was 20 min. Detection of OVA-C was performed using UV absorbance at 220 nm with a reference wavelength of 240 nm, and the corresponding bandwidths were 4 nm and 20 nm.
4.4. Biodistribution
C57BL/6 mice were intravenously injected (lateral caudal vein) with 100 μL of either MPLA + OVA-C liposomes, OVA-C-only liposomes, or control liposomes (no OPSS or MPLA), each containing rhodamine. After one hour, mice were sacrificed, and blood and spleen were collected. Blood was collected with a heparinized syringe and incubated with Red Blood Cell lysis buffer at room temperature for 5 min before PBS was added to stop the lysis. The tube was centrifuged at 420 g for 5 min, the supernatant was discarded, and the remaining cells were resuspended in 10 mL of 1× PBS. Spleens were incubated in 1 mL of 1 mg/mL collagenase for 10 min at 37 °C, 5% CO2. The collagenase was deactivated by adding 10 mL of RPMI 1640 + 10% FBS + 1% penicillin–streptomycin. Spleen cells were then filtered through a 100 μm cell strainer, centrifuged at 420 g for 5 min, and resuspended in 10 mL of 1× PBS. Blood and spleen cells were imaged with a Leica DMI6000B inverted fluorescence microscope using a 20× objective (Leica Microsystems, Buffalo Grove, IL, USA). Photos were taken using Leica Application Suite, version 3.7.0 (Leica Microsystems Inc., Wetzlar, Germany).
4.5. Flow Cytometry
Flow cytometry was performed to determine which cells had internalized rhodamine-labeled liposomes. Cells were plated in a 96-well V-bottom polystyrene microplate and centrifuged for 5 min at 420 g at 4 °C in a Sorvall Legend X1R centrifuge. After centrifugation, the cells were resuspended in 100 μL of FACS buffer (1× PBS +1% bovine serum albumin) that contained antibodies against CD45 and CD11b. Cells were incubated with the antibodies for 10 min at 21 °C in the dark to allow for conjugation before pelleting by centrifugation, as described above. The cells were then resuspended in 120 μL of FACS buffer before analysis by flow cytometry, using a Beckman Coulter CytoFLEX flow cytometer with the CytExpert software version 2.0.0.153 (Beckman Coulter, Brea, CA, USA). Myeloid cells were identified as CD45+ and CD11b+, and the percentage of myeloid cells that had internalized liposomes was determined by gating for the rhodamine label. Data are presented as mean ± standard error (n = 6).
4.6. Vaccination
Six cohorts of C57BL/6 mice were injected subcutaneously with 100 μL of either free OVA-C, OVA-C liposomes, or MPLA + OVA-C liposomes at days 0 and 7 of the 14-day vaccination experiments. In total, 4 cohorts consisted of 3 male and 3 female mice, with 1 male and 1 female per vaccination group; 2 additional cohorts had 8 and 5 mice. The ELISpot plate was overdeveloped in the 5-mouse cohort and was unusable; however, the ELISA data from the same cohort were retained.
4.7. Tissue Preparation for ELISA and ELISPOT
Mice were sacrificed, and whole blood and spleens were harvested. Blood was centrifuged at 420 g for 7 min to collect serum. Spleens were treated as above in the biodistribution studies but were resuspended in 10 mL of RPMI 1640 + 10% FBS + 1% penicillin–streptomycin instead of PBS for ELISpot analysis.
4.8. ELISA
Wells in a 96-well flat-bottom plate (MaxiSorp Thermo Scientific) were incubated with 500 ng of streptavidin (Thermo Scientific) for 1 h at 21 °C to coat wells. After coating, 500 ng of SMPH (succinimidyl 6-((beta-maleimidopropionamido)hexanoate)) was added to the wells and incubated for 1 h at 21 °C. In total, 500 ng of OVA-C peptide (ISQAVHAAHAEINEAGRGGGC) was then added to the wells and incubated for two hours at 21 °C. Wells were blocked for 1 h with 100 μL of PBS + 0.5% milk at 21 °C, after which, primary antibody or serum from the experimental mice diluted to 1:40 was added to the wells and incubated at 4 °C overnight. Murine anti-OVA IgG (Sigma-Aldrich) was used to produce a standard curve, and a 3-parameter exponential curve was used to fit. One female MPLA + OVA-C sample with an optical density beyond the highest standard concentration of the standard curve was recorded as the highest standard concentration. Horseradish HRP-goat anti-murine IgG was used as a secondary antibody, diluted 1:4000 in 0.5% PBS milk. A volume of 100 μL of TMB was added as substrate for the enzyme and was halted by the addition of 1% HCl, and the plate absorbance was analyzed at 450 nm. This method was developed and kindly shared by Dr. Alex Francian [
14].
4.9. ELISpot
Spleen cells were plated in ImmunoSpot anti-murine IFN-γ strips at 300,000 cells per well in 200 μL of RPMI 1640 +10% FBS + 1% penicillin–streptomycin. The wells contained either media alone (negative control), 6 μg of OVA-C (antigen), or PMA (positive control). Cells were incubated for 24 h at 37 °C and 9% CO2 before the wells were developed according to ImmunoSpot’s protocol. Spots were counted using an ImunoSpot S6 Micro analyzer (ImmunoSpot, Shaker Heights, OH, USA). Quality control included negative controls to ascertain baseline IFN-y release of each sample, positive controls to determine cell and plate development viability, and duplicate wells at varying cell densities to determine appropriate paracrine stimulation. Negative control well counts were subtracted from the average of the duplicate antigen well counts.
4.10. Statistical Analysis
Statistical analyses were performed by JMP Pro 17 and R (version 4.4.2) [
22]. Normalcy was tested using the Shapiro–Wilk test. An unpaired two-tailed
t-test assuming unequal variance was used to compare normally distributed datasets, while a nonparametric comparison (Wilcoxon method) was used for nonparametric datasets. Alpha values of less than 0.05 were considered significant and are indicated by an asterisk, while alpha values of less than 0.01 are indicated by two asterisks.
Figure 2 was generated using
ggplot2 [
23],
ggpubr [
24], and
cowplot [
25].


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}