
Nonadiabatic Surface Hopping Dynamics of Photocatalytic Water Splitting Process with Heptazine–(H2O)4 Chromophore



Abstract
1. Introduction
2. Results
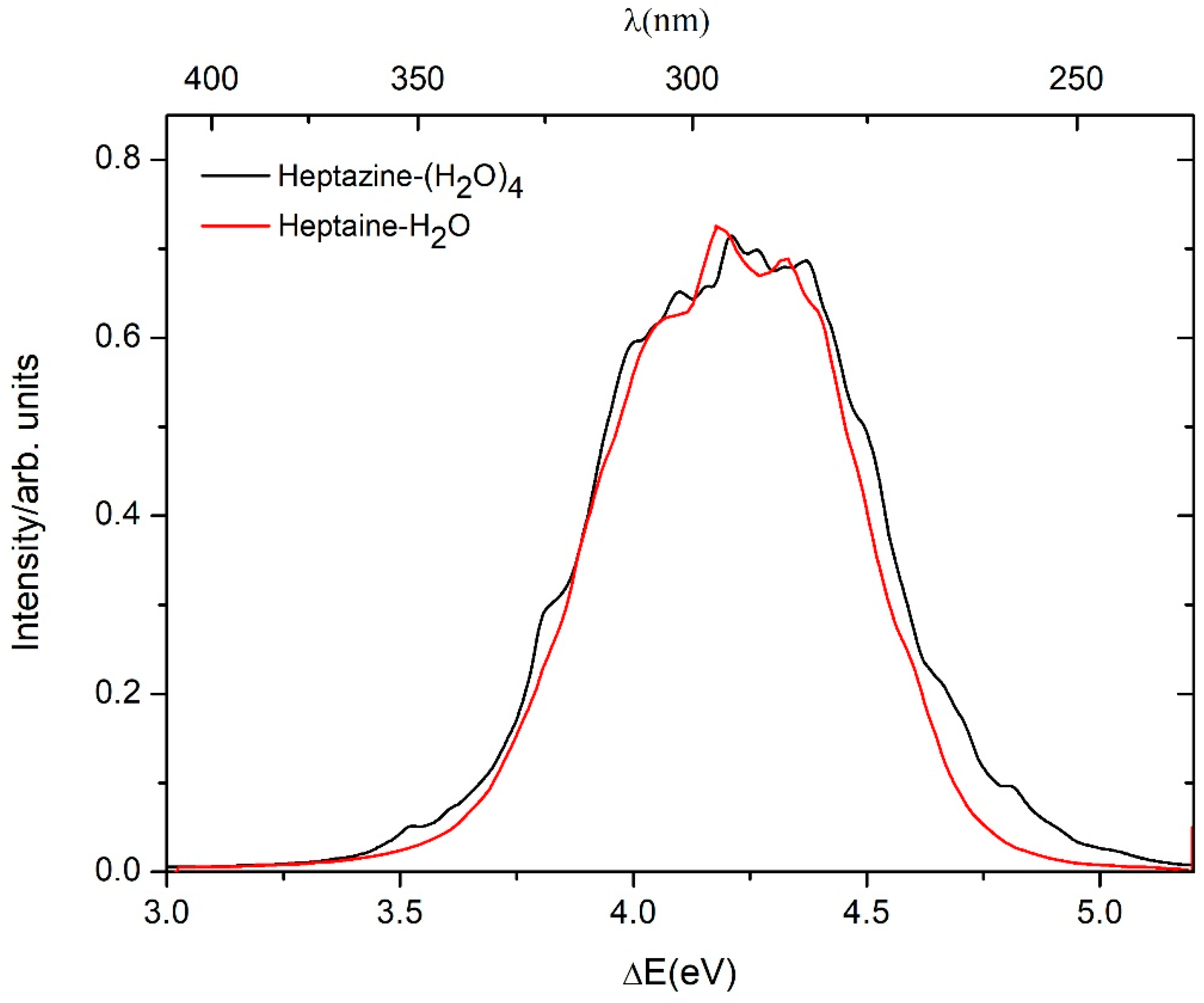
2.1. Ground State Equilibrium Structures and Absorption Spectrum
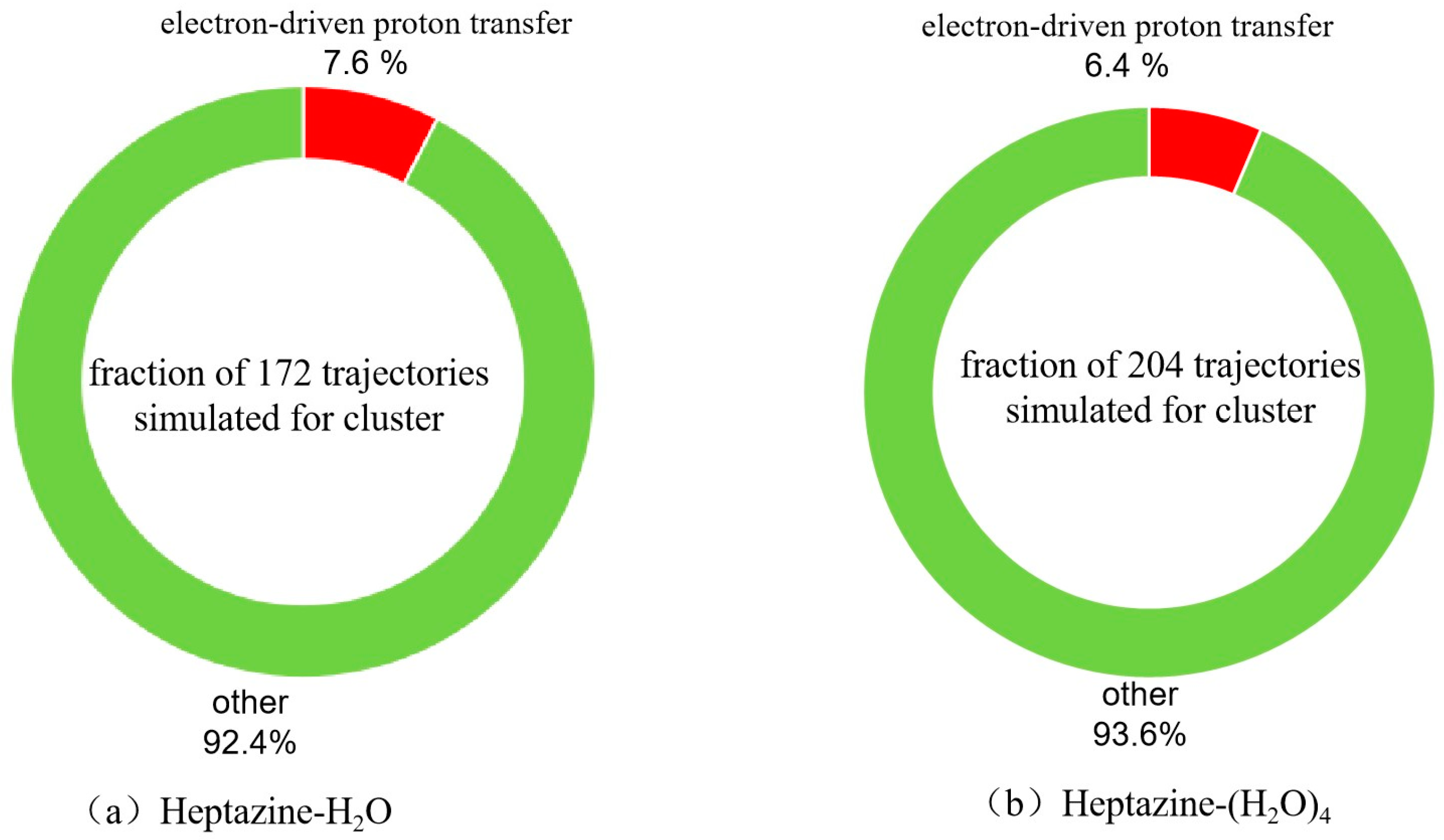
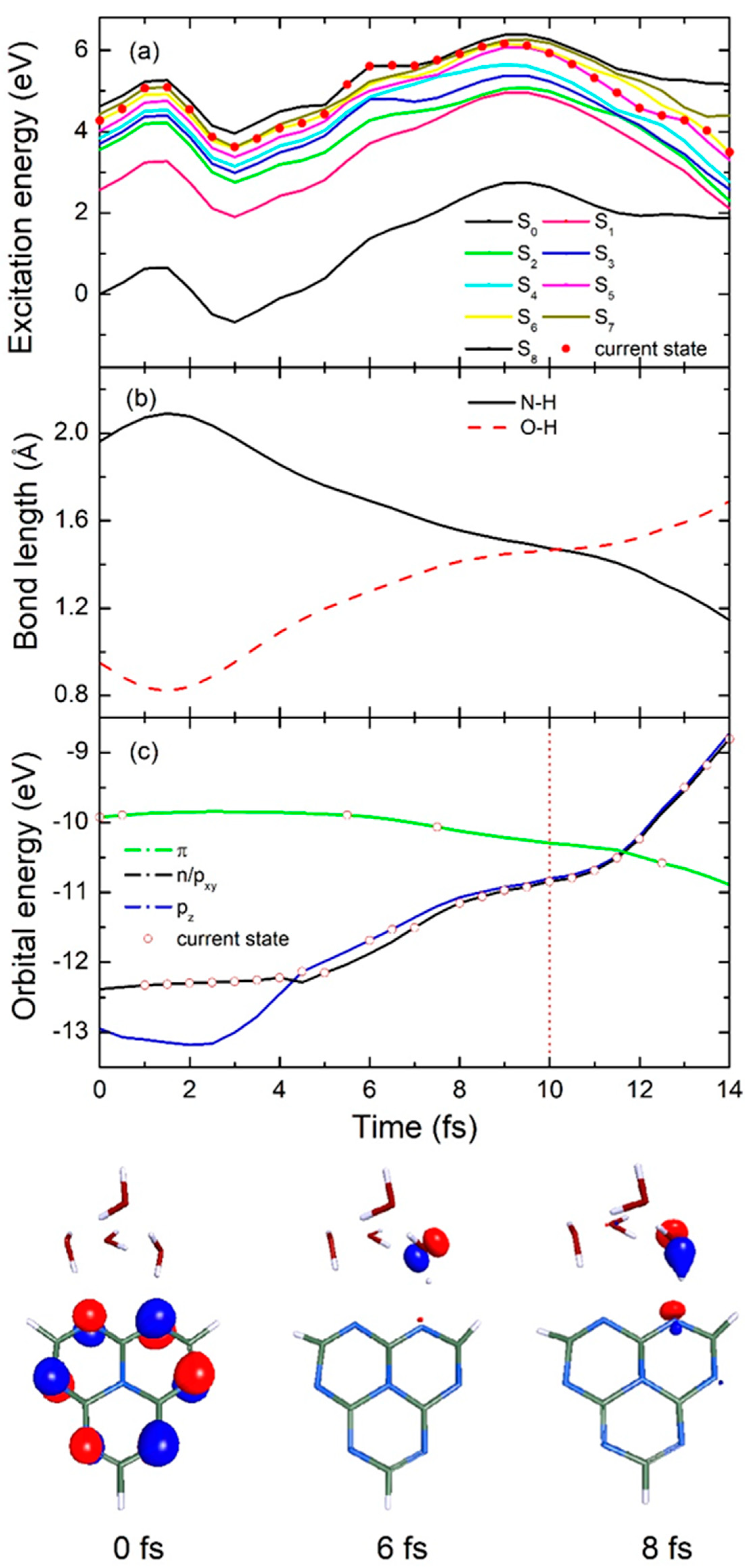
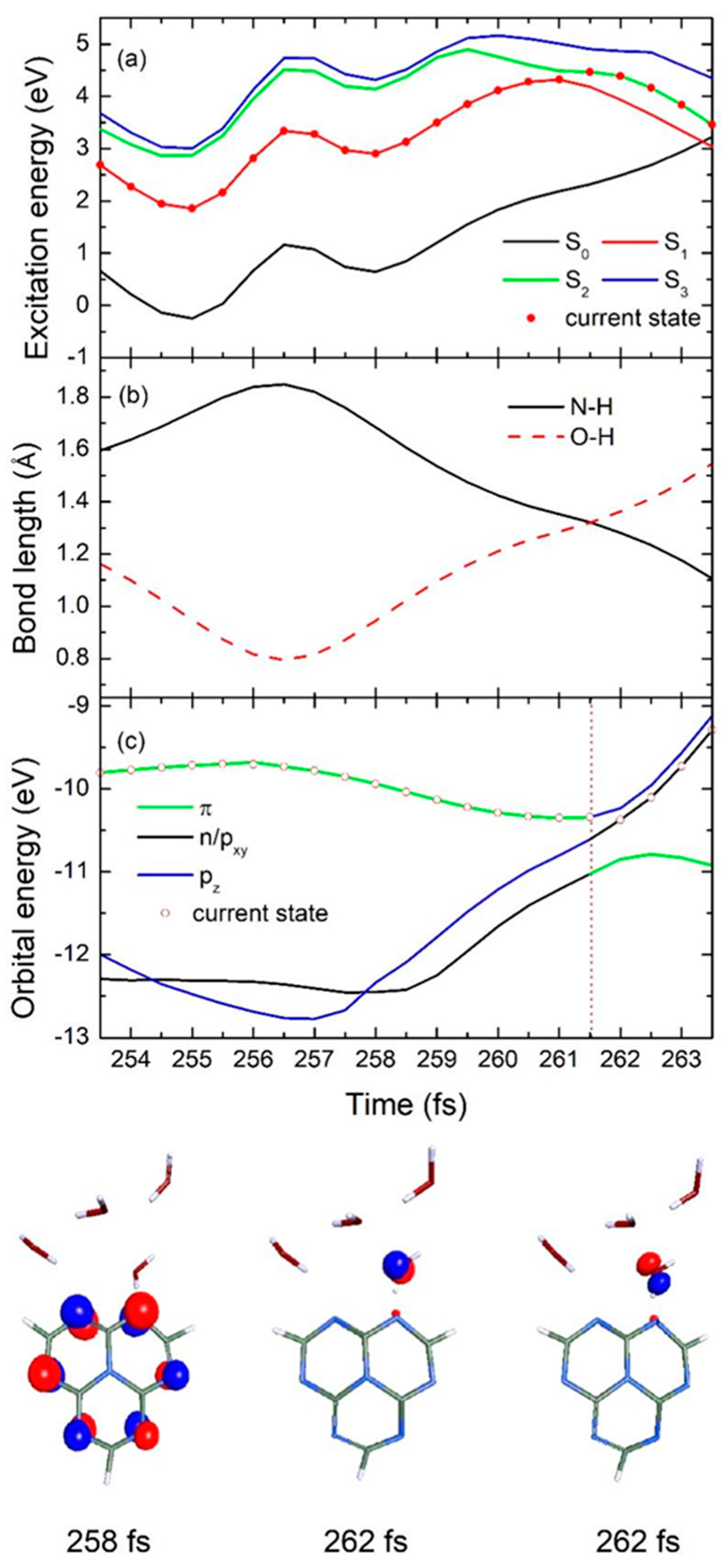
2.2. Nonadiabatic Dynamics Simulations
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EDPT | Electron-driven proton transfer |
References
- Weinberg, D.R.; Gagliardi, C.J.; Hull, J.F.; Murphy, C.F.; Kent, C.A.; Westlake, B.C.; Paul, A.; Ess, D.H.; McCafferty, D.G.; Meyer, T.J. Proton-Coupled Electron Transfer. Chem. Rev. 2012, 112, 4016–4093. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W. Computational Model of Photocatalytic Water Splitting. J. Phys. Chem. A 2008, 112, 7311–7313. [Google Scholar] [CrossRef]
- Fujishima, A.; Kenichi, H. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Maeda, K.; Kazunari, D. Photocatalytic Water Splitting: Recent Progress and Future Challenges. J. Phys. Chem. Lett. 2010, 1, 2655–2661. [Google Scholar] [CrossRef]
- Zou, Z.; Ye, J.; Sayama, K.; Arakawa, H. Direct Splitting of Water under Visible Light Irradiation with an Oxide Semiconductor Photocatalyst. Nature 2001, 414, 625–627. [Google Scholar] [CrossRef]
- Gust, D.; Moore, T.A.; Moore, A.L. Solar Fuels via Artificial Photosynthesis. Acc. Chem. Res. 2009, 42, 1890–1898. [Google Scholar] [CrossRef]
- Moore, G.F.; Brudvig, G.W. Energy Conversion in Photosynthesis: A Paradigm for Solar Fuel Production. Annu. Rev. Condens. Matter Phys. 2011, 2, 303–327. [Google Scholar] [CrossRef]
- Kärkäs, M.D.; Verho, O.; Johnston, E.V.; Åkermark, B. Artificial Photosynthesis: Molecular Systems for Catalytic Water Oxidation. Chem. Rev. 2014, 114, 11863–12001. [Google Scholar] [CrossRef]
- Domcke, W.; Ehrmaier, J.; Sobolewski, A.L. Solar Energy Harvesting with Carbon Nitrides and N-Heterocyclic Frameworks: Do We Understand the Mechanism? ChemPhotoChem 2019, 3, 10–23. [Google Scholar] [CrossRef]
- Liu, X.; Sobolewski, A.L.; Borrelli, R.; Domcke, W. Computational Investigation of the Photoinduced Homolytic Dissociation of Water in the Pyridine—Water Complex. Phys. Chem. Chem. Phys. 2013, 15, 5957–5966. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Antonietti, M. Polymeric Graphitic Carbon Nitride as a Heterogeneous Organocatalyst: From Photochemistry to Multipurpose Catalysis to Sustainable Chemistry. Angew. Chem. Int. Ed. 2012, 51, 68–89. [Google Scholar] [CrossRef]
- Wang, X.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J.M.; Domen, K.; Antonietti, M. A Metal-Free Polymeric Photocatalyst for Hydrogen Production from Water under Visible Light. Nat. Mater. 2009, 8, 76–80. [Google Scholar] [CrossRef]
- Wang, X.; Blechert, S.; Antonietti, M. Polymeric Graphitic Carbon Nitride for Heterogeneous Photocatalysis. ACS Catal. 2012, 2, 1596–1606. [Google Scholar] [CrossRef]
- Lin, Z.; Wang, X. Nanostructure Engineering and Doping of Conjugated Carbon Nitride Semiconductors for Hydrogen Photosynthesis. Angew. Chem. Int. Ed. Engl. 2013, 52, 1735–1738. [Google Scholar] [CrossRef]
- Chu, S.; Wang, Y.; Guo, Y.; Feng, J.; Wang, C.; Luo, W.; Fan, X.; Zou, Z. Band Structure Engineering of Carbon Nitride: In Search of a Polymer Photocatalyst with High Photooxidation Property. ACS Catal. 2013, 3, 912–919. [Google Scholar] [CrossRef]
- Martin, D.J.; Qiu, K.; Shevlin, S.A.; Handoko, A.D.; Chen, X.; Guo, Z.; Tang, J. Highly Efficient Photocatalytic H2Evolution from Water using Visible Light and Structure-Controlled Graphitic Carbon Nitride. Angew. Chem. Int. Ed. 2014, 53, 9240–9245. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Liu, N.; Han, Y.; Zhang, X.; Huang, H.; Lifshitz, Y.; Lee, S.-T.; Zhong, J.; Kang, Z. Metal-Free Efficient Photocatalyst for Stable Visible Water Splitting via a Two-Electron Pathway. Science 2015, 347, 970–974. [Google Scholar] [CrossRef]
- Zhang, G.; Lan, Z.-A.; Lin, L.; Lin, S.; Wang, X. Overall Water Splitting by Pt/Gc 3 N 4 Photocatalysts without Using Sacrificial Agents. Chem. Sci. 2016, 7, 3062–3066. [Google Scholar] [CrossRef]
- Ehrmaier, J.; Karsili, T.N.V.; Sobolewski, A.L.; Domcke, W. Mechanism of Photocatalytic Water Splitting with Graphitic Carbon Nitride: Photochemistry of the Heptazine–Water Complex. J. Phys. Chem. A 2017, 121, 4754–4764. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, S.; Zhou, J.; Zhang, R. Synergistic Effect of Carbon Dopants on Photoinduced Water−Splitting on Graphitic Carbon Nitride. Int. J. Hydrogen Energy 2024, 71, 709–717. [Google Scholar] [CrossRef]
- Liu, X.; Sobolewski, A.L.; Wolfgang, D. Photoinduced Oxidation of Water in the Pyridine-Water Complex: Comparison of the Singlet and Triplet Photochemistries. J. Phys. Chem. A 2014, 118, 7788–7795. [Google Scholar] [CrossRef]
- Esteves-López, N.; Coussan, S.; Dedonder-Lardeux, C.; Jouvet, C. Photoinduced Water Splitting in Pyridine Water Clusters. Phys. Chem. Chem. Phys. 2016, 18, 25637–25644. [Google Scholar] [CrossRef]
- Pang, X.; Ehrmaier, J.; Wu, X.; Jiang, C.; Xie, W.; Sobolewski, A.L.; Domcke, W. Photoinduced Hydrogen-Transfer Reactions in Pyridine-Water Clusters: Insights from Excited-State Electronic-Structure Calculations. Chem. Phys. 2018, 515, 550–556. [Google Scholar] [CrossRef]
- Pang, X.; Jiang, C.; Xie, W.; Domcke, W. Photoinduced Electron-Driven Proton Transfer from Water to an N-Heterocyclic Chromophore: Nonadiabatic Dynamics Studies for Pyridine—Water Clusters. Phys. Chem. Chem. Phys. 2019, 21, 14073–14079. [Google Scholar] [CrossRef]
- Rabe, E.J.; Corp, K.L.; Sobolewski, A.L.; Domcke, W.; Schlenker, C.W. Proton-Coupled Electron Transfer from Water to a Model Heptazine-Based Molecular Photocatalyst. J. Phys. Chem. Lett. 2018, 9, 6257–6261. [Google Scholar] [CrossRef]
- Ullah, N.; Chen, S.; Zhao, Y.; Zhang, R. Photoinduced Water–Heptazine Electron-Driven Proton Transfer: Perspective for Water Splitting with G-C3n4. J. Phys. Chem. Lett. 2019, 10, 4310–4316. [Google Scholar] [CrossRef]
- Huang, X.; Domcke, W. Ab Initio Nonadiabatic Surface-Hopping Trajectory Simulations of Photocatalytic Water Oxidation and Hydrogen Evolution with the Heptazine Chromophore. J. Phys. Chem. A 2021, 125, 9917–9931. [Google Scholar] [CrossRef]
- Ehrmaier, J.; Rabe, E.J.; Pristash, S.R.; Corp, K.L.; Schlenker, C.W.; Sobolewski, A.L.; Domcke, W. Singlet–Triplet Inversion in Heptazine and in Polymeric Carbon Nitrides. J. Phys. Chem. A 2019, 123, 8099–8108. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W. Are Heptazine-Based Organic Light-Emitting Diode Chromophores Thermally Activated Delayed Fluorescence or Inverted Singlet-Triplet Systems? J. Phys. Chem. Lett. 2021, 12, 6852–6860. [Google Scholar] [CrossRef]
- Lau, V.W.; Klose, D.; Kasap, H.; Podjaski, F.; Pignié, M.; Reisner, E.; Jeschke, G.; Lotsch, B.V. Dark Photocatalysis: Storage of Solar Energy in Carbon Nitride for Time-Delayed Hydrogen Generation. Angew. Chem. 2016, 129, 525–529. [Google Scholar] [CrossRef]
- Lin, B.; Yang, G.; Wang, L. Stacking-Layer-Number Dependence of Water Adsorption in 3d Ordered Close-Packed G-C3n4 Nanosphere Arrays for Photocatalytic Hydrogen Evolution. Angew. Chem. 2019, 131, 4635–4639. [Google Scholar] [CrossRef]
- Ullah, N.; Chen, S.; Zhang, R. Excited-State Nonadiabatic Dynamics Simulations on the Heptazine and Adenine in a Water Environment: A Mini Review. J. Chin. Chem. Soc. 2023, 70, 195–208. [Google Scholar] [CrossRef]
- You, P.; Lian, C.; Chen, D.; Xu, J.; Zhang, C.; Meng, S.; Wang, E. Nonadiabatic Dynamics of Photocatalytic Water Splitting on a Polymeric Semiconductor. Nano Lett. 2021, 21, 6449–6455. [Google Scholar] [CrossRef]
- Yang, H.; Wu, R.; Li, W.; Wen, J. Ultrafast Hydrogen Production in Boron/Oxygen-Codoped Graphitic Carbon Nitride Revealed by Nonadiabatic Dynamics Simulations. Phys. Chem. Chem. Phys. 2024, 26, 14205–14215. [Google Scholar] [CrossRef]
- Aragó, J.; Sancho-García, J.C.; Ortí, E.; Beljonne, D. Ab Initio Modeling of Donor–Acceptor Interactions and Charge-Transfer Excitations in Molecular Complexes: The Case of Terthiophene–Tetracyanoquinodimethane. J. Chem. Theory Comput. 2011, 7, 2068–2077. [Google Scholar] [CrossRef]
- Granucci, G.; Persico, M. Critical Appraisal of the Fewest Switches Algorithm for Surface Hopping. J. Chem. Phys. 2007, 126, 134114. [Google Scholar] [CrossRef]
- Ehrmaier, J.; Janicki, M.J.; Sobolewski, A.L.; Domcke, W. Mechanism of Photocatalytic Water Splitting with Triazine-Based Carbon Nitrides: Insights from Ab Initio Calculations for the Triazine—Water Complex. Phys. Chem. Chem. Phys. 2018, 20, 14420–14430. [Google Scholar] [CrossRef]
- Wu, H.-Z.; Liu, L.-M.; Zhao, S.-J. The Effect of Water on the Structural, Electronic and Photocatalytic Properties of Graphitic Carbon Nitride. Phys. Chem. Chem. Phys. 2014, 16, 3299–3304. [Google Scholar] [CrossRef]
- Jonas, W.; Rainer, N.; Markus, A.; Peter, S. Adsorption and Photocatalytic Splitting of Water on Graphitic Carbon Nitride: A Combined First Principles and Semiempirical Study. Phys. Chem. Chem. Phys. 2014, 16, 15917–15926. [Google Scholar]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. B 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W.; Dedonder-Lardeux, C.; Jouvet, C. Excited-State Hydrogen Detachment and Hydrogen Transfer Driven by Repulsive 1πσ* States: A New Paradigm for Nonradiative Decay in Aromatic Biomolecules. Phys. Chem. Chem. Phys. 2002, 4, 1093–1100. [Google Scholar] [CrossRef]
- Schirmer, J. Beyond the Random-Phase Approximation: A New Approximation Scheme for the Polarization Propagator. Phys. Rev. A 1982, 26, 2395–2416. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bar, M.; Baron, H.P.; Bauernschmitt, R.; Bocker, S.; Ehrig, M.; Eichkorn, K.; Elliot, S.; Furche, F.; Haase, F. 6.6, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH; Turbomole: Karlsruhe, Germany, 2014. [Google Scholar]
- Barbatti, M.; Ruckenbauer, M.; Plasser, F.; Pittner, J.; Granucci, G.; Persico, M.; Lischka, H. Newton-X: A Surface-Hopping Program for Nonadiabatic Molecular Dynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 26–33. [Google Scholar] [CrossRef]
- Belyaev, A.K.; Domcke, W.; Lasser, C.; Trigila, G. Nonadiabatic Nuclear Dynamics of the Ammonia Cation Studied by Surface Hopping Classical Trajectory Calculations. J. Chem. Phys. 2015, 142, 104307. [Google Scholar] [CrossRef]
- Xie, W.; Domcke, W. Accuracy of Trajectory Surface-Hopping Methods: Test for a Two-Dimensional Model of the Photodissociation of Phenol. J. Chem. Phys. 2017, 147, 184114. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Heptazine–H2O | Heptazine–(H2O)4 | ||||
|---|---|---|---|---|---|
| State | Energy | f | State | Energy | f |
| S1 | 2.60 | 0.00 | S1 | 2.63 | 0.00 |
| S2 | 3.72 | 0.00 | S2 | 3.74 | 0.00 |
| S3 | 3.80 | 0.00 | S3 | 3.85 | 0.00 |
| S4 | 3.89 | 0.00 | S4 | 4.00 | 0.00 |
| S5 | 4.20 | 0.26 | S5 | 4.38 | 0.25 |
| S6 | 4.22 | 0.25 | S6 | 4.40 | 0.30 |
| S7 | 4.75 | 0.00 | S7 | 4.83 | 0.00 |
| S8 | 4.80 | 0.00 | S8 | 4.97 | 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, X.; Yang, C.; Zhang, N.; Jiang, C. Nonadiabatic Surface Hopping Dynamics of Photocatalytic Water Splitting Process with Heptazine–(H2O)4 Chromophore. Int. J. Mol. Sci. 2025, 26, 4549. https://doi.org/10.3390/ijms26104549
Pang X, Yang C, Zhang N, Jiang C. Nonadiabatic Surface Hopping Dynamics of Photocatalytic Water Splitting Process with Heptazine–(H2O)4 Chromophore. International Journal of Molecular Sciences. 2025; 26(10):4549. https://doi.org/10.3390/ijms26104549
Chicago/Turabian StylePang, Xiaojuan, Chenghao Yang, Ningbo Zhang, and Chenwei Jiang. 2025. "Nonadiabatic Surface Hopping Dynamics of Photocatalytic Water Splitting Process with Heptazine–(H2O)4 Chromophore" International Journal of Molecular Sciences 26, no. 10: 4549. https://doi.org/10.3390/ijms26104549
APA StylePang, X., Yang, C., Zhang, N., & Jiang, C. (2025). Nonadiabatic Surface Hopping Dynamics of Photocatalytic Water Splitting Process with Heptazine–(H2O)4 Chromophore. International Journal of Molecular Sciences, 26(10), 4549. https://doi.org/10.3390/ijms26104549