
Long-Read Sequencing Identifies Mosaic Sequence Variations in Friedreich’s Ataxia-GAA Repeats

 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
Abstract
1. Introduction
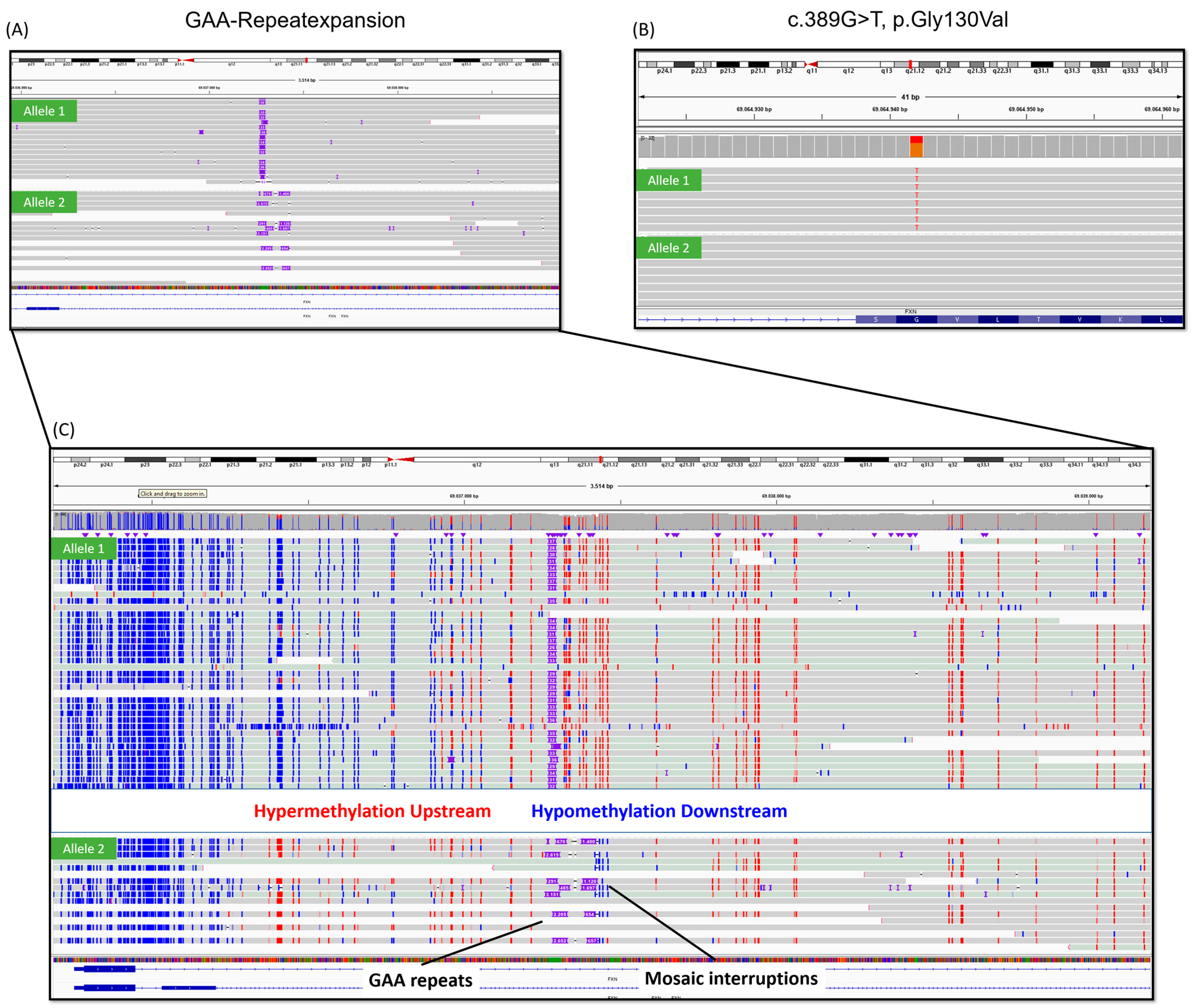
2. Results
2.1. Case Description
2.2. Genetic Analysis
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FRDA | Friedreich’s ataxia |
| GS | Genome sequencing |
| STR | Short tandem repeats |
| TR-PCR | Triple-repeat-primed PCR |
| LR-PCR | Long-range PCR |
References
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Durr, A.; Cossee, M.; Agid, Y.; Campuzano, V.; Mignard, C.; Penet, C.; Mandel, J.L.; Brice, A.; Koenig, M. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N. Engl. J. Med. 1996, 335, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Bidichandani, S.I.; Delatycki, M.B. Friedreich Ataxia. In GeneReviews((R)) 1993–2025; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2025. [Google Scholar]
- Mateo, I.; Llorca, J.; Volpini, V.; Corral, J.; Berciano, J.; Combarros, O. Expanded GAA repeats and clinical variation in Friedreich’s ataxia. Acta Neurol. Scand. 2004, 109, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Fleszar, Z.; Dufke, C.; Sturm, M.; Schule, R.; Schols, L.; Haack, T.B.; Synofzik, M. Short-read genome sequencing allows “en route” diagnosis of patients with atypical Friedreich ataxia. J. Neurol. 2023, 270, 4112–4117. [Google Scholar] [CrossRef]
- Harding, A.E. Friedreich’s ataxia: A clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 1981, 104, 589–620. [Google Scholar] [CrossRef]
- McDaniel, D.O.; Keats, B.; Vedanarayanan, V.V.; Subramony, S.H. Sequence variation in GAA repeat expansions may cause differential phenotype display in Friedreich’s ataxia. Mov. Disord. 2001, 16, 1153–1158. [Google Scholar] [CrossRef]
- Galea, C.A.; Huq, A.; Lockhart, P.J.; Tai, G.; Corben, L.A.; Yiu, E.M.; Gurrin, L.C.; Lynch, D.R.; Gelbard, S.; Durr, A.; et al. Compound heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann. Neurol. 2016, 79, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Candayan, A.; Yunisova, G.; Cakar, A.; Durmus, H.; Basak, A.N.; Parman, Y.; Battaloglu, E. The first biallelic missense mutation in the FXN gene in a consanguineous Turkish family with Charcot-Marie-Tooth-like phenotype. Neurogenetics 2020, 21, 73–78. [Google Scholar] [CrossRef]
- Evans-Galea, M.V.; Carrodus, N.; Rowley, S.M.; Corben, L.A.; Tai, G.; Saffery, R.; Galati, J.C.; Wong, N.C.; Craig, J.M.; Lynch, D.R.; et al. FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann. Neurol. 2012, 71, 487–497. [Google Scholar] [CrossRef]
- Long, A.; Napierala, J.S.; Polak, U.; Hauser, L.; Koeppen, A.H.; Lynch, D.R.; Napierala, M. Somatic instability of the expanded GAA repeats in Friedreich’s ataxia. PLoS ONE 2017, 12, e0189990. [Google Scholar] [CrossRef]
- Nethisinghe, S.; Kesavan, M.; Ging, H.; Labrum, R.; Polke, J.M.; Islam, S.; Garcia-Moreno, H.; Callaghan, M.F.; Cavalcanti, F.; Pook, M.A.; et al. Interruptions of the FXN GAA Repeat Tract Delay the Age at Onset of Friedreich’s Ataxia in a Location Dependent Manner. Int. J. Mol. Sci. 2021, 22, 7507. [Google Scholar] [CrossRef]
- Dolzhenko, E.; Deshpande, V.; Schlesinger, F.; Krusche, P.; Petrovski, R.; Chen, S.; Emig-Agius, D.; Gross, A.; Narzisi, G.; Bowman, B.; et al. ExpansionHunter: A sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 2019, 35, 4754–4756. [Google Scholar] [CrossRef] [PubMed]
- Al-Mahdawi, S.; Ging, H.; Bayot, A.; Cavalcanti, F.; La Cognata, V.; Cavallaro, S.; Giunti, P.; Pook, M.A. Large Interruptions of GAA Repeat Expansion Mutations in Friedreich Ataxia Are Very Rare. Front. Cell Neurosci. 2018, 12, 443. [Google Scholar] [CrossRef] [PubMed]
- Uppili, B.; Sharma, P.; Ahmad, I.; Sahni, S.; Asokachandran, V.; Nagaraja, A.B.; Srivastava, A.K.; Faruq, M. Sequencing through hyperexpanded Friedreich’s ataxia-GAA repeats by nanopore technology: Implications in genotype-phenotype correlation. Brain Commun. 2023, 5, fcad020. [Google Scholar] [CrossRef]
- Loomis, E.W.; Eid, J.S.; Peluso, P.; Yin, J.; Hickey, L.; Rank, D.; McCalmon, S.; Hagerman, R.J.; Tassone, F.; Hagerman, P.J. Sequencing the unsequenceable: Expanded CGG-repeat alleles of the fragile X gene. Genome Res. 2013, 23, 121–128. [Google Scholar] [CrossRef]
- Mangin, A.; de Pontual, L.; Tsai, Y.C.; Monteil, L.; Nizon, M.; Boisseau, P.; Mercier, S.; Ziegle, J.; Harting, J.; Heiner, C.; et al. Robust Detection of Somatic Mosaicism and Repeat Interruptions by Long-Read Targeted Sequencing in Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2021, 22, 2616. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.; Luth, T.; Schaake, S.; Laabs, B.H.; Schluter, K.; Labeta, J.; Pozojevic, J.; Tse, R.; Konig, I.; Jamora, R.D.; et al. Mosaic divergent repeat interruptions in XDP influence repeat stability and disease onset. Brain 2023, 146, 1075–1082. [Google Scholar] [CrossRef]
- Depienne, C.; Mandel, J.L. 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? Am. J. Hum. Genet. 2021, 108, 764–785. [Google Scholar] [CrossRef]
- Park, J.; Sturm, M.; Seibel-Kelemen, O.; Ossowski, S.; Haack, T.B. Lessons Learned from Translating Genome Sequencing to Clinical Routine: Understanding the Accuracy of a Diagnostic Pipeline. Genes 2024, 15, 136. [Google Scholar] [CrossRef]
- Figueroa, K.P.; Gross, C.; Buena-Atienza, E.; Paul, S.; Gandelman, M.; Kakar, N.; Sturm, M.; Casadei, N.; Admard, J.; Park, J.; et al. A GGC-repeat expansion in ZFHX3 encoding polyglycine causes spinocerebellar ataxia type 4 and impairs autophagy. Nat. Genet. 2024, 56, 1080–1089. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Chen, L.C.; Yu, S.C.; Huang, Y.T. LongPhase: An ultra-fast chromosome-scale phasing algorithm for small and large variants. Bioinformatics 2022, 38, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Li, S.; Su, J.; Leung, A.W.; Lam, T.W.; Luo, R. Symphonizing pileup and full-alignment for deep learning-based long-read variant calling. Nat. Comput. Sci. 2022, 2, 797–803. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.; Dufke, C.; Fleszar, Z.; Schlotterbek, M.; Buena-Atienza, E.; Stühn, L.G.; Gross, C.; Sturm, M.; Ossowski, S.; Schöls, L.; et al. Long-Read Sequencing Identifies Mosaic Sequence Variations in Friedreich’s Ataxia-GAA Repeats. Int. J. Mol. Sci. 2025, 26, 4969. https://doi.org/10.3390/ijms26114969
Park J, Dufke C, Fleszar Z, Schlotterbek M, Buena-Atienza E, Stühn LG, Gross C, Sturm M, Ossowski S, Schöls L, et al. Long-Read Sequencing Identifies Mosaic Sequence Variations in Friedreich’s Ataxia-GAA Repeats. International Journal of Molecular Sciences. 2025; 26(11):4969. https://doi.org/10.3390/ijms26114969
Chicago/Turabian StylePark, Joohyun, Claudia Dufke, Zofia Fleszar, Michael Schlotterbek, Elena Buena-Atienza, Lara G. Stühn, Caspar Gross, Marc Sturm, Stephan Ossowski, Ludger Schöls, and et al. 2025. "Long-Read Sequencing Identifies Mosaic Sequence Variations in Friedreich’s Ataxia-GAA Repeats" International Journal of Molecular Sciences 26, no. 11: 4969. https://doi.org/10.3390/ijms26114969
APA StylePark, J., Dufke, C., Fleszar, Z., Schlotterbek, M., Buena-Atienza, E., Stühn, L. G., Gross, C., Sturm, M., Ossowski, S., Schöls, L., Riess, O., & Haack, T. B. (2025). Long-Read Sequencing Identifies Mosaic Sequence Variations in Friedreich’s Ataxia-GAA Repeats. International Journal of Molecular Sciences, 26(11), 4969. https://doi.org/10.3390/ijms26114969