
Synergistic Effects of Weight Loss and Catheter Ablation: Can microRNAs Serve as Predictive Biomarkers for the Prevention of Atrial Fibrillation Recurrence?




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Risk Factors for the Development of AF and the Possible Use of Circulating Associated miR as Biomarker for Pathological Remodeling and Therapeutic Reverse Modeling
2.1. Obesity, Adipose Tissue, and AF
2.2. Hypertension
2.3. Sex Differences in AF
2.4. Obstructive Sleep Apnea (OSA)
2.5. Aging/Inflammaging
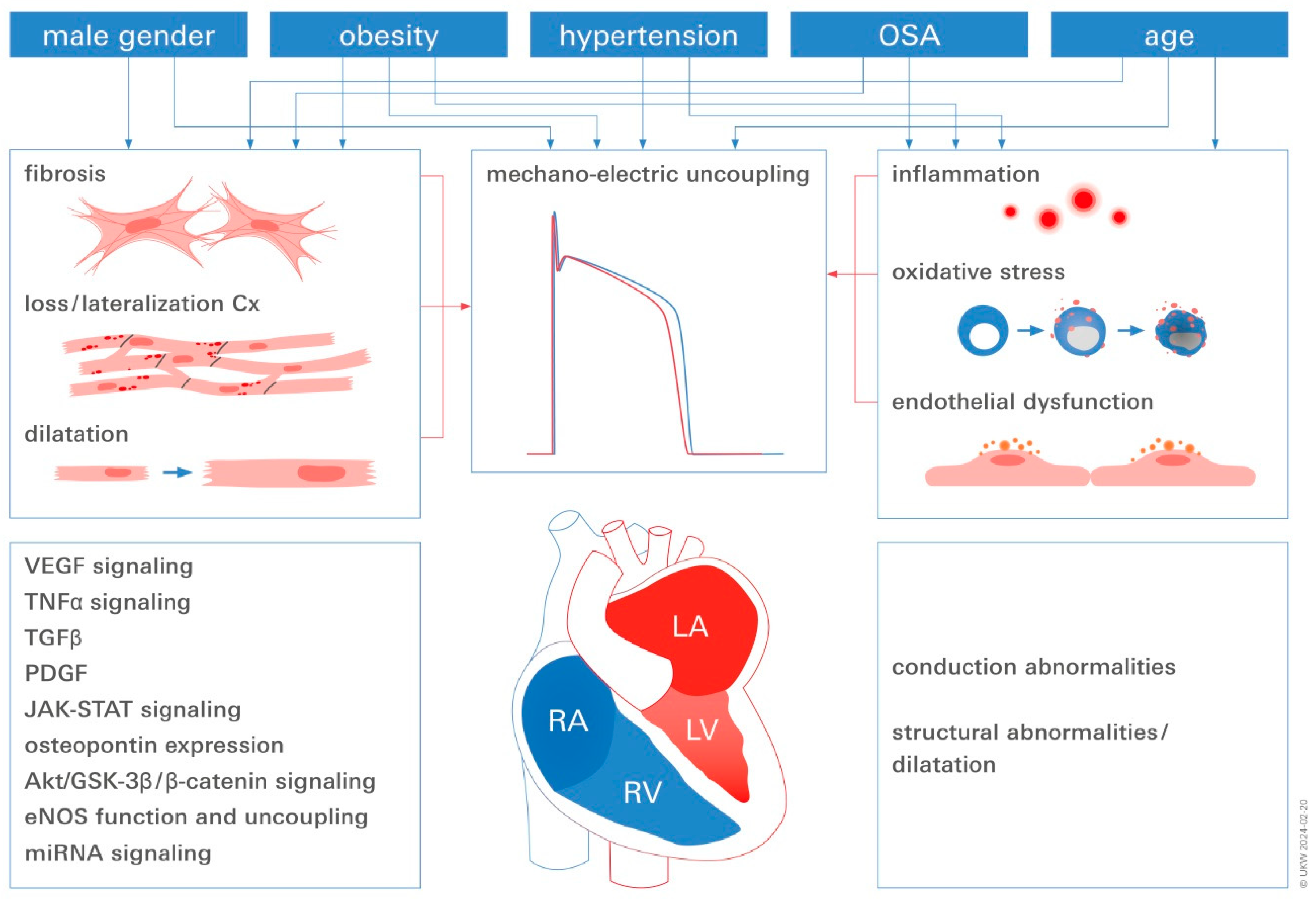
3. Converging Central Pathomechanisms Underlying AF Pathogenesis and Progression
3.1. Inflammation
3.2. Endothelial Dysfunction (ED)
3.3. AF and Mechano-Electric Coupling
3.4. Fibrosis in Obese AF Patients
3.5. The Relationship between Obesity, Oxidative Stress, and AF
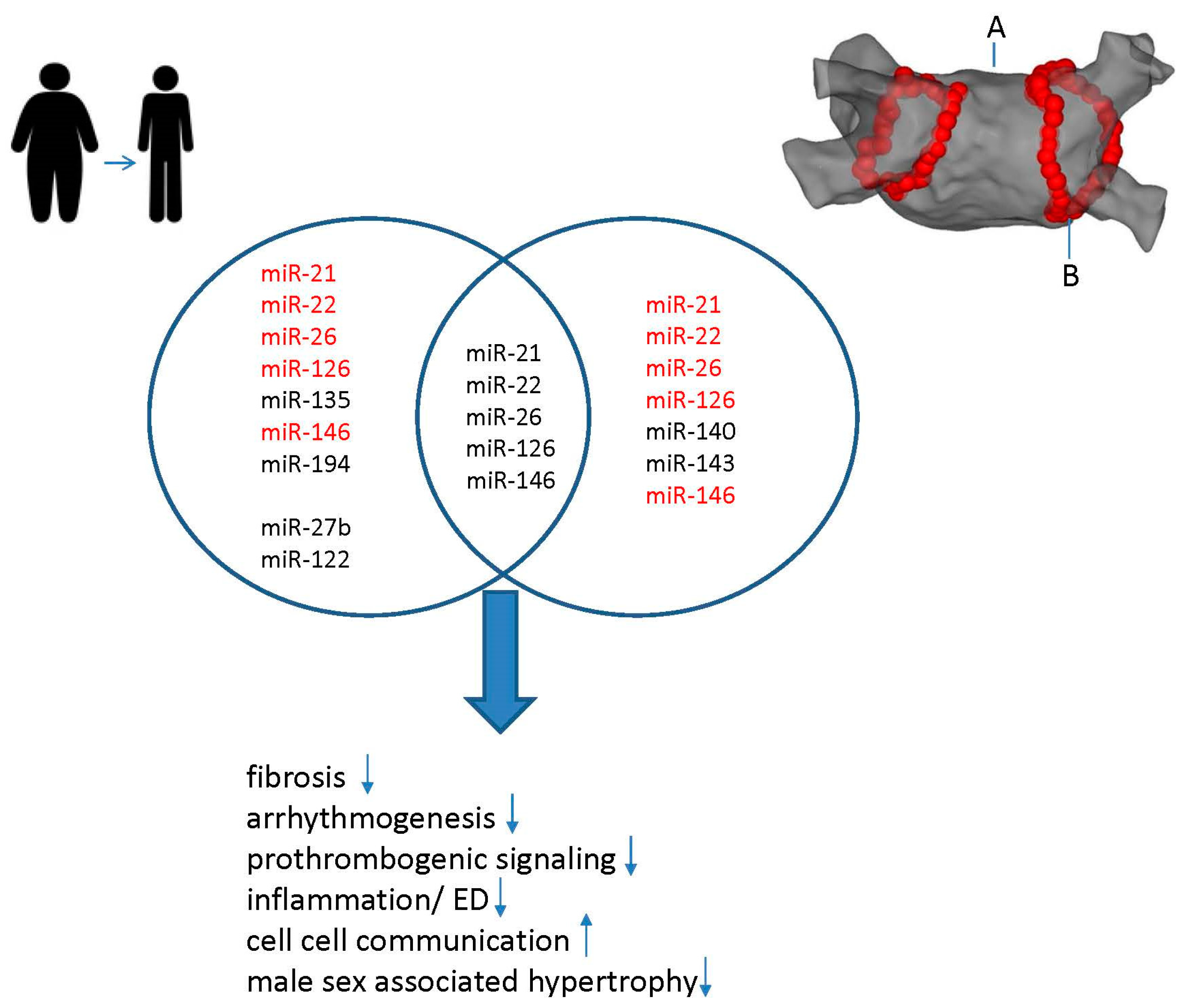
4. Synergistic Effects of Weight Loss and CA on miR Modulation in AF
Effects of CA on miR Modulation
5. Concluding Remarks
Limitations
6. Materials and Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Shu, H.; Cheng, J.; Li, N.; Zhang, Z.; Nie, J.; Peng, Y.; Wang, Y.; Wang, D.W.; Zhou, N. Obesity and atrial fibrillation: A narrative review from arrhythmogenic mechanisms to clinical significance. Cardiovasc. Diabetol. 2023, 22, 192. [Google Scholar] [CrossRef]
- Donnellan, E.; Wazni, O.M.; Elshazly, M.; Kanj, M.; Hussein, A.A.; Baranowski, B.; Kochar, A.; Trulock, K.; Aminian, A.; Schauer, P.; et al. Impact of Bariatric Surgery on Atrial Fibrillation Type. Circ. Arrhythmia Electrophysiol. 2020, 13, e007626. [Google Scholar] [CrossRef]
- Ahammed, M.R.; Ananya, F.N. Impact of Weight Loss on Atrial Fibrillation. Cureus 2023, 15, e46232. [Google Scholar] [CrossRef]
- Mahajan, R.; Lau, D.H.; Brooks, A.G.; Shipp, N.J.; Wood, J.P.M.; Manavis, J.; Samuel, C.S.; Patel, K.P.; Finnie, J.W.; Alasady, M.; et al. Atrial Fibrillation and Obesity: Reverse Remodeling of Atrial Substrate with Weight Reduction. JACC Clin. Electrophysiol. 2021, 7, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Joglar, J.A.; Chung, M.K.; Armbruster, A.L.; Benjamin, E.J.; Chyou, J.Y.; Cronin, E.M.; Deswal, A.; Eckhardt, L.L.; Goldberger, Z.D.; Gopinathannair, R.; et al. 2023 ACC/AHA/ACCP/HRS Guideline for the Diagnosis and Management of Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2024, 149, e1–e156. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, K.H.; Jafry, A.H.; Beard, C.; Nasir, Y.M.; Agarwal, S.; Khan, J.; Clifton, S.; Reece, J.; Munir, M.B.; Deshmukh, A.; et al. The effect of weight loss on recurrence of atrial fibrillation after catheter ablation: A systematic review and meta-analysis. J. Cardiovasc. Electrophysiol. 2023, 34, 2514–2526. [Google Scholar] [CrossRef]
- Dyleva, Y.A.; Gruzdeva, O.V. [MicroRNA and obesity. A modern view of the problem (review of literature).]. Klin. Lab. Diagn. 2020, 65, 411–417. [Google Scholar] [CrossRef]
- Heyn, G.S.; Correa, L.H.; Magalhaes, K.G. The Impact of Adipose Tissue-Derived miRNAs in Metabolic Syndrome, Obesity, and Cancer. Front. Endocrinol. 2020, 11, 563816. [Google Scholar] [CrossRef] [PubMed]
- Suthahar, N.; Meijers, W.C.; Sillje, H.H.W.; de Boer, R.A. From Inflammation to Fibrosis-Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr. Heart Fail. Rep. 2017, 14, 235–250. [Google Scholar] [CrossRef]
- Kunzel, S.R.; Hoffmann, M.; Weber, S.; Kunzel, K.; Kammerer, S.; Gunscht, M.; Klapproth, E.; Rausch, J.S.E.; Sadek, M.S.; Kolanowski, T.; et al. Diminished PLK2 Induces Cardiac Fibrosis and Promotes Atrial Fibrillation. Circ. Res. 2021, 129, 804–820. [Google Scholar] [CrossRef]
- Iacobellis, G. Epicardial adipose tissue in contemporary cardiology. Nat. Rev. Cardiol. 2022, 19, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Elsanhoury, A.; Nelki, V.; Kelle, S.; Van Linthout, S.; Tschope, C. Epicardial Fat Expansion in Diabetic and Obese Patients With Heart Failure and Preserved Ejection Fraction-A Specific HFpEF Phenotype. Front. Cardiovasc. Med. 2021, 8, 720690. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, S.; Da, S.; Aronow, W.S. Risk factors modification in atrial fibrillation: A brief review. Expert Rev. Cardiovasc. Ther. 2023, 22, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Middeldorp, M.E.; Sandhu, R.K.; Mao, J.; Gencer, B.; Danik, J.S.; Moorthy, V.; Cook, N.R.; Albert, C.M. Risk Factors for the Development of New-Onset Persistent Atrial Fibrillation: Subanalysis of the VITAL Study. Circ. Arrhythmia Electrophysiol. 2023, 16, e012334. [Google Scholar] [CrossRef] [PubMed]
- Jamaly, S.; Carlsson, L.; Peltonen, M.; Jacobson, P.; Sjostrom, L.; Karason, K. Bariatric Surgery and the Risk of New-Onset Atrial Fibrillation in Swedish Obese Subjects. J. Am. Coll. Cardiol. 2016, 68, 2497–2504. [Google Scholar] [CrossRef] [PubMed]
- Folli, F.; Centofanti, L.; Magnani, S.; Tagliabue, E.; Bignotto, M.; La Sala, L.; Pontiroli, A.E. Obesity effect on newly diagnosed and recurrent post-ablation atrial fibrillation: A systematic review and meta-analysis. J. Endocrinol. Investig. 2023, 47, 1051–1066. [Google Scholar] [CrossRef]
- Gaborit, B.; Venteclef, N.; Ancel, P.; Pelloux, V.; Gariboldi, V.; Leprince, P.; Amour, J.; Hatem, S.N.; Jouve, E.; Dutour, A.; et al. Human epicardial adipose tissue has a specific transcriptomic signature depending on its anatomical peri-atrial, peri-ventricular, or peri-coronary location. Cardiovasc. Res. 2015, 108, 62–73. [Google Scholar] [CrossRef]
- Haemers, P.; Hamdi, H.; Guedj, K.; Suffee, N.; Farahmand, P.; Popovic, N.; Claus, P.; LePrince, P.; Nicoletti, A.; Jalife, J.; et al. Atrial fibrillation is associated with the fibrotic remodelling of adipose tissue in the subepicardium of human and sheep atria. Eur. Heart J. 2017, 38, 53–61. [Google Scholar] [CrossRef]
- Kusayama, T.; Furusho, H.; Kashiwagi, H.; Kato, T.; Murai, H.; Usui, S.; Kaneko, S.; Takamura, M. Inflammation of left atrial epicardial adipose tissue is associated with paroxysmal atrial fibrillation. J. Cardiol. 2016, 68, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Goeller, M.; Achenbach, S.; Marwan, M.; Doris, M.K.; Cadet, S.; Commandeur, F.; Chen, X.; Slomka, P.J.; Gransar, H.; Cao, J.J.; et al. Epicardial adipose tissue density and volume are related to subclinical atherosclerosis, inflammation and major adverse cardiac events in asymptomatic subjects. J. Cardiovasc. Comput. Tomogr. 2018, 12, 67–73. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, F.; Yang, M.; Zhong, J. Increasing Level of Interleukin-1beta in Epicardial Adipose Tissue Is Associated with Persistent Atrial Fibrillation. J. Interferon Cytokine Res. 2020, 40, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Everett, T.H.t.; Olgin, J.E. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm 2007, 4, S24–S27. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shen, H.; Min, J.; Gao, Y.; Liu, K.; Xi, W.; Yang, J.; Yin, L.; Xu, J.; Xiao, J.; et al. YKL-40 is highly expressed in the epicardial adipose tissue of patients with atrial fibrillation and associated with atrial fibrosis. J. Transl. Med. 2018, 16, 229. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xi, W.; Yin, L.; Wang, J.; Shen, H.; Gao, Y.; Min, J.; Zhang, Y.; Wang, Z. Human Epicardial Adipose Tissue cTGF Expression is an Independent Risk Factor for Atrial Fibrillation and Highly Associated with Atrial Fibrosis. Sci. Rep. 2018, 8, 3585. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Guzik, T.J.; Zhang, Y.H.; Zhang, M.H.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res. 2005, 97, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Voigt, N.; Heijman, J.; Wang, Q.; Chiang, D.Y.; Li, N.; Karck, M.; Wehrens, X.H.T.; Nattel, S.; Dobrev, D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 2014, 129, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Farhat, K.; Po, S.S.; Armoundas, A.A.; Stavrakis, S. Autonomic Nervous System and Cardiac Metabolism: Links Between Autonomic and Metabolic Remodeling in Atrial Fibrillation. JACC Clin. Electrophysiol. 2023, 9, 1196–1206. [Google Scholar] [CrossRef]
- Kallistratos, M.S.; Poulimenos, L.E.; Manolis, A.J. Atrial fibrillation and arterial hypertension. Pharmacol. Res. 2018, 128, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Carey, R.M.; Calhoun, D.A.; Bakris, G.L.; Brook, R.D.; Daugherty, S.L.; Dennison-Himmelfarb, C.R.; Egan, B.M.; Flack, J.M.; Gidding, S.S.; Judd, E.; et al. Resistant Hypertension: Detection, Evaluation, and Management: A Scientific Statement From the American Heart Association. Hypertension 2018, 72, e53–e90. [Google Scholar] [CrossRef]
- Dzeshka, M.S.; Shantsila, A.; Shantsila, E.; Lip, G.Y.H. Atrial Fibrillation and Hypertension. Hypertension 2017, 70, 854–861. [Google Scholar] [CrossRef]
- Mills, K.T.; Bundy, J.D.; Kelly, T.N.; Reed, J.E.; Kearney, P.M.; Reynolds, K.; Chen, J.; He, J. Global Disparities of Hypertension Prevalence and Control: A Systematic Analysis of Population-Based Studies From 90 Countries. Circulation 2016, 134, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Assaf, A.Y.; Noujaim, C.; Mekhael, M.; Younes, H.; Chouman, N.; Dhore-Patil, A.; Donnellan, E.; Feng, H.; Shan, B.; Kholmovski, E.G.; et al. Early Remodeling of the Left Atrium Following Catheter Ablation of Atrial Fibrillation: Insights from DECAAFII. JACC Clin. Electrophysiol. 2023, 9, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Pislaru, S.V.; Lin, G.; Scott, C.G.; Lee, A.T.; Asirvatham, S.J.; Pellikka, P.A.; Kane, G.C.; Pislaru, C. Association of Postprocedural Left Atrial Volume and Reservoir Function with Outcomes in Patients with Atrial Fibrillation Undergoing Catheter Ablation. J. Am. Soc. Echocardiogr. 2022, 35, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.M.; Meoni, L.A.; Chu, A.Y.; Wang, N.Y.; Ford, D.E.; Liang, K.Y.; Gallo, J.J.; Klag, M.J. Body mass index and risk of incident hypertension over the life course: The Johns Hopkins Precursors Study. Circulation 2012, 126, 2983–2989. [Google Scholar] [CrossRef] [PubMed]
- Landsberg, L.; Aronne, L.J.; Beilin, L.J.; Burke, V.; Igel, L.I.; Lloyd-Jones, D.; Sowers, J. Obesity-related hypertension: Pathogenesis, cardiovascular risk, and treatment—A position paper of the The Obesity Society and The American Society of Hypertension. Obesity 2013, 21, 8–24. [Google Scholar] [CrossRef]
- Bramlage, P.; Pittrow, D.; Wittchen, H.U.; Kirch, W.; Boehler, S.; Lehnert, H.; Hoefler, M.; Unger, T.; Sharma, A.M. Hypertension in overweight and obese primary care patients is highly prevalent and poorly controlled. Am. J. Hypertens. 2004, 17, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Kockskamper, J.; Pluteanu, F. Left Atrial Myocardium in Arterial Hypertension. Cells 2022, 11, 3157. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Arroyo, O.; Ortega, A.; Redon, J.; Cortes, R. Therapeutic Potential of Extracellular Vesicles in Hypertension-Associated Kidney Disease. Hypertension 2021, 77, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.L.; Armugam, A.; Sepramaniam, S.; Karolina, D.S.; Lim, K.Y.; Lim, J.Y.; Chong, J.P.; Ng, J.Y.; Chen, Y.T.; Chan, M.M.; et al. Circulating microRNAs in heart failure with reduced and preserved left ventricular ejection fraction. Eur. J. Heart Fail. 2015, 17, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Gildea, J.J.; Carlson, J.M.; Schoeffel, C.D.; Carey, R.M.; Felder, R.A. Urinary exosome miRNome analysis and its applications to salt sensitivity of blood pressure. Clin. Biochem. 2013, 46, 1131–1134. [Google Scholar] [CrossRef]
- Perez-Hernandez, J.; Olivares, D.; Forner, M.J.; Ortega, A.; Solaz, E.; Martinez, F.; Chaves, F.J.; Redon, J.; Cortes, R. Urinary exosome miR-146a is a potential marker of albuminuria in essential hypertension. J. Transl. Med. 2018, 16, 228. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wei, Y.; Wang, Z. microRNA-21 and hypertension. Hypertens. Res. 2018, 41, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Desantis, V.; Potenza, M.A.; Sgarra, L.; Nacci, C.; Scaringella, A.; Cicco, S.; Solimando, A.G.; Vacca, A.; Montagnani, M. microRNAs as Biomarkers of Endothelial Dysfunction and Therapeutic Target in the Pathogenesis of Atrial Fibrillation. Int. J. Mol. Sci. 2023, 24, 5307. [Google Scholar] [CrossRef]
- Westerman, S.; Wenger, N. Gender Differences in Atrial Fibrillation: A Review of Epidemiology, Management, and Outcomes. Curr. Cardiol. Rev. 2019, 15, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Linde, C.; Bongiorni, M.G.; Birgersdotter-Green, U.; Curtis, A.B.; Deisenhofer, I.; Furokawa, T.; Gillis, A.M.; Haugaa, K.H.; Lip, G.Y.H.; Van Gelder, I.; et al. Sex differences in cardiac arrhythmia: A consensus document of the European Heart Rhythm Association, endorsed by the Heart Rhythm Society and Asia Pacific Heart Rhythm Society. EP Eur. 2018, 20, 1565–1565ao. [Google Scholar] [CrossRef]
- Friberg, L.; Bergfeldt, L. Atrial fibrillation prevalence revisited. J. Intern. Med. 2013, 274, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Chugh, S.S.; Havmoeller, R.; Narayanan, K.; Singh, D.; Rienstra, M.; Benjamin, E.J.; Gillum, R.F.; Kim, Y.H.; McAnulty, J.H., Jr.; Zheng, Z.J.; et al. Worldwide epidemiology of atrial fibrillation: A Global Burden of Disease 2010 Study. Circulation 2014, 129, 837–847. [Google Scholar] [CrossRef]
- Akoum, N.; Mahnkopf, C.; Kholmovski, E.G.; Brachmann, J.; Marrouche, N.F. Age and sex differences in atrial fibrosis among patients with atrial fibrillation. EP Eur. 2018, 20, 1086–1092. [Google Scholar] [CrossRef]
- Ehdaie, A.; Cingolani, E.; Shehata, M.; Wang, X.; Curtis, A.B.; Chugh, S.S. Sex Differences in Cardiac Arrhythmias: Clinical and Research Implications. Circ. Arrhythmia Electrophysiol. 2018, 11, e005680. [Google Scholar] [CrossRef]
- Staerk, L.; Sherer, J.A.; Ko, D.; Benjamin, E.J.; Helm, R.H. Atrial Fibrillation: Epidemiology, Pathophysiology, and Clinical Outcomes. Circ. Res. 2017, 120, 1501–1517. [Google Scholar] [CrossRef]
- Nattel, S. New ideas about atrial fibrillation 50 years on. Nature 2002, 415, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Burstein, B.; Dobrev, D. Atrial remodeling and atrial fibrillation: Mechanisms and implications. Circ. Arrhythmia Electrophysiol. 2008, 1, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Wijffels, M.C.; Kirchhof, C.J.; Dorland, R.; Allessie, M.A. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995, 92, 1954–1968. [Google Scholar] [CrossRef] [PubMed]
- Duffy, H.S.; Wit, A.L. Is there a role for remodeled connexins in AF? No simple answers. J. Mol. Cell. Cardiol. 2008, 44, 4–13. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kato, T.; Iwasaki, Y.K.; Nattel, S. Connexins and atrial fibrillation: Filling in the gaps. Circulation 2012, 125, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Saffitz, J.E. Connexins, conduction, and atrial fibrillation. N. Engl. J. Med. 2006, 354, 2712–2714. [Google Scholar] [CrossRef] [PubMed]
- Chaldoupi, S.M.; Loh, P.; Hauer, R.N.; de Bakker, J.M.; van Rijen, H.V. The role of connexin40 in atrial fibrillation. Cardiovasc. Res. 2009, 84, 15–23. [Google Scholar] [CrossRef]
- Nattel, S.; Dobrev, D. Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat. Rev. Cardiol. 2016, 13, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Asatryan, B.; Yee, L.; Ben-Haim, Y.; Dobner, S.; Servatius, H.; Roten, L.; Tanner, H.; Crotti, L.; Skinner, J.R.; Remme, C.A.; et al. Sex-Related Differences in Cardiac Channelopathies: Implications for Clinical Practice. Circulation 2021, 143, 739–752. [Google Scholar] [CrossRef]
- Thibault, S.; Ton, A.T.; Huynh, F.; Fiset, C. Connexin Lateralization Contributes to Male Susceptibility to Atrial Fibrillation. Int. J. Mol. Sci. 2022, 23, 106696. [Google Scholar] [CrossRef]
- Wijesurendra, R.S.; Casadei, B. Mechanisms of atrial fibrillation. Heart 2019, 105, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.; Rahman, F.; Schnabel, R.B.; Yin, X.; Benjamin, E.J.; Christophersen, I.E. Atrial fibrillation in women: Epidemiology, pathophysiology, presentation, and prognosis. Nat. Rev. Cardiol. 2016, 13, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Grimaldi, T.; Origliani, G.; Fantini, G.; Coppi, F.; Modena, M.G. Menopause and cardiovascular risk. Pathophysiol. Haemost. Thromb. 2002, 32, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Magnani, J.W.; Moser, C.B.; Murabito, J.M.; Nelson, K.P.; Fontes, J.D.; Lubitz, S.A.; Sullivan, L.M.; Ellinor, P.T.; Benjamin, E.J. Age of natural menopause and atrial fibrillation: The Framingham Heart Study. Am. Heart, J. 2012, 163, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Magnani, J.W.; Moser, C.B.; Murabito, J.M.; Sullivan, L.M.; Wang, N.; Ellinor, P.T.; Vasan, R.S.; Benjamin, E.J.; Coviello, A.D. Association of sex hormones, aging, and atrial fibrillation in men: The Framingham Heart Study. Circ. Arrhythmia Electrophysiol. 2014, 7, 307–312. [Google Scholar] [CrossRef]
- Lai, J.; Zhou, D.; Xia, S.; Shang, Y.; Want, L.; Zheng, L.; Zhu, J. Reduced testosterone levels in males with lone atrial fibrillation. Clin. Cardiol. 2009, 32, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.A.; Shores, M.M.; Matsumoto, A.M.; Buzkova, P.; Lange, L.A.; Kronmal, R.A.; Heckbert, S.R.; Mukamal, K.J. Serum androgens and risk of atrial fibrillation in older men: The Cardiovascular Health Study. Clin. Cardiol. 2018, 41, 830–836. [Google Scholar] [CrossRef]
- Zeller, T.; Schnabel, R.B.; Appelbaum, S.; Ojeda, F.; Berisha, F.; Schulte-Steinberg, B.; Brueckmann, B.E.; Kuulasmaa, K.; Jousilahti, P.; Blankenberg, S.; et al. Low testosterone levels are predictive for incident atrial fibrillation and ischaemic stroke in men, but protective in women—Results from the FINRISK study. Eur. J. Prev. Cardiol. 2018, 25, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Jennings, M.M.; Donahue, J.K. Connexin Remodeling Contributes to Atrial Fibrillation. J. Atr. Fibrillation 2013, 6, 839. [Google Scholar] [CrossRef]
- Li, S.; Jiang, Z.; Wen, L.; Feng, G.; Zhong, G. MicroRNA-208a-3p contributes to connexin40 remolding in human chronic atrial fibrillation. Exp. Ther. Med. 2017, 14, 5355–5362. [Google Scholar] [CrossRef]
- Lu, Z.; Tilly, M.J.; Geurts, S.; Aribas, E.; Roeters van Lennep, J.; de Groot, N.M.S.; Ikram, M.A.; van Rosmalen, J.; Kavousi, M. Sex-specific anthropometric and blood pressure trajectories and risk of incident atrial fibrillation: The Rotterdam Study. Eur. J. Prev. Cardiol. 2022, 29, 1744–1755. [Google Scholar] [CrossRef]
- Gami, A.S.; Hodge, D.O.; Herges, R.M.; Olson, E.J.; Nykodym, J.; Kara, T.; Somers, V.K. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J. Am. Coll. Cardiol. 2007, 49, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Kanagala, R.; Murali, N.S.; Friedman, P.A.; Ammash, N.M.; Gersh, B.J.; Ballman, K.V.; Shamsuzzaman, A.S.; Somers, V.K. Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation 2003, 107, 2589–2594. [Google Scholar] [CrossRef] [PubMed]
- Congrete, S.; Bintvihok, M.; Thongprayoon, C.; Bathini, T.; Boonpheng, B.; Sharma, K.; Chokesuwattanaskul, R.; Srivali, N.; Tanawuttiwat, T.; Cheungpasitporn, W. Effect of obstructive sleep apnea and its treatment of atrial fibrillation recurrence after radiofrequency catheter ablation: A meta-analysis. J. Evid.-Based Med. 2018, 11, 145–151. [Google Scholar] [CrossRef]
- Lopez-Galvez, R.; Rivera-Caravaca, J.M.; Mandaglio-Collados, D.; Orenes-Pinero, E.; Lahoz, A.; Hernandez-Romero, D.; Martinez, C.M.; Carpes, M.; Arribas, J.M.; Canovas, S.; et al. Molecular mechanisms of postoperative atrial fibrillation in patients with obstructive sleep apnea. FASEB J. 2023, 37, e22941. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Stampfer-Kountchev, M.; Robatscher, P.; Veerhuis, R.; Eikelenboom, P.; Grubeck-Loebenstein, B. How chronic inflammation can affect the brain and support the development of Alzheimer’s disease in old age: The role of microglia and astrocytes. Aging Cell 2004, 3, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Shityakov, S.; Nagai, M.; Ergun, S.; Braunger, B.M.; Forster, C.Y. The Protective Effects of Neurotrophins and MicroRNA in Diabetic Retinopathy, Nephropathy and Heart Failure via Regulating Endothelial Function. Biomolecules 2022, 12, 1113. [Google Scholar] [CrossRef]
- Karnati, S.; Guntas, G.; Rajendran, R.; Shityakov, S.; Horing, M.; Liebisch, G.; Kosanovic, D.; Ergun, S.; Nagai, M.; Forster, C.Y. Quantitative Lipidomic Analysis of Takotsubo Syndrome Patients’ Serum. Front. Cardiovasc. Med. 2022, 9, 797154. [Google Scholar] [CrossRef]
- Robles, N.R.; Macias, J.F. Hypertension in the elderly. Cardiovasc. Hematol. Agents Med. Chem. 2015, 12, 136–145. [Google Scholar] [CrossRef]
- Baylis, D.; Bartlett, D.B.; Patel, H.P.; Roberts, H.C. Understanding how we age: Insights into inflammaging. Longev. Health 2013, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Buckley, L.; Sun, C.; Al Rifai, M.; Yu, B.; Nambi, V.; Virani, S.S.; Selvin, E.; Matsushita, K.; Hoogeveen, R.C.; et al. Association of interleukin-6 and interleukin-18 with cardiovascular disease in older adults: Atherosclerosis Risk in Communities study. Eur. J. Prev. Cardiol. 2023, 30, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Petraglia, L.; Poggio, P.; Valerio, V.; Cabaro, S.; Campana, P.; Comentale, G.; Attena, E.; Russo, V.; Pilato, E.; et al. Inflammation and Cardiovascular Diseases in the Elderly: The Role of Epicardial Adipose Tissue. Front. Med. 2022, 9, 844266. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, T.; Grune, J.; Landmesser, U.; Attanasio, P. Detection and modification of biomarkers of inflammation determining successful rhythm control in patients with atrial fibrillation. Biomarkers 2023, 28, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple facets of NF-kappaB in the heart: To be or not to NF-kappaB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Accardi, G.; Caruso, C. Immune-inflammatory responses in the elderly: An update. Immun. Ageing 2018, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Leite, A.R.; Borges-Canha, M.; Cardoso, R.; Neves, J.S.; Castro-Ferreira, R.; Leite-Moreira, A. Novel Biomarkers for Evaluation of Endothelial Dysfunction. Angiology 2020, 71, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Constans, J.; Conri, C. Circulating markers of endothelial function in cardiovascular disease. Clin. Chim. Acta 2006, 368, 33–47. [Google Scholar] [CrossRef]
- Khan, A.A.; Thomas, G.N.; Lip, G.Y.H.; Shantsila, A. Endothelial function in patients with atrial fibrillation. Ann. Med. 2020, 52, 1–11. [Google Scholar] [CrossRef]
- Rossi, V.A.; Laptseva, N.; Nebunu, D.; Haider, T.; Nagele, M.P.; Ruschitzka, F.; Sudano, I.; Flammer, A.J. Impaired retinal micro-vascular function in patients with atrial fibrillation. Int. J. Cardiol. 2023, 398, 131592. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Eickhoff, C.; Garcia-Agundez, A.; Bertone, P.; Paudel, S.S.; Tambe, D.T.; Litzky, L.A.; Cox-Flaherty, K.; Klinger, J.R.; Monaghan, S.F.; et al. Transcriptional profiles of pulmonary artery endothelial cells in pulmonary hypertension. Sci. Rep. 2023, 13, 22534. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.L. Arterial stiffness and hypertension. Clin. Hypertens. 2023, 29, 31. [Google Scholar] [CrossRef] [PubMed]
- Camargo, L.L.; Wang, Y.; Rios, F.J.; McBride, M.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Endoplasmic Reticular Stress Interplay in the Vasculopathy of Hypertension. Can. J. Cardiol. 2023, 39, 1874–1887. [Google Scholar] [CrossRef] [PubMed]
- Piao, X.; Ma, L.; Xu, Q.; Zhang, X.; Jin, C. Noncoding RNAs: Versatile regulators of endothelial dysfunction. Life Sci. 2023, 334, 122246. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.A.; Yamakuchi, M.; Ferlito, M.; Mendell, J.T.; Lowenstein, C.J. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc. Natl. Acad. Sci. USA 2008, 105, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Fan, Z.; Han, Y.; Sun, X.; Dong, C.; Liu, G.; Yin, X.; Liu, L.; Bai, Y.; Yang, B. miR-135 protects against atrial fibrillation by suppressing intracellular calcium-mediated NLRP3 inflammasome activation. J. Cell Commun. Signal. 2023, 17, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Xie, X.; Sun, Y.; He, H.; Huang, H.; Liu, Y.; Wu, H.; Dai, M. Paeonol inhibits NLRP3 mediated inflammation in rat endothelial cells by elevating hyperlipidemic rats plasma exosomal miRNA-223. Eur. J. Pharmacol. 2020, 885, 173473. [Google Scholar] [CrossRef]
- De Jong, A.M.; Maass, A.H.; Oberdorf-Maass, S.U.; Van Veldhuisen, D.J.; Van Gilst, W.H.; Van Gelder, I.C. Mechanisms of atrial structural changes caused by stretch occurring before and during early atrial fibrillation. Cardiovasc. Res. 2011, 89, 754–765. [Google Scholar] [CrossRef]
- Eckstein, J.; Verheule, S.; de Groot, N.M.; Allessie, M.; Schotten, U. Mechanisms of perpetuation of atrial fibrillation in chronically dilated atria. Prog. Biophys. Mol. Biol. 2008, 97, 435–451. [Google Scholar] [CrossRef]
- Ravelli, F. Mechano-electric feedback and atrial fibrillation. Prog. Biophys. Mol. Biol. 2003, 82, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Yang, B.; Nattel, S. MicroRNAs and atrial fibrillation: Mechanisms and translational potential. Nat. Rev. Cardiol. 2015, 12, 80–90. [Google Scholar] [CrossRef]
- Ravelli, F.; Mase, M. MicroRNAs: New contributors to mechano-electric coupling and atrial fibrillation. Prog. Biophys. Mol. Biol. 2021, 159, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hou, S.; Huang, D.; Luo, X.; Zhang, J.; Chen, J.; Xu, W. Expression profile analysis of circulating microRNAs and their effects on ion channels in Chinese atrial fibrillation patients. Int. J. Clin. Exp. Med. 2015, 8, 845–853. [Google Scholar]
- Huang, H.; Chen, H.; Liang, X.; Chen, X.; Chen, C. Upregulated miR-328-3p and its high risk in atrial fibrillation: A systematic review and meta-analysis with meta-regression. Medicine 2022, 101, e28980. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, H. Individual Contributions of Cardiac Ion Channels on Atrial Repolarization and Reentrant Waves: A Multiscale In-Silico Study. J. Cardiovasc. Dev. Dis. 2022, 9, 28. [Google Scholar] [CrossRef]
- Menezes Junior, A.D.S.; Ferreira, L.C.; Barbosa, L.J.V.; Silva, D.M.E.; Saddi, V.A.; Silva, A. Circulating MicroRNAs as Specific Biomarkers in Atrial Fibrillation: A Meta-Analysis. Non-Coding RNA 2023, 9, 13. [Google Scholar] [CrossRef]
- Cao, Y.; Cui, L. Identifying the key microRNAs implicated in atrial fibrillation. Anatol. J. Cardiol. 2021, 25, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Pan, Z.; Shan, H.; Xiao, J.; Sun, X.; Wang, N.; Lin, H.; Xiao, L.; Maguy, A.; Qi, X.Y.; et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J. Clin. Investig. 2013, 123, 1939–1951. [Google Scholar] [CrossRef]
- Jalife, J.; Kaur, K. Atrial remodeling, fibrosis, and atrial fibrillation. Trends Cardiovasc. Med. 2015, 25, 475–484. [Google Scholar] [CrossRef]
- Poulet, C.; Kunzel, S.; Buttner, E.; Lindner, D.; Westermann, D.; Ravens, U. Altered physiological functions and ion currents in atrial fibroblasts from patients with chronic atrial fibrillation. Physiol. Rep. 2016, 4, e12681. [Google Scholar] [CrossRef] [PubMed]
- Xintarakou, A.; Tzeis, S.; Psarras, S.; Asvestas, D.; Vardas, P. Atrial fibrosis as a dominant factor for the development of atrial fibrillation: Facts and gaps. EP Eur. 2020, 22, 342–351. [Google Scholar] [CrossRef]
- Nattel, S. Electrical coupling between cardiomyocytes and fibroblasts: Experimental testing of a challenging and important concept. Cardiovasc. Res. 2018, 114, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Launbo, N.; Zobel, E.H.; von Scholten, B.J.; Faerch, K.; Jorgensen, P.G.; Christensen, R.H. Targeting epicardial adipose tissue with exercise, diet, bariatric surgery or pharmaceutical interventions: A systematic review and meta-analysis. Obes. Rev. 2021, 22, e13136. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, X.; Jia, H.; Wang, H.; Shui, G.; Qin, Y.; Shu, X.; Wang, Y.; Dong, J.; Liu, G. Nuclear Factor E2-Related Factor 2 Mediates Oxidative Stress-Induced Lipid Accumulation in Adipocytes by Increasing Adipogenesis and Decreasing Lipolysis. Antioxid. Redox Signal. 2020, 32, 173–192. [Google Scholar] [CrossRef] [PubMed]
- Samman Tahhan, A.; Sandesara, P.B.; Hayek, S.S.; Alkhoder, A.; Chivukula, K.; Hammadah, M.; Mohamed-Kelli, H.; O’Neal, W.T.; Topel, M.; Ghasemzadeh, N.; et al. Association between oxidative stress and atrial fibrillation. Heart Rhythm 2017, 14, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Yamamoto, M.; Honjo, H.; Kodama, I.; Kamiya, K. The role of gap junctions in stretch-induced atrial fibrillation. Cardiovasc. Res. 2014, 104, 364–370. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pezhouman, A.; Cao, H.; Fishbein, M.C.; Belardinelli, L.; Weiss, J.N.; Karagueuzian, H.S. Atrial Fibrillation Initiated by Early Afterdepolarization-Mediated Triggered Activity during Acute Oxidative Stress: Efficacy of Late Sodium Current Blockade. J. Heart Health 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Choi, E.; Cha, M.J.; Hwang, K.C. Looking into a conceptual framework of ROS-miRNA-atrial fibrillation. Int. J. Mol. Sci. 2014, 15, 21754–21776. [Google Scholar] [CrossRef]
- Liang, X.; Zhang, Q.; Wang, X.; Yuan, M.; Zhang, Y.; Xu, Z.; Li, G.; Liu, T. Reactive oxygen species mediated oxidative stress links diabetes and atrial fibrillation. Mol. Med. Rep. 2018, 17, 4933–4940. [Google Scholar] [CrossRef]
- Filardi, T.; Sabato, C.; Lubrano, C.; Santangelo, C.; Morano, S.; Lenzi, A.; Migliaccio, S.; Ferretti, E.; Catanzaro, G. MicroRNA Modulation by Dietary Supplements in Obesity. Biomedicines 2020, 8, 545. [Google Scholar] [CrossRef] [PubMed]
- Creta, A.; Elliott, P.; Earley, M.J.; Dhinoja, M.; Finlay, M.; Sporton, S.; Chow, A.; Hunter, R.J.; Papageorgiou, N.; Lowe, M.; et al. Catheter ablation of atrial fibrillation in patients with hypertrophic cardiomyopathy: A European observational multicentre study. EP Eur. 2021, 23, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Providencia, R.; Adragao, P.; de Asmundis, C.; Chun, J.; Chierchia, G.; Defaye, P.; Anselme, F.; Creta, A.; Lambiase, P.D.; Schmidt, B.; et al. Impact of Body Mass Index on the Outcomes of Catheter Ablation of Atrial Fibrillation: A European Observational Multicenter Study. J. Am. Heart Assoc. 2019, 8, e012253. [Google Scholar] [CrossRef] [PubMed]
- Marsetti, P.S.; Milagro, F.I.; Zulet, M.A.; Martinez, J.A.; Lorente-Cebrian, S. Changes in miRNA expression with two weight-loss dietary strategies in a population with metabolic syndrome. Nutrition 2021, 83, 111085. [Google Scholar] [CrossRef] [PubMed]
- Garnier, L.F.; Rouesnel, P.; Espitalier, F. Atrial fibrillation and anticoagulation. Arch. Des Mal. Du Coeur Et Des Vaiss. 2004, 97, 1001–1005. [Google Scholar]
- Ahn, H.J.; Lee, S.R.; Choi, E.K.; Han, K.D.; Rhee, T.M.; Kwon, S.; Kim, S.; Oh, S.; Lip, G.Y.H. Associations between obesity parameters and the risk of incident atrial fibrillation and ischaemic stroke in the different age groups. Front. Cardiovasc. Med. 2022, 9, 906844. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Iaccarino, G.; De Luca, N.; Trimarco, B.; Condorelli, G. Atrial fibrillation and microRNAs. Front. Physiol. 2014, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Veie, C.H.B.; Nielsen, I.M.T.; Frisk, N.L.S.; Dalgaard, L.T. Extracellular microRNAs in Relation to Weight Loss-A Systematic Review and Meta-Analysis. Non-Coding RNA 2023, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Wang, Y.; Gao, Y.; Wang, X.; Cui, X.; Zhang, Y.; Pang, L.; Ji, C.; Guo, X.; Chi, X. The role of microRNA-23b-5p in regulating brown adipogenesis and thermogenic program. Endocr. Connect. 2020, 9, 457–470. [Google Scholar] [CrossRef]
- Garcia-Elias, A.; Tajes, M.; Yanez-Bisbe, L.; Enjuanes, C.; Comin-Colet, J.; Serra, S.A.; Fernandez-Fernandez, J.M.; Aguilar-Agon, K.W.; Reilly, S.; Marti-Almor, J.; et al. Atrial Fibrillation in Heart Failure Is Associated with High Levels of Circulating microRNA-199a-5p and 22-5p and a Defective Regulation of Intracellular Calcium and Cell-to-Cell Communication. Int. J. Mol. Sci. 2021, 22, 10377. [Google Scholar] [CrossRef]
- Yue, Q.; Liu, Y.; Ji, J.; Hu, T.; Lin, T.; Yu, S.; Li, S.; Wu, N. Down-regulation of OIP5-AS1 inhibits obesity-induced myocardial pyroptosis and miR-22/NLRP3 inflammasome axis. Immun. Inflamm. Dis. 2023, 11, e1066. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, H.X.; Jena, P.K.; Sheng, L.; Ali, M.R.; Wan, Y.Y. miR-22 inhibition reduces hepatic steatosis via FGF21 and FGFR1 induction. JHEP Rep. Innov. Hepatol. 2020, 2, 100093. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, D.; Ding, C.Z.; Guo, F.; Wu, L.N.; Huang, F.J.; Liu, Y.L.; Zhao, S.Y.; Xin, Y.; Ma, S.N.; et al. MicroRNA-194: A novel regulator of glucagon-like peptide-1 synthesis in intestinal L cells. Cell Death Dis. 2021, 12, 113. [Google Scholar] [CrossRef] [PubMed]
- Ait-Aissa, K.; Nguyen, Q.M.; Gabani, M.; Kassan, A.; Kumar, S.; Choi, S.K.; Gonzalez, A.A.; Khataei, T.; Sahyoun, A.M.; Chen, C.; et al. MicroRNAs and obesity-induced endothelial dysfunction: Key paradigms in molecular therapy. Cardiovasc. Diabetol. 2020, 19, 136. [Google Scholar] [CrossRef] [PubMed]
- Siwaponanan, P.; Kaewkumdee, P.; Sudcharee, P.; Udompunturak, S.; Chomanee, N.; Udol, K.; Pattanapanyasat, K.; Krittayaphong, R. Increased small extracellular vesicle levels and decreased miR-126 levels associated with atrial fibrillation and coexisting diabetes mellitus. Clin. Cardiol. 2023, 46, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Pereira-da-Silva, T.; Napoleao, P.; Costa, M.C.; Gabriel, A.F.; Selas, M.; Silva, F.; Enguita, F.J.; Ferreira, R.C.; Carmo, M.M. Circulating miRNAs Are Associated with the Systemic Extent of Atherosclerosis: Novel Observations for miR-27b and miR-146. Diagnostics 2021, 11, 318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jing, W. Upregulation of miR-122 is associated with cardiomyocyte apoptosis in atrial fibrillation. Mol. Med. Rep. 2018, 18, 1745–1751. [Google Scholar] [CrossRef]
- Takahashi, K.; Sasano, T.; Sugiyama, K.; Kurokawa, J.; Tamura, N.; Soejima, Y.; Sawabe, M.; Isobe, M.; Furukawa, T. High-fat diet increases vulnerability to atrial arrhythmia by conduction disturbance via miR-27b. J. Mol. Cell. Cardiol. 2016, 90, 38–46. [Google Scholar] [CrossRef]
- Dai, M.; Jiang, T.; Luo, C.D.; Du, W.; Wang, M.; Qiu, Q.Y.; Wang, H. Radiofrequency ablation reduces expression of SELF by upregulating the expression of microRNA-26a/b in the treatment of atrial fibrillation. J. Interv. Card. Electrophysiol. 2022, 65, 663–673. [Google Scholar] [CrossRef]
- Sardu, C.; Santamaria, M.; Paolisso, G.; Marfella, R. microRNA expression changes after atrial fibrillation catheter ablation. Pharmacogenomics 2015, 16, 1863–1877. [Google Scholar] [CrossRef]
- Zhou, Q.; Maleck, C.; von Ungern-Sternberg, S.N.I.; Neupane, B.; Heinzmann, D.; Marquardt, J.; Duckheim, M.; Scheckenbach, C.; Stimpfle, F.; Gawaz, M.; et al. Circulating MicroRNA-21 Correlates with Left Atrial Low-Voltage Areas and Is Associated with Procedure Outcome in Patients Undergoing Atrial Fibrillation Ablation. Circ. Arrhythmia Electrophysiol. 2018, 11, e006242. [Google Scholar] [CrossRef] [PubMed]
- Namino, F.; Yamakuchi, M.; Iriki, Y.; Okui, H.; Ichiki, H.; Maenosono, R.; Oketani, N.; Masamoto, I.; Miyata, M.; Horiuchi, M.; et al. Dynamics of Soluble Thrombomodulin and Circulating miRNAs in Patients with Atrial Fibrillation Undergoing Radiofrequency Catheter Ablation. Clin. Appl. Thromb./Hemost. 2019, 25, 3. [Google Scholar] [CrossRef] [PubMed]
- Kiyosawa, N.; Watanabe, K.; Morishima, Y.; Yamashita, T.; Yagi, N.; Arita, T.; Otsuka, T.; Suzuki, S. Exploratory Analysis of Circulating miRNA Signatures in Atrial Fibrillation Patients Determining Potential Biomarkers to Support Decision-Making in Anticoagulation and Catheter Ablation. Int. J. Mol. Sci. 2020, 21, 2444. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Förster, C.Y.; Künzel, S.R.; Shityakov, S.; Stavrakis, S. Synergistic Effects of Weight Loss and Catheter Ablation: Can microRNAs Serve as Predictive Biomarkers for the Prevention of Atrial Fibrillation Recurrence? Int. J. Mol. Sci. 2024, 25, 4689. https://doi.org/10.3390/ijms25094689
Förster CY, Künzel SR, Shityakov S, Stavrakis S. Synergistic Effects of Weight Loss and Catheter Ablation: Can microRNAs Serve as Predictive Biomarkers for the Prevention of Atrial Fibrillation Recurrence? International Journal of Molecular Sciences. 2024; 25(9):4689. https://doi.org/10.3390/ijms25094689
Chicago/Turabian StyleFörster, Carola Y., Stephan R. Künzel, Sergey Shityakov, and Stavros Stavrakis. 2024. "Synergistic Effects of Weight Loss and Catheter Ablation: Can microRNAs Serve as Predictive Biomarkers for the Prevention of Atrial Fibrillation Recurrence?" International Journal of Molecular Sciences 25, no. 9: 4689. https://doi.org/10.3390/ijms25094689
APA StyleFörster, C. Y., Künzel, S. R., Shityakov, S., & Stavrakis, S. (2024). Synergistic Effects of Weight Loss and Catheter Ablation: Can microRNAs Serve as Predictive Biomarkers for the Prevention of Atrial Fibrillation Recurrence? International Journal of Molecular Sciences, 25(9), 4689. https://doi.org/10.3390/ijms25094689