
Identification of Susceptibility Genes Underlying Bovine Respiratory Disease in Xinjiang Brown Cattle Based on DNA Methylation


Abstract
1. Introduction
2. Results
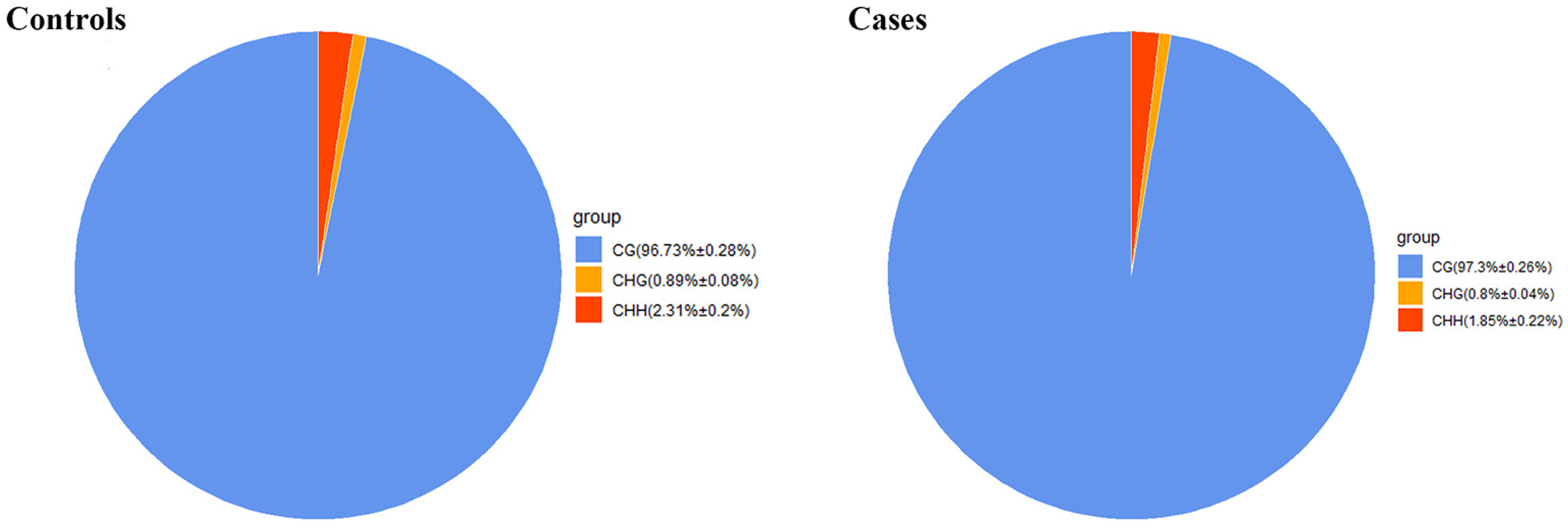
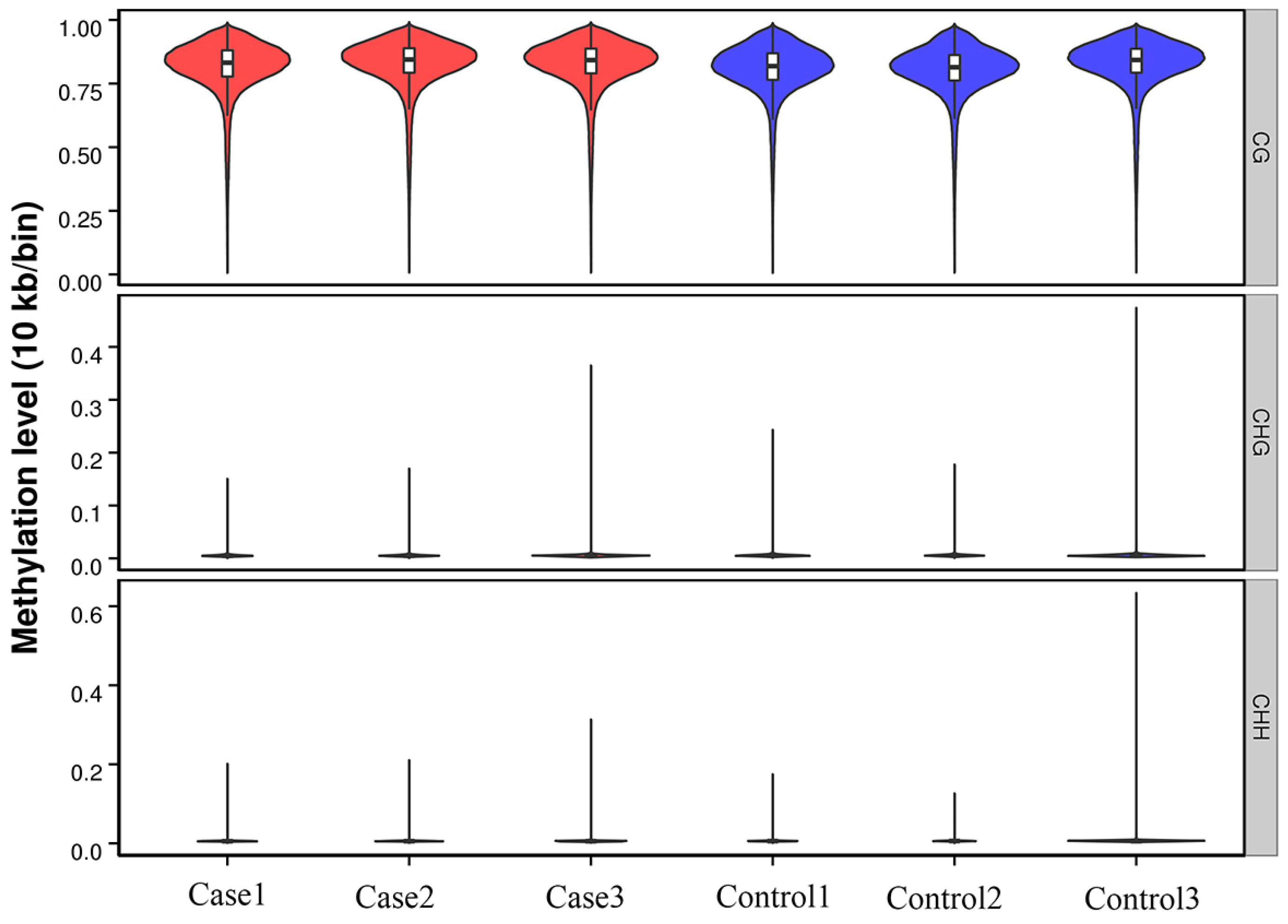
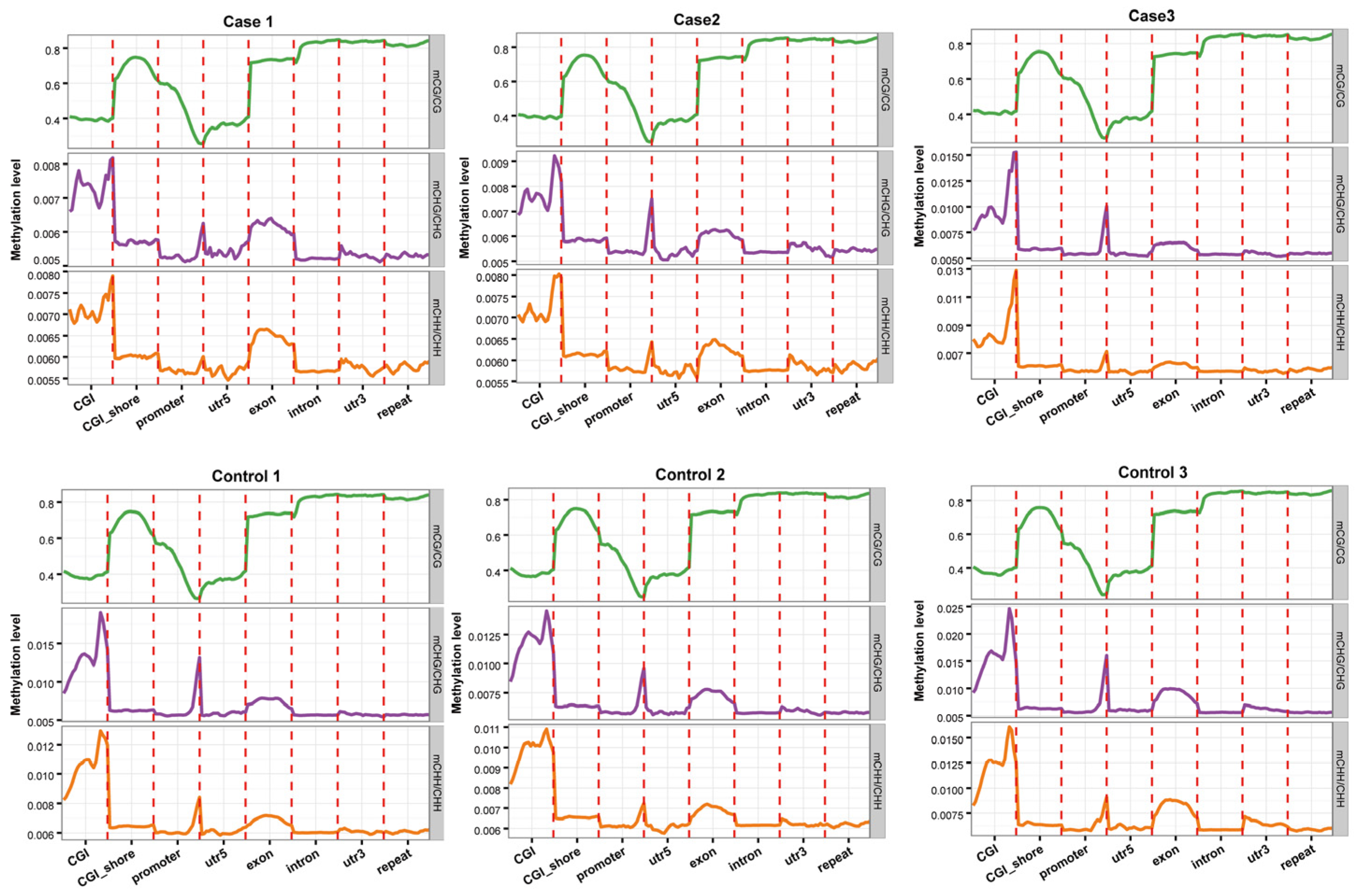
2.1. Genome-Wide DNA Methylation Profile Analyses
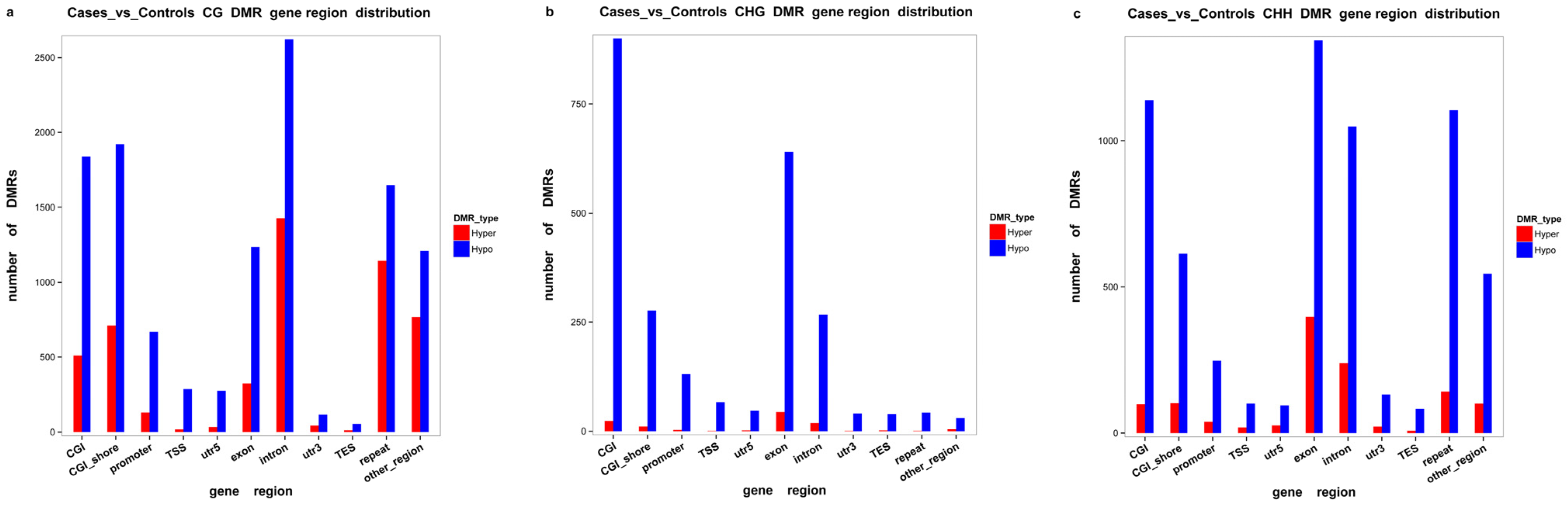
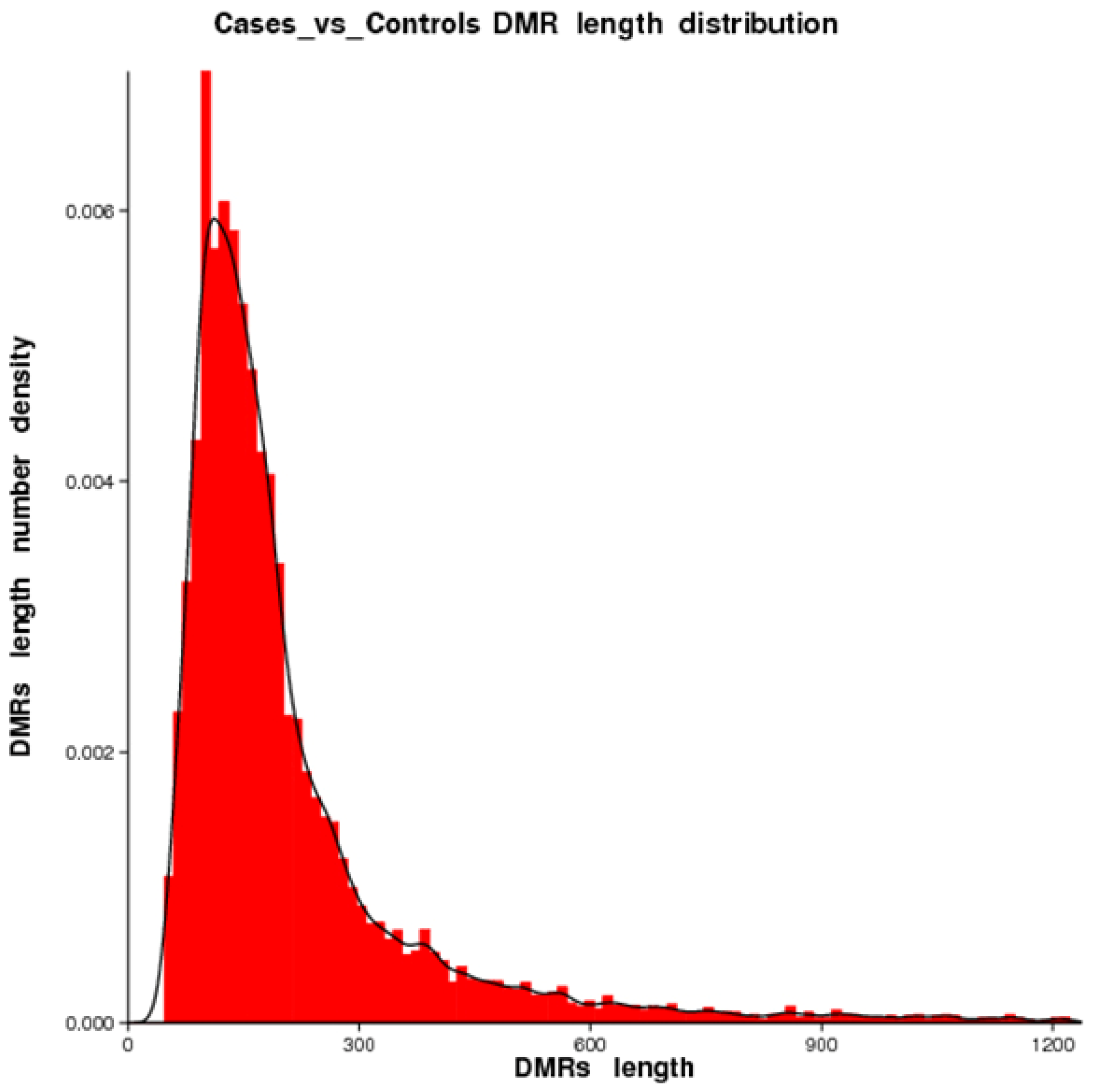
2.2. DMR Profiling
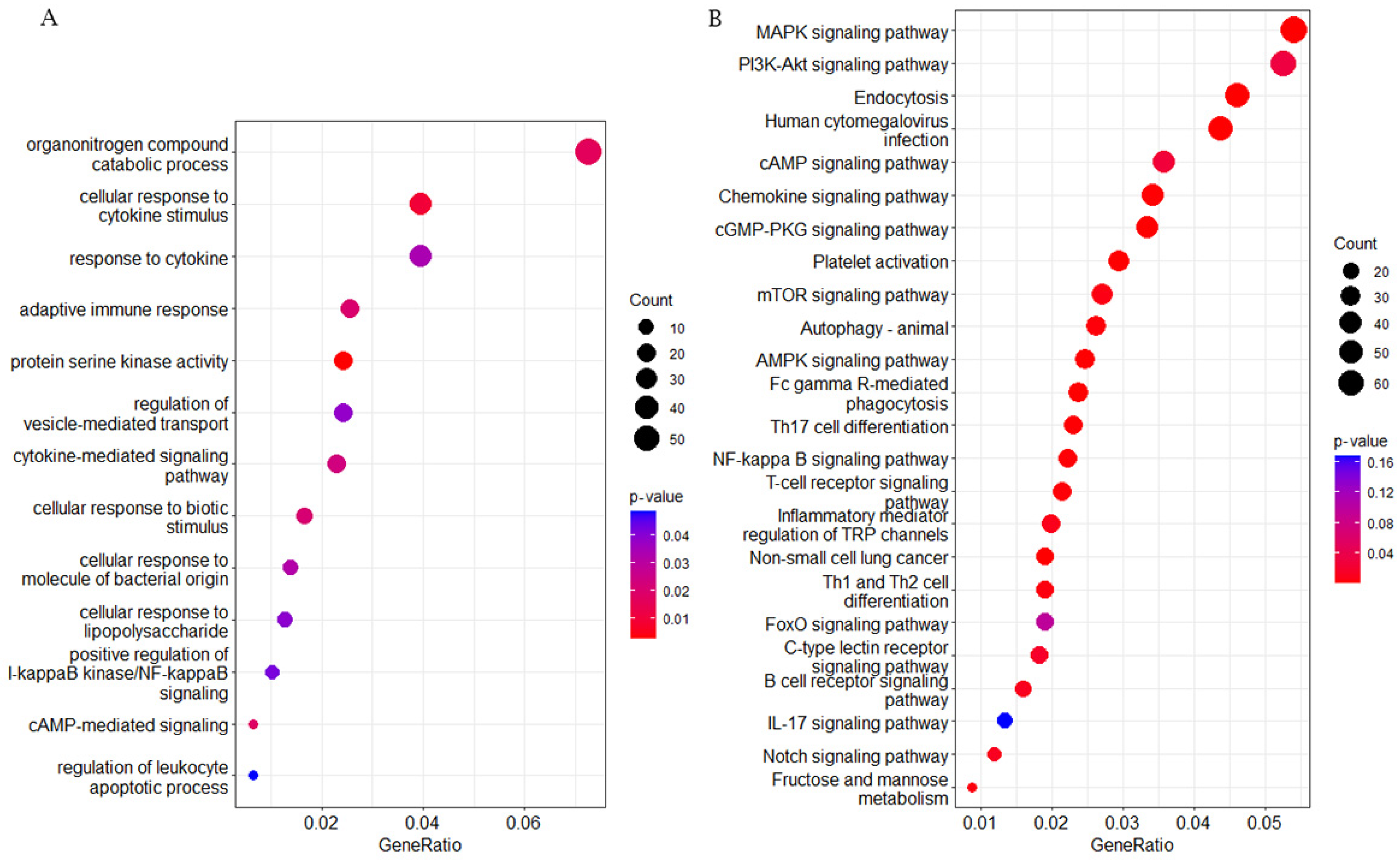
2.3. GO/KEGG Enrichment Analysis for DMGs
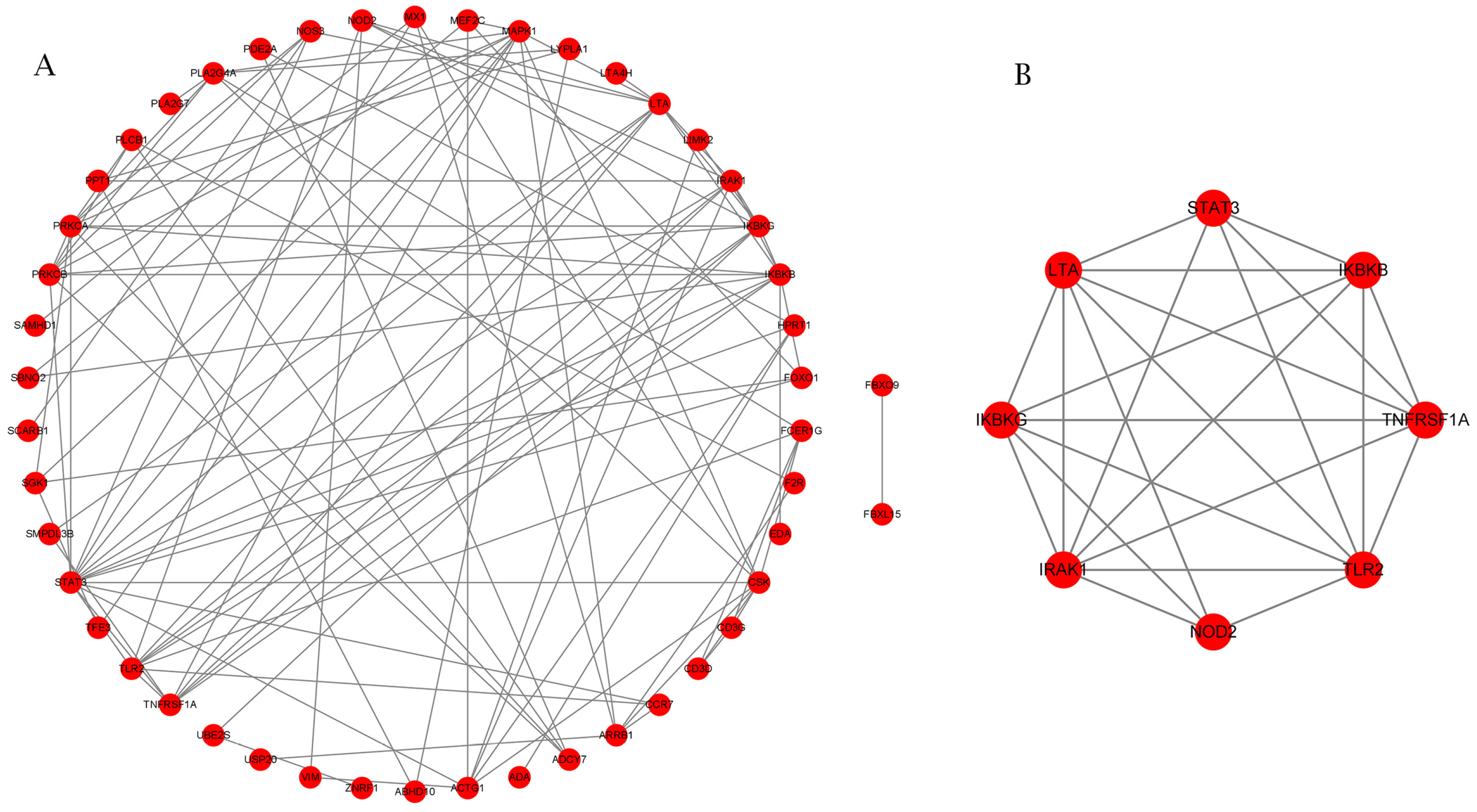
2.4. Screening of Possibly Valuable DMGs Linked to Immune Function
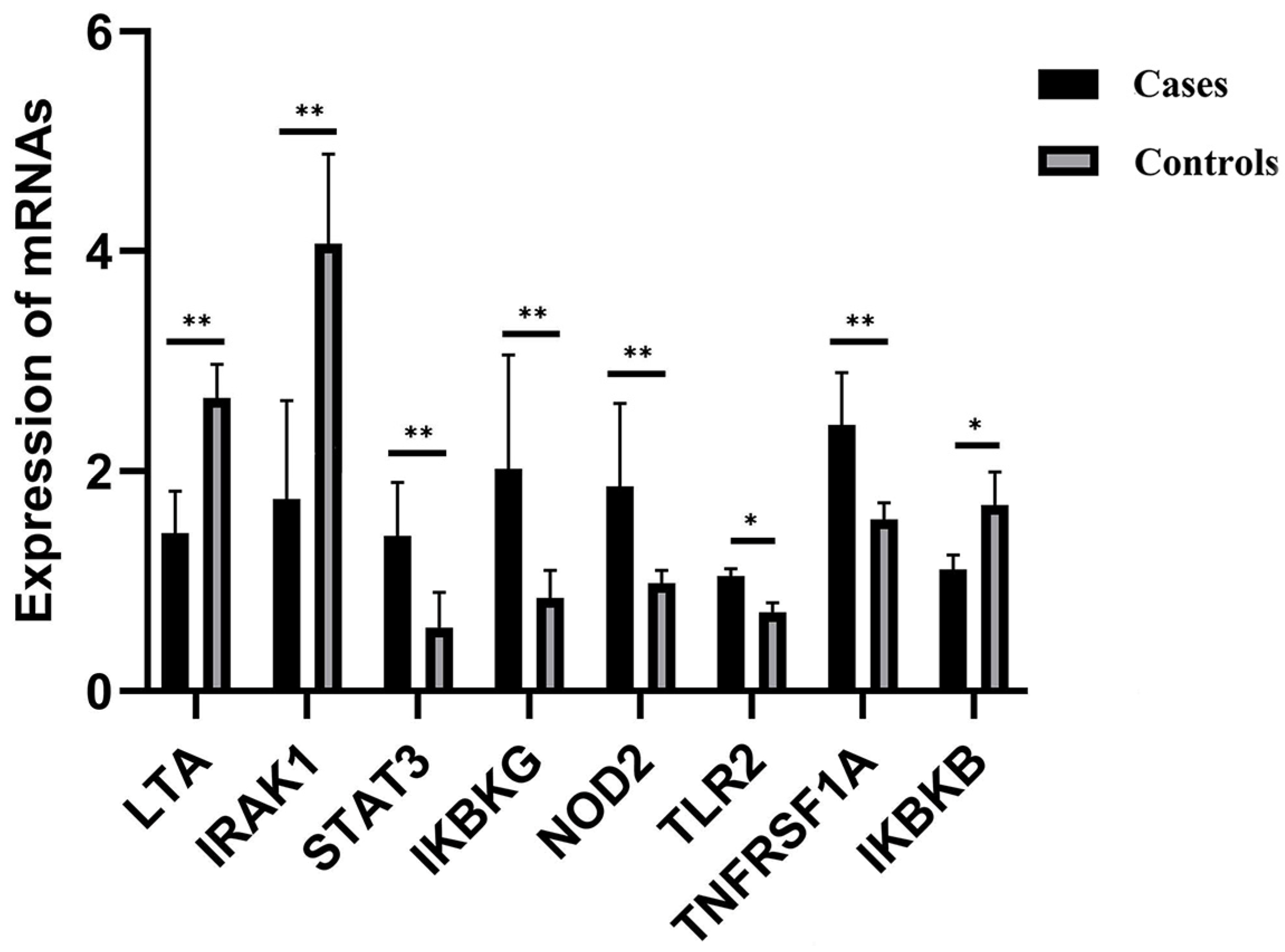
2.5. Differential Gene Methylation Regulating Influence upon the Immunology of Cattle
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Library Construction
4.3. Whole-Genome Bisulfite Sequencing (WGBS) and Differentially Methylated Region (DMR) Determination
4.4. Functional Enrichment Analysis
4.5. Quantitative Reverse Transcription-PCR
4.6. RNA-Seq Data Analysis
4.7. Correlation Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arcangioli, M.A.; Duet, A.; Meyer, G.; Dernburg, A.; Bézille, P.; Poumarat, F.; Le Grand, D. The role of Mycoplasma bovis in bovine respiratory disease outbreaks in veal calf feedlots. Vet. J. 2008, 177, 89–93. [Google Scholar] [CrossRef]
- Kiser, J.N.; Lawrence, T.E.; Neupane, M.; Seabury, C.M.; Taylor, J.F.; Womack, J.E.; Neibergs, H.L. Rapid Communication: Subclinical bovine respiratory disease: Loci and pathogens associated with lung lesions in feedlot cattle. J. Anim. Sci. 2017, 95, 2726–2731. [Google Scholar] [CrossRef]
- Keele, J.W.; Kuehn, L.A.; McDaneld, T.G.; Tait, R.G.; Jones, S.A.; Smith, T.P.; Shackelford, S.D.; King, D.A.; Wheeler, T.L.; Lindholm-Perry, A.K.; et al. Genomewide association study of lung lesions in cattle using sample pooling. J. Anim. Sci. 2015, 93, 956–964. [Google Scholar] [CrossRef]
- Neupane, M.; Kiser, J.N.; Neibergs, H.L. Gene set enrichment analysis of SNP data in dairy and beef cattle with bovine respiratory disease. Anim. Genet. 2018, 49, 527–538. [Google Scholar] [CrossRef]
- Kiser, J.N.; Neupane, M.; White, S.N.; Neibergs, H.L. Identification of genes associated with susceptibility to Mycobacterium avium ssp. Paratuberculosis (Map) tissue infection in Holstein cattle using gene set enrichment analysis–SNP. Mamm. Genome 2018, 29, 539–549. [Google Scholar] [CrossRef]
- Jang, H.; Serra, C. Nutrition, epigenetics, and diseases. Clin. Nutr. Res. 2014, 3, 1–8. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Hannan Parker, A.; Wilkinson, S.W.; Ton, J. Epigenetics: A catalyst of plant immunity against pathogens. New Phytol. 2022, 233, 66–83. [Google Scholar] [CrossRef]
- Guo, K.; Eid, S.A.; Elzinga, S.E.; Pacut, C.; Feldman, E.L.; Hur, J. Genome-wide profiling of DNA methylation and gene expression identifies candidate genes for human diabetic neuropathy. Clin. Epigenetics 2020, 12, 1–16. [Google Scholar]
- Takahashi, K.; Sugi, Y.; Nakano, K.; Kobayakawa, T.; Nakanishi, Y.; Hosono, A.; Kaminogawa, S. Regulation of gene expression through gut microbiota-dependent DNA methylation in colonic epithelial cells. ImmunoHorizons 2020, 4, 178–190. [Google Scholar] [CrossRef]
- Seutter, S.; Winfield, J.; Esbitt, A.; Snyder, S.; Magner, A.; Kim, K.; Carcuffe, C.; Schmoyer, J.; Kamrani, P.; Mercando, J.; et al. Interleukin 1 beta and prostaglandin E2 affect expression of DNA methylating and demethylating enzymes in human gingival fibroblasts. Int. Immunopharmacol. 2020, 78, 105920. [Google Scholar] [CrossRef]
- Wang, X.; Yu, Y. Regulation mechanisms of epigenetics on inflammation and its perspective on breeding for mastitis resistance in dairy cattle. Yi Chuan Hered. 2010, 32, 663–669. [Google Scholar] [CrossRef]
- Lee, C.C.; Avalos, A.M.; Ploegh, H.L. Accessory molecules for Toll-like receptors and their function. Nat. Rev. Immunol. 2012, 12, 168–179. [Google Scholar] [CrossRef]
- Patrat, C.; Ouimette, J.F.; Rougeulle, C. X chromosome inactivation in human development. Development 2020, 147, 183095. [Google Scholar] [CrossRef]
- Singmann, P.; Shem-Tov, D.; Wahl, S.; Grallert, H.; Fiorito, G.; Shin, S.Y.; Schramm, K.; Wolf, P.; Kunze, S.; Baran, Y.; et al. Characterization of whole-genome autosomal differences of DNA methylation between men and women. Epigenetics Chromatin 2015, 8, 43. [Google Scholar] [CrossRef]
- Simons, R.L.; Leim, M.K.; Beach, S.R.; Philibert, R.A.; Cutrona, C.E.; Gibbons, F.X.; Barr, A. Economic hardship and biological weathering: The epigenetics of aging in a U.S. sample of black women. Soc. Sci Med. 2016, 150, 192–200. [Google Scholar] [CrossRef]
- McGill, J.L.; Sacco, R.E. The immunology of bovine respiratory disease. Vet. Clin. North Am. Food Anim. Pract. 2020, 36, 333–348. [Google Scholar] [CrossRef]
- Kurdyukov, S.; Bullock, M. DNA methylation analysis: Choosing the right method. Biology 2016, 5, 3. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Bandettini, W.P.; Kellman, P.; Mancini, C.; Booker, O.J.; Vasu, S.; Leung, S.W.; Wilson, J.R.; Shanbhag, S.M.; Chen, M.Y.; Arai, E.A. MultiContrast Delayed Enhancement (MCODE) improves detection of subendocardial myocardial infarction by late gadolinium enhancement cardiovascular magnetic resonance: A clinical validation study. J. Cardiovasc. Magn. Reson. 2012, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Y.; Zhang, S.X.; Xu, Y.Y.; Ma, Y.L.; Zhang, D.X.; Li, X.Y.; Zhao, S. The DNA methylation state of wnt and tgf beta signals is a key factor on functional regulation of skeletal muscle satellite cell development. Front. Genet. 2019, 10, 220. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sun, J.; Zhang, L.; Li, C.; Womack, J.E.; Li, Z.; Lan, X.; Lei, C.; Zhang, C.; Zhao, X.; et al. Genome-wide DNA methylation profiles and their relationships with mRNA and the microRNA transcriptome in bovine muscle tissue (Bos taurine). Sci. Rep. 2014, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ponsuksili, S.; Trakooljul, N.; Basavaraj, S.; Hadlich, F.; Murani, E.; Wimmers, K. Epigenome-wide skeletal muscle DNA methylation profiles at the background of distinct metabolic types and ryanodine receptor variation in pigs. BMC Genom. 2019, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gensous, N.; Bacalini, M.G.; Franceschi, C.; Meskers, C.; Maier, A.B.; Garagnani, P. Age-related DNA methylation changes: Potential impact on skeletal muscle aging in humans. Front. Physiol. 2019, 10, 465621. [Google Scholar] [CrossRef] [PubMed]
- Namous, H.; Penagaricano, F.; Del Corvo, M.; Capra, E.; Thomas, D.L.; Stella, A.; Williams, J.L.; Marsan, P.A.; Khatib, H. Integrative analysis of methylomic and tran-scriptomic data in fetal sheep muscle tissues in response to maternal diet during pregnancy. BMC Genom. 2018, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Srithayakumar, V.; Jiminez, J.; Jin, W.; Hosseini, A.; Raszek, M.; Orsel, K.; Guan, L.L.; Plastow, G. Longitudinal blood transcriptomic analysis to identify molecular regulatory patterns of bovine respiratory disease in beef cattle. Genomics 2020, 112, 3968–3977. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Liang, Y.; Deng, K.; Zhang, Z.; Zhang, G.; Zhang, Y.; Wang, F. Analysis of DNA methylation profiles during sheep skeletal muscle development using whole-genome bisulfite sequencing. BMC Genom. 2020, 21, 1–15. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Li, F.Z.; Feng, X.; Yang, H.; Zhu, A.X.; Pang, J.; Han, L.; Zhang, T.; Yao, X.; Wang, F. Genome-wide analysis of DNA Methylation profiles on sheep ovaries associated with prolificacy using whole-genome Bisulfite sequencing. BMC Genom. 2017, 18, 1–17. [Google Scholar]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Coto, E.; Díaz-Corte, C.; Tranche, S.; Gómez, J.; Alonso, B.; Iglesias, S.; Reguero, J.R.; López-Larrea, C.; Coto-Segura, P. Gene variants in the NF-KB pathway (NFKB1, NFKBIA, NFKBIZ) and their association with type 2 diabetes and impaired renal function. Hum. Immunol. 2018, 79, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [PubMed]
- Behl, J.D.; Mishra, P.; Verma, N.K.; Niranjan, S.K.; Dangi, P.S.; Sharma, R.; Behl, R. Nucleotide polymorphisms in the bovine lymphotoxin a gene and their distribution among Bos indicus zebu cattle breeds. Gene 2016, 579, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Frey-Jakobs, S.; Hartberger, J.M.; Fliegauf, M.; Bossen, C.; Wehmeyer, M.L.; Neubauer, J.C.; Bulashevska, A.; Proietti, M.; Fröbel, P.; Nöltner, C.; et al. ZNF341 controls STAT3 expression and thereby immunocompetence. Sci. Immunol. 2018, 3, eaat4941. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yu, H.; Liu, Y.; Gibson, S.A.; Yan, Z.; Xu, X.; Gaggar, A.; Li, P.-K.; Li, C.; Wei, S.; et al. Protective effect of suppressing STAT3 activity in LPS-induced acute lung injury. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 311, L868–L880. [Google Scholar] [CrossRef] [PubMed]
- Gavino, A.C.; Nahmod, K.; Bharadwaj, U.; Makedonas, G.; Tweardy, D.J. STAT3 inhibition prevents lung inflammation, remodeling, and accumulation of Th2 and Th17 cells in a murine asthma model. Allergy 2016, 71, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Menendez, M.; Kurylowicz, K.; Griffin, C.T. Genomic locus proteomic screening identifies the NF-kappa B signaling pathway components NF kappa B1 and IKBKG as transcriptional regulators of Ripk3 in endothelial cells. PLoS ONE 2021, 16, e0253519. [Google Scholar]
- Manjarrez-Orduno, N.; Marasco, E.; Chung, S.A.; Katz, M.S.; Kiridly, J.F.; Simpfendorfer, K.R.; Freudenberg, J.; Ballard, D.H.; Nashi, E.; Hopkins, T.J. CSK regulatory polymorphism is associated with systemic lupus erythematosus and influences B-cell signaling and activation. Nat. Genet. 2012, 44, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Vang, T.; Torgersen, K.M.; Sundvold, V.; Saxena, M.; Levy, F.O.; Skalhegg, B.S.; Hansson, V.; Mustelin, T.; Taskén, K. Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J. Exp. Med. 2001, 193, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dang, Y.; Li, P.; Rong, M.; Chen, G. Expression of IRAK1 in lung cancer tissues and its clinicopathological significance: A microarray study. Int. J. Clin. Exp. Patho. 2014, 7, 8096. [Google Scholar]
- Liu, Y.; Tsai, M.; Wu, S.; Chang, T.; Tsai, T.; Gow, C.; Wang, H.; Shih, J. MiR-146b-5p enhances the sensitivity of NSCLC to EGFR tyrosine kinase inhibitors by regulating the IRAK1/NF-kappa b pathway. Mol. Ther.-Nucleic Acids 2020, 22, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Sun, R.; Liu, G.; Peng, L.; Zheng, L.; Xie, P.; Lin, S.T.; Mei, Y.; Qiang, Y.Y.; Li, C.Z. S100A14 suppresses metastasis of nasopharyngeal carcinoma by inhibition of NF-kB signaling through degradation of IRAK1. Oncogene 2020, 39, 5307–5322. [Google Scholar] [CrossRef] [PubMed]
- Kale, S.D.; Dikshit, N.; Kumar, P.; Balamuralidhar, V.; Khameneh, H.J.; Malik, N.B.A.; Koh, T.H.; Tan, G.G.Y.; Tan, T.T.; Mortellaro, A.; et al. Nod2 is required for the early innate immune clearance of Acinetobacter baumannii from the lungs. Sci. Rep. 2017, 7, 1–9. [Google Scholar]
- Shimada, K.; Chen, S.; Dempsey, P.W.; Sorrentino, R.; Alsabeh, R.; Slepenkin, A.V.; Peterson, E.; Doherty, T.M.; Underhill, D.; Crother, T.R.; et al. The NOD/RIP2 pathway is essential for host defenses against chlamydophila pneumoniae lung infection. PLoS Pathog. 2009, 5, e1000379. [Google Scholar] [CrossRef]
- Gandotra, S.; Jang, S.; Murray, P.J.; Salgame, P.; Ehrt, S. Nucleotide-binding oligomerization domain protein 2-deficient mice control infection with Mycobactefium tuberculosis. Infect. Immun. 2007, 75, 5127–5134. [Google Scholar] [CrossRef] [PubMed]
- Divangahi, M.; Mostowy, S.; Coulombe, F.; Kozak, R.; Guillot, L.; Veyrier, F.; Kobayashi, K.S.; Flavell, R.A.; Gros, P.; Behr, M.A. NOD2-deficient mice have impaired resistance to mycobacterium tuberculosis infection through defective innate and adaptive immunity. J. Immunol. 2008, 181, 7157–7165. [Google Scholar] [CrossRef]
- Re, F.; Jack, L.; Strominger, Y. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J. Leukoc. Biol. 2001, 276, 37692–37699. [Google Scholar] [CrossRef]
- Girkin, J.; Loo, S.; Esneau, C.; Maltby, S.; Mercuri, F.; Chua, B.; Reid, A.T.; Veerati, P.C.; Grainge, C.L.; Wark, P.A.; et al. TLR2-mediated innate immune priming boosts lung anti-viral immunity. Eur. Respir. J. 2021, 58, 2001584. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Guo, J.; Dong, L.; Wang, J. Cerebellar fastigial nucleus stimulation in a chronic unpredictable mild stress rat model reduces Post-Stroke depression by suppressing brain inflammation via the microRNA-29c/TNFRSF1A signaling pathway. Med. Sci. Monitor 2019, 25, 5594–5605. [Google Scholar] [CrossRef]
- Egusquiaguirre, S.P.; Yeh, J.E.; Walker, S.R.; Liu, S.; Frank, D.A. The STAT3 target gene TNFRSF1A modulates the NF-kappa b pathway in breast cancer cells. Neoplasia 2018, 20, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Schmid, J.A.; Birbach, A. I kappa B kinase beta (1KK beta/1KK2/1KBKB): A key molecule in signaling to the transcription factor NF-kappa B. Cytokine Growth Factor Rev. 2008, 19, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, Y.; Wu, H.; Song, Y.L.; Shivalila, C.S.; Markoulaki, S.; Jaenisch, R. Parent-of-origin DNA methylation dynamics during mouse development. Cell Rep. 2016, 16, 3167–3180. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, X.W.; Zhou, Y.B.; Lee, M.J.; Guo, L.; Han, W.; Mo, W.; Cao, W.-M.; Sun, D.; Xie, R.; et al. Decoding the dynamic DNA methylation and hydroxymethylation landscapes in endodermal lineage intermediates during pancreatic differentiation of hESC. Nucleic Acids Res. 2018, 46, 2883–2900. [Google Scholar] [CrossRef]
- Fang, X.; Zhao, Z.; Yu, H.; Li, G.; Jiang, P.; Yang, Y.; Yang, R.; Yu, X. Comparative genome-wide methylation analysis of longissimus dorsi muscles between Japanese black (Wagyu) and Chinese Red Steppes cattle. PLoS ONE 2017, 12, e0182492. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.R.; Busche, S.; Ge, B.; Kwan, T.; Pastinen, T.; Blanchette, M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014, 15, 1–17. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Zhang, J.; Han, B.; Zheng, W.; Lin, S.; Li, H.; Gao, Y.; Sun, D. Genome-Wide DNA Methylation Profile in Jejunum Reveals the Potential Genes Associated With Paratuberculosis in Dairy Cattle. Front. Genet. 2021, 12, 735147. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 2019, 35, 421–432. [Google Scholar] [CrossRef]
- Park, Y.; Hao, W. Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics 2016, 32, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; Von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Z.; You, J.H.; Zhong, B.S.; Ren, C.F.; Zhang, Y.L.; Meng, L.; Zhang, G.; Jia, R.; Ying, S.; Wang, F. Scd1 mammary-specific vector constructed and overexpressed in goat fibroblast cells resulting in an increase of palmitoleic acid and oleic acid. Biochem. Bioph. Res. Commun. 2014, 443, 389–394. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Samples | Clean Bases (Gb) | Clean Reads | GC (%) | Q 30 (%) | Mapping Rate (%) | Bs Conversion Rate (%) | mC Percent (%) |
|---|---|---|---|---|---|---|---|---|
| Cases | Case1 | 76.94 | 280,373,522 | 23.22 | 91.9 | 72.05 | 99.742 | 3.13 |
| Case2 | 78.94 | 287,142,601 | 22.86 | 92.59 | 72.12 | 99.717 | 3.21 | |
| Case3 | 78.90 | 287,493,079 | 22.81 | 91.37 | 72.05 | 99.703 | 3.19 | |
| Controls | Control1 | 75.60 | 277,034,171 | 22.77 | 89.35 | 70.79 | 99.702 | 3.05 |
| Control2 | 74.03 | 272,716,162 | 22.73 | 88.91 | 70.42 | 99.673 | 3.07 | |
| Control3 | 76.73 | 279,618,157 | 23.05 | 91.34 | 73.19 | 99.707 | 3.39 |
| RNA-Seq | WGBS-Seq | |||
|---|---|---|---|---|
| Gene | Regulation | Meth Chr | Annotation | Stat |
| LTA | Underexpressed | 23 | exon, utr5, TSS, promoter, | hyper |
| IRAK1 | Underexpressed | X | exon, intron, utr5, TSS promoter | hyper |
| CSK | Underexpressed | 21 | intron, exon, utr5 | hyper |
| STAT3 | Overexpressed | 19 | intron | hypo |
| IKBKG | Overexpressed | X | TSS, exon, utr5, intron, promoter | hypo |
| NOD2 | Overexpressed | 18 | exon | hypo |
| TLR2 | Overexpressed | 17 | exon | hypo |
| TNFRSF1A | Overexpressed | 5 | promoter, intron, exon, utr3 | hypo |
| IKBKB | Underexpressed | 27 | intron | hypo |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, H.; Fang, C.; Liu, L.-L.; Farnir, F.; Liu, W.-J. Identification of Susceptibility Genes Underlying Bovine Respiratory Disease in Xinjiang Brown Cattle Based on DNA Methylation. Int. J. Mol. Sci. 2024, 25, 4928. https://doi.org/10.3390/ijms25094928
Cao H, Fang C, Liu L-L, Farnir F, Liu W-J. Identification of Susceptibility Genes Underlying Bovine Respiratory Disease in Xinjiang Brown Cattle Based on DNA Methylation. International Journal of Molecular Sciences. 2024; 25(9):4928. https://doi.org/10.3390/ijms25094928
Chicago/Turabian StyleCao, Hang, Chao Fang, Ling-Ling Liu, Frederic Farnir, and Wu-Jun Liu. 2024. "Identification of Susceptibility Genes Underlying Bovine Respiratory Disease in Xinjiang Brown Cattle Based on DNA Methylation" International Journal of Molecular Sciences 25, no. 9: 4928. https://doi.org/10.3390/ijms25094928
APA StyleCao, H., Fang, C., Liu, L.-L., Farnir, F., & Liu, W.-J. (2024). Identification of Susceptibility Genes Underlying Bovine Respiratory Disease in Xinjiang Brown Cattle Based on DNA Methylation. International Journal of Molecular Sciences, 25(9), 4928. https://doi.org/10.3390/ijms25094928