

Vitamin C as Scavenger of Reactive Oxygen Species during Healing after Myocardial Infarction

Abstract
1. Introduction
2. Metabolism of Vitamin C

3. The Role of Vitamin C in Cardiovascular Pathologies
3.1. Role of ROS in the Heart
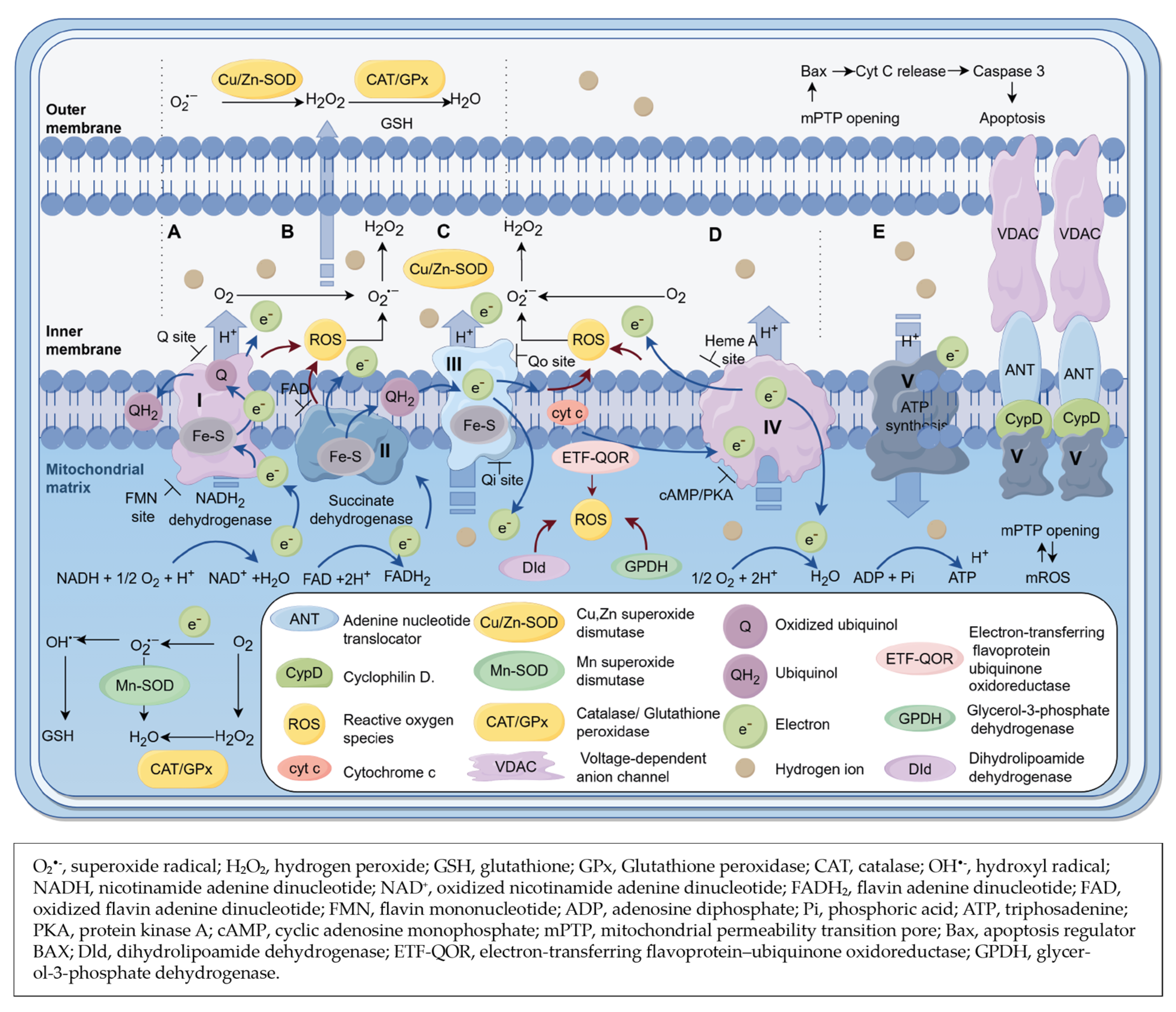
3.2. Cytosolic Sources of ROS (ctROS) in the Heart
3.3. Mitochondrial Sources of ROS (mtROS) in Heart

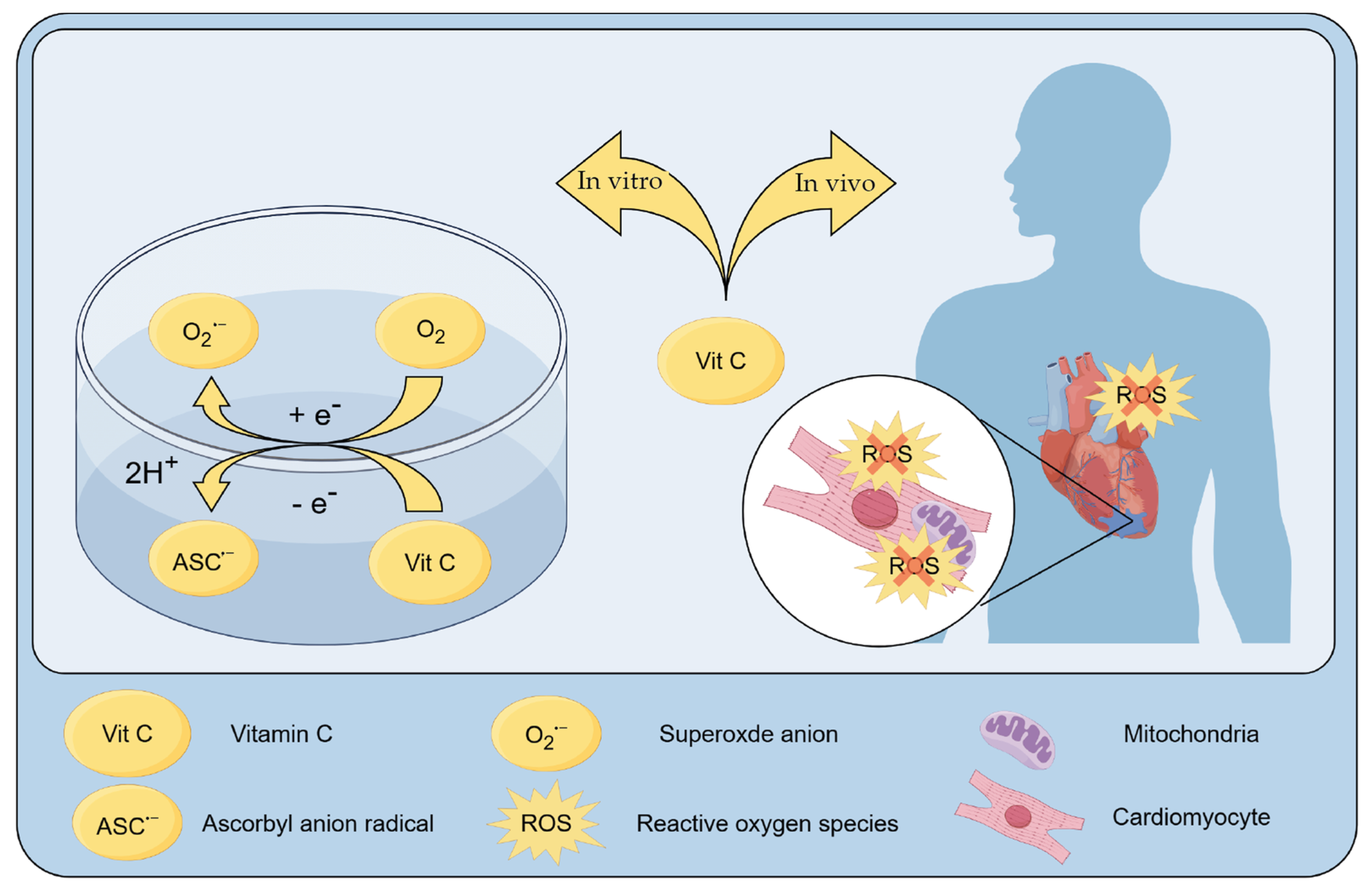
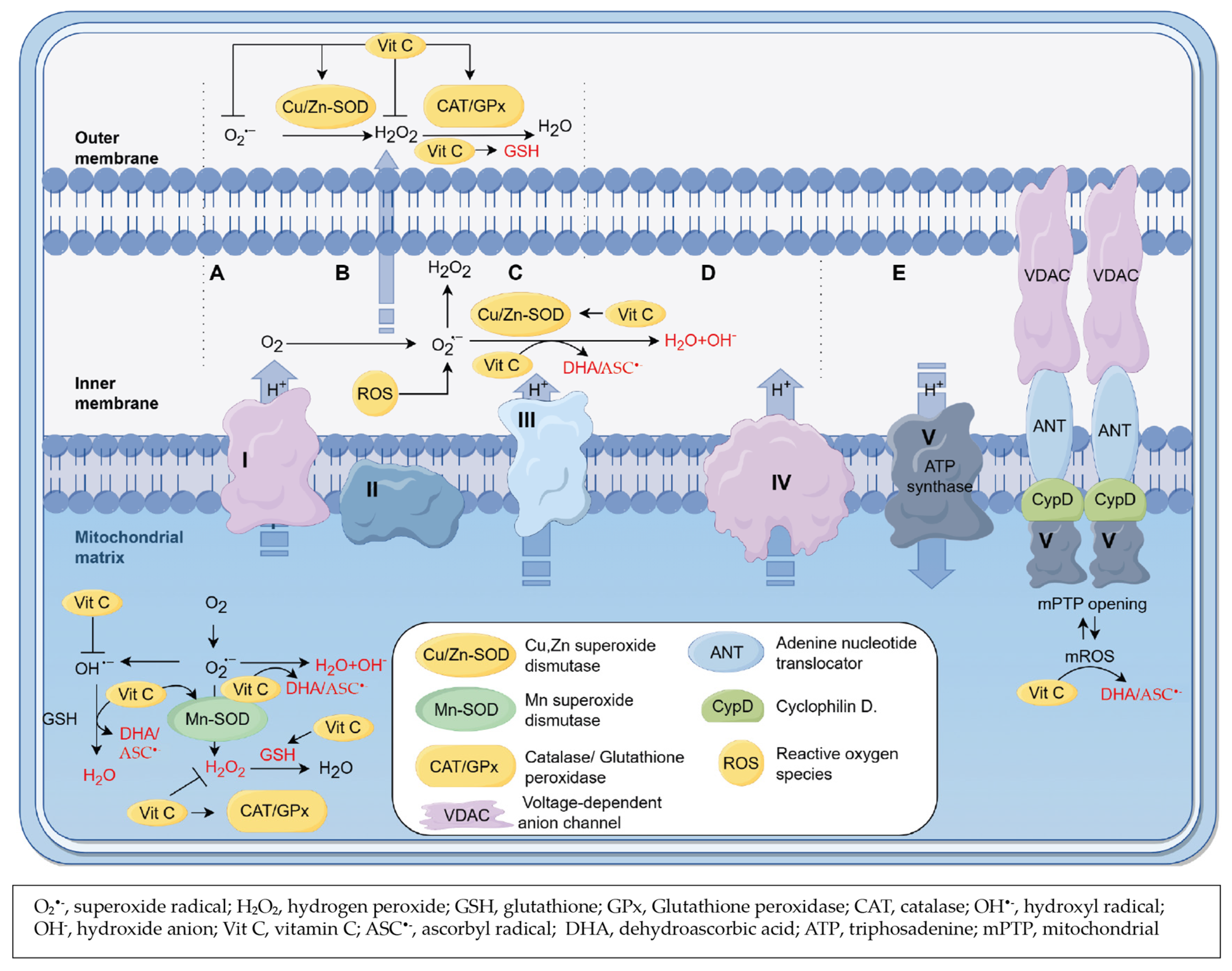
3.4. Role of Vitamin C as a Scavenger of ROS


{kind=link}
{kind=link}
{kind=link}
| Population | Study Size (n) | Age (Years) | Intervention (Doses of Vitamin C) | Trial Duration | Outcomes | Reference |
|---|---|---|---|---|---|---|
| Male with the first MI | 180 | <65 | Habitual | 3 months | Low plasma concentration had no benefit on MI | [35] |
| Patients with sepsis and ARDS present for less than 24 h | 167 | 50 mg/kg in dextrose 5% in water, n = 84 (iv) | 96 h | No improvement of organ dysfunction sores, inflammation and vascular injury | [36] | |
| Male with no prevalent HF | 3919 | 60–79 | Habitual | 11 years | High plasma concentration reduced the risk of HF with and without myocardial infarction | [39] |
| Postmenopausal women with diabetes | 1923 | 55–69 | ≥300 mg/d (orally) | a mean time of 15 years | Higher cardiovascular mortality | [183] |
| Healthy adults | 13421 | a mean of 61 | 320–1110 mg/d (orally) | a mean time of 11 years | Lower cardiovascular mortality | [187] |
| Patients with clinically stable class I or II effort angina pectoris | 56 | a mean of 67 (50–84) years | 16.6 mg/min over 1 h before PCI (iv) | - | Microcirculatory reperfusion improved; Oxidative stress decreased | [188] |
| Patients with asymptomatic adults with elevated coronary calcium scores | 1005 | 50–70 | Combination of vitamin C 1 g, vitamin E 1000 U and atorvastatin 20 mg daily (orally) | a mean duration of 4.3 years | Levels of total cholesterol, low-density lipoprotein cholesterol and triglycerides reduced | [189] |
| Patients with MI | 800 | A mean of 62 | 1000 mg in 12 h (iv) followed by 1200 mg/day combined with vitamin E 600 mg/day (orally) | For 30 days | In-hospital cardiac mortality, non-fatal new myocardial infarction, shock/pulmonary edema, etc. decreased. | [190] |
| Adults without MI or stroke | 14641 | ≥50 | 500 mg/day (orally) | For 8 years | No benefit on MI, stroke or cardiovascular mortality. | [191] |
| Patients with MI | 84 | 53.1 ± 11.2 in the antioxidant treatment group, 55.1 ± 8.4 years in the control group | 250 mL 20% mannitol (iv) over the first hour, 1000 mg of vitamin C in 500 mL 5% glucose (iv) over the first 4 h | - | Short term (e.g., cardiogenic shock), and long term (e.g., left ventricular insufficiency) complications decreased. | [192] |
| Patients with MI | 120 | - | 500 mg, twice per day after MI (orally) | 5 days | Levels of SOD and total thiols increased Levels of XO and malondialdehyde decreased. | [193] |
| Male with MI | 21 | 60.5 ± 6.8 | 2000 mg after MI (orally) | - | Oxidative stress decreased. | [194] |
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hartikainen, T.S.; Sörensen, N.A.; Haller, P.M.; Goßling, A.; Lehmacher, J.; Zeller, T.; Blankenberg, S.; Westermann, D.; Neumann, J.T. Clinical application of the 4th Universal Definition of Myocardial Infarction. Eur. Heart J. 2020, 41, 2209–2216. [Google Scholar] [CrossRef]
- Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [CrossRef]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef]
- Barnett, R. Acute myocardial infarction. Lancet 2019, 393, 2580. [Google Scholar] [CrossRef]
- Fox, K.A.A.; Steg, P.G.; Eagle, K.A.; Goodman, S.G.; Anderson, F.A.; Granger, C.B.; Flather, M.D.; Budaj, A.; Quill, A.; Gore, J.M. Decline in rates of death and heart failure in acute coronary syndromes, 1999–2006. JAMA 2007, 297, 1892–1900. [Google Scholar] [CrossRef]
- Furman, M.I.; Dauerman, H.L.; Goldberg, R.J.; Yarzebski, J.; Lessard, D.; Gore, J.M. Twenty-two year (1975 to 1997) trends in the incidence, in-hospital and long-term case fatality rates from initial Q-wave and non-Q-wave myocardial infarction: A multi-hospital, community-wide perspective. J. Am. Coll. Cardiol. 2001, 37, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Liew, R.; Sulfi, S.; Ranjadayalan, K.; Cooper, J.; Timmis, A.D. Declining case fatality rates for acute myocardial infarction in South Asian and white patients in the past 15 years. Heart 2006, 92, 1030–1034. [Google Scholar] [CrossRef]
- Mandelzweig, L.; Battler, A.; Boyko, V.; Bueno, H.; Danchin, N.; Filippatos, G.; Gitt, A.; Hasdai, D.; Hasin, Y.; Marrugat, J.; et al. The second Euro Heart Survey on acute coronary syndromes: Characteristics, treatment, and outcome of patients with ACS in Europe and the Mediterranean Basin in 2004. Eur. Heart J. 2006, 27, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Latif, F.; Uyeda, L.; Edson, R.; Bhatt, D.L.; Goldman, S.; Holmes, D.R.; Rao, S.V.; Shunk, K.; Aggarwal, K.; Uretsky, B.; et al. Stent-Only Versus Adjunctive Balloon Angioplasty Approach for Saphenous Vein Graft Percutaneous Coronary Intervention: Insights from DIVA Trial. Circ. Cardiovasc. Interv. 2020, 13, e008494. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, G.; Kharbanda, R.K.; Crea, F.; Banning, A.P. No-reflow: Again prevention is better than treatment. Eur. Heart J. 2010, 31, 2449–2455. [Google Scholar] [CrossRef]
- Fan, Q.; Tao, R.; Zhang, H.; Xie, H.; Lu, L.; Wang, T.; Su, M.; Hu, J.; Zhang, Q.; Chen, Q.; et al. Dectin-1 Contributes to Myocardial Ischemia/Reperfusion Injury by Regulating Macrophage Polarization and Neutrophil Infiltration. Circulation 2019, 139, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Peet, C.; Ivetic, A.; Bromage, D.I.; Shah, A.M. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc. Res. 2020, 116, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Feng, Y.; Gong, C.; Bao, X.; Wei, Z.; Chang, L.; Chen, H.; Xu, B. Hypertension-Driven Regulatory T-Cell Perturbations Accelerate Myocardial Ischemia-Reperfusion Injury. Hypertension 2023, 80, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- Mathes, D.; Weirather, J.; Nordbeck, P.; Arias-Loza, A.-P.; Burkard, M.; Pachel, C.; Kerkau, T.; Beyersdorf, N.; Frantz, S.; Hofmann, U. CD4+ Foxp3+ T-cells contribute to myocardial ischemia-reperfusion injury. J. Mol. Cell Cardiol. 2016, 101, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Zhou, R.; Qu, Y.; Yang, Y.; Wang, Z.; Song, N.; Liang, R.; Qian, J. Dexmedetomidine preconditioning mitigates myocardial ischemia/reperfusion injury via inhibition of mast cell degranulation. Biomed. Pharmacother. 2021, 141, 111853. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, S.R. Anti-oxidant therapy: Does it have a role in the treatment of human disease? Expert Opin. Investig. Drugs 1997, 6, 211–236. [Google Scholar] [CrossRef]
- Lim, S.L.; Lam, C.S.P.; Segers, V.F.M.; Brutsaert, D.L.; De Keulenaer, G.W. Cardiac endothelium-myocyte interaction: Clinical opportunities for new heart failure therapies regardless of ejection fraction. Eur. Heart J. 2015, 36, 2050–2060. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Guo, Y.; Pu, W.T. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef]
- Wang, J.; Huang, P.; Yu, Q.; Lu, J.; Liu, P.; Yang, Y.; Feng, Z.; Cai, J.; Yang, G.; Yuan, H.; et al. Epilepsy and long-term risk of arrhythmias. Eur. Heart J. 2023, 44, 3374–3382. [Google Scholar] [CrossRef]
- Luxán, G.; Stewen, J.; Díaz, N.; Kato, K.; Maney, S.K.; Aravamudhan, A.; Berkenfeld, F.; Nagelmann, N.; Drexler, H.C.; Zeuschner, D.; et al. Endothelial EphB4 maintains vascular integrity and transport function in adult heart. Elife 2019, 8, e45863. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Bisaccia, G.; Ricci, F.; Gallina, S.; Di Baldassarre, A.; Ghinassi, B. Mitochondrial Dysfunction and Heart Disease: Critical Appraisal of an Overlooked Association. Int. J. Mol. Sci. 2021, 22, 614. [Google Scholar] [CrossRef] [PubMed]
- Faust, O.; Hong, W.; Loh, H.W.; Xu, S.; Tan, R.-S.; Chakraborty, S.; Barua, P.D.; Molinari, F.; Acharya, U.R. Heart rate variability for medical decision support systems: A review. Comput. Biol. Med. 2022, 145, 105407. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Bungau, S.; Kumar, K.; Zengin, G.; Khan, F.; Kumar, A.; Kaur, R.; Venkatachalam, T.; Tit, D.M.; Vesa, C.M.; et al. Pleotropic Effects of Polyphenols in Cardiovascular System. Biomed. Pharmacother. 2020, 130, 110714. [Google Scholar] [CrossRef]
- Miyazawa, T.; Burdeos, G.C.; Itaya, M.; Nakagawa, K.; Miyazawa, T. Vitamin E: Regulatory Redox Interactions. IUBMB Life 2019, 71, 430–441. [Google Scholar] [CrossRef]
- Berretta, M.; Quagliariello, V.; Maurea, N.; Di Francia, R.; Sharifi, S.; Facchini, G.; Rinaldi, L.; Piezzo, M.; Manuela, C.; Nunnari, G.; et al. Multiple Effects of Ascorbic Acid against Chronic Diseases: Updated Evidence from Preclinical and Clinical Studies. Antioxidants 2020, 9, 1182. [Google Scholar] [CrossRef]
- Salonen, R.M.; Nyyssönen, K.; Kaikkonen, J.; Porkkala-Sarataho, E.; Voutilainen, S.; Rissanen, T.H.; Tuomainen, T.-P.; Valkonen, V.-P.; Ristonmaa, U.; Lakka, H.-M.; et al. Six-year effect of combined vitamin C and E supplementation on atherosclerotic progression: The Antioxidant Supplementation in Atherosclerosis Prevention (ASAP) Study. Circulation 2003, 107, 947–953. [Google Scholar] [CrossRef]
- Bechara, N.; Flood, V.M.; Gunton, J.E. A Systematic Review on the Role of Vitamin C in Tissue Healing. Antioxidants 2022, 11, 1605. [Google Scholar] [CrossRef] [PubMed]
- Tomşa, A.M.; Răchişan, A.L.; Pandrea, S.L.; Benea, A.; Uifălean, A.; Toma, C.; Popa, R.; Pârvu, A.E.; Junie, L.M. Curcumin and Vitamin C Attenuate Gentamicin-Induced Nephrotoxicity by Modulating Distinctive Reactive Species. Metabolites 2022, 13, 49. [Google Scholar] [CrossRef]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Engl. J. Med. 1989, 320, 915–924. [Google Scholar]
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. 1999, 13, 1007–1024. [Google Scholar] [CrossRef]
- Pfister, R.; Sharp, S.J.; Luben, R.; Wareham, N.J.; Khaw, K.T. Plasma vitamin C predicts incident heart failure in men and women in European Prospective Investigation into Cancer and Nutrition-Norfolk prospective study. Am. Heart J. 2011, 162, 246–253. [Google Scholar] [CrossRef]
- Nyyssonen, K.; Parviainen, M.T.; Salonen, R.; Tuomilehto, J.; Salonen, J.T. Vitamin C deficiency and risk of myocardial infarction: Prospective population study of men from eastern Finland. BMJ 1997, 314, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Riemersma, R.A.; Carruthers, K.F.; Elton, R.A.; Fox, K.A. Vitamin C and the risk of acute myocardial infarction. Am. J. Clin. Nutr. 2000, 71, 1181–1186. [Google Scholar] [CrossRef]
- Fowler, A.A., 3rd; Truwit, J.D.; Hite, R.D.; Morris, P.E.; DeWilde, C.; Priday, A.; Fisher, B.; Thacker, L.R., 2nd; Natarajan, R.; Brophy, D.F.; et al. Effect of Vitamin C Infusion on Organ Failure and Biomarkers of Inflammation and Vascular Injury in Patients with Sepsis and Severe Acute Respiratory Failure: The CITRIS-ALI Randomized Clinical Trial. JAMA 2019, 322, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Kushi, L.H.; Folsom, A.R.; Prineas, R.J.; Mink, P.J.; Wu, Y.; Bostick, R.M. Dietary antioxidant vitamins and death from coronary heart disease in postmenopausal women. N. Engl. J. Med. 1996, 334, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Loria, C.M.; Klag, M.J.; Caulfield, L.E.; Whelton, P.K. Vitamin C status and mortality in US adults. Am. J. Clin. Nutr. 2000, 72, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Wannamethee, S.G.; Bruckdorfer, K.R.; Shaper, A.G.; Papacosta, O.; Lennon, L.; Whincup, P.H. Plasma vitamin C, but not vitamin E, is associated with reduced risk of heart failure in older men. Circ. Heart Fail. 2013, 6, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Bánhegyi, G.; Braun, L.; Csala, M.; Puskás, F.; Mandl, J. Ascorbate metabolism and its regulation in animals. Free Radic. Biol. Med. 1997, 23, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, J.C. Dehydroascorbic acid. J. Chromatogr. A 2000, 881, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Cunningham, L.; Rose, R.C. Spontaneous decay of oxidized ascorbic acid (dehydro-L-ascorbic acid) evaluated by high-pressure liquid chromatography. Clin. Chem. 1990, 36, 1807–1809. [Google Scholar] [CrossRef] [PubMed]
- Linster, C.L.; Van Schaftingen, E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007, 274, 1–22. [Google Scholar] [CrossRef]
- Kanfer, J.; Ashwell, G.; Burns, J.J. Formation of L-lyxonic and L-xylonic acids from L-ascorbic acid in rat kidney. J. Biol. Chem. 1960, 235, 2518–2521. [Google Scholar] [CrossRef]
- Buettner, G.R. Ascorbate autoxidation in the presence of iron and copper chelates. Free Radic. Res. Commun. 1986, 1, 349–353. [Google Scholar] [CrossRef]
- Hata, R.; Senoo, H. L-ascorbic acid 2-phosphate stimulates collagen accumulation, cell proliferation, and formation of a three-dimensional tissuelike substance by skin fibroblasts. J. Cell Physiol. 1989, 138, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Frikke-Schmidt, H.; Lykkesfeldt, J. Keeping the intracellular vitamin C at a physiologically relevant level in endothelial cell culture. Anal. Biochem. 2010, 397, 135–137. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Determination of ascorbic acid and dehydroascorbic acid in biological samples by high-performance liquid chromatography using subtraction methods: Reliable reduction with tris[2-carboxyethyl]phosphine hydrochloride. Anal. Biochem. 2000, 282, 89–93. [Google Scholar] [CrossRef]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef]
- Ramachandra, C.J.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Lin, Y.-H.; Hausenloy, D.J. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 2020, 57, 102884. [Google Scholar] [CrossRef]
- Lassegue, B.; Martin, A.S.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef]
- Gurusamy, N.; Mukherjee, S.; Lekli, I.; Bearzi, C.; Bardelli, S.; Das, D.K. Inhibition of ref-1 stimulates the production of reactive oxygen species and induces differentiation in adult cardiac stem cells. Antioxid. Redox Signal. 2009, 11, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Buggisch, M.; Ateghang, B.; Ruhe, C.; Strobel, C.; Lange, S.; Wartenberg, M.; Sauer, H. Stimulation of ES-cell-derived cardiomyogenesis and neonatal cardiac cell proliferation by reactive oxygen species and NADPH oxidase. J. Cell Sci. 2007, 120, 885–894. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.N.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Yun, J.; Rocic, P.; Pung, Y.F.; Belmadani, S.; Carrao, A.C.R.; Ohanyan, V.; Chilian, W.M. Redox-dependent mechanisms in coronary collateral growth: The “redox window” hypothesis. Antioxid. Redox Signal. 2009, 11, 1961–1974. [Google Scholar] [CrossRef]
- Su, B.; Bu, S.D.; Kong, B.H.; Dai, R.X.; Su, Q. Cystatin C alleviates H2O2-induced H9c2 cell injury. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6360–6370. [Google Scholar]
- Jiang, F.; Xu, X.R.; Li, W.M.; Xia, K.; Wang, L.F.; Yang, X.C. Monotropein alleviates H2O2-induced inflammation, oxidative stress and apoptosis via NF-kappaB/AP-1 signaling. Mol. Med. Rep. 2020, 22, 4828–4836. [Google Scholar] [CrossRef] [PubMed]
- Othmene, Y.B.; Monceaux, K.; Karoui, A.; Salem, I.B.; Belhadef, A.; Abid-Essefi, S.; Lemaire, C. Tebuconazole induces ROS-dependent cardiac cell toxicity by activating DNA damage and mitochondrial apoptotic pathway. Ecotoxicol. Environ. Saf. 2020, 204, 111040. [Google Scholar] [CrossRef]
- Elorza, A.A.; Soffia, J.P. mtDNA Heteroplasmy at the Core of Aging-Associated Heart Failure. An Integrative View of OXPHOS and Mitochondrial Life Cycle in Cardiac Mitochondrial Physiology. Front. Cell Dev. Biol. 2021, 9, 625020. [Google Scholar] [CrossRef]
- Francois, A.; Canella, A.; Marcho, L.M.; Stratton, M.S. Protein acetylation in cardiac aging. J. Mol. Cell Cardiol. 2021, 157, 90–97. [Google Scholar] [CrossRef]
- Moris, D.; Spartalis, M.; Tzatzaki, E.; Spartalis, E.; Karachaliou, G.-S.; Triantafyllis, A.S.; Karaolanis, G.I.; Tsilimigras, D.I.; Theocharis, S. The role of reactive oxygen species in myocardial redox signaling and regulation. Ann. Transl. Med. 2017, 5, 324. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Dulak-Lis, M.; Tsiropoulou, S.; Harvey, A.; Briones, A.M.; Touyz, R.M. Oxidative stress and human hypertension: Vascular mechanisms, biomarkers, and novel therapies. Can. J. Cardiol. 2015, 31, 631–641. [Google Scholar] [CrossRef]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Bernal, M.E.; Varon, J.; Acosta, P.; Montagnier, L. Oxidative stress in critical care medicine. Int. J. Clin. Pract. 2010, 64, 1480–1488. [Google Scholar] [CrossRef]
- Weiss, J.N.; Korge, P.; Honda, H.M.; Ping, P. Role of the mitochondrial permeability transition in myocardial disease. Circ. Res. 2003, 93, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yan, Y.; Niu, F.; Wang, Y.; Chen, X.; Su, G.; Liu, Y.; Zhao, X.; Qian, L.; Liu, P.; et al. Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021, 7, 193. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Park, T.-J.; Park, J.H.; Lee, G.S.; Lee, J.-Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.-Y.; Oh, K.-J.; Han, B.-S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 835. [Google Scholar] [CrossRef]
- Chung, B.; Wang, Y.; Thiel, M.; Rostami, F.; Rogoll, A.; Hirsch, V.G.; Malik, Z.; Bührke, A.; Bär, C.; Klintschar, M.; et al. Pre-emptive iron supplementation prevents myocardial iron deficiency and attenuates adverse remodelling after myocardial infarction. Cardiovasc. Res. 2023, 119, 1969–1980. [Google Scholar] [CrossRef]
- Ju, J.; Li, X.-M.; Zhao, X.-M.; Li, F.-H.; Wang, S.-C.; Wang, K.; Li, R.-F.; Zhou, L.-Y.; Liang, L.; Wang, Y.; et al. Circular RNA FEACR inhibits ferroptosis and alleviates myocardial ischemia/reperfusion injury by interacting with NAMPT. J. Biomed. Sci. 2023, 30, 45. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Bernardo, A.; Davies, K.J.A. What is the concentration of hydrogen peroxide in blood and plasma? Arch. Biochem. Biophys. 2016, 603, 48–53. [Google Scholar] [CrossRef]
- Chen, Z.; Oberley, T.D.; Ho, Y.-S.; Chua, C.C.; Siu, B.; Hamdy, R.C.; Epstein, C.J.; Chua, B.H. Overexpression of CuZnSOD in coronary vascular cells attenuates myocardial ischemia/reperfusion injury. Free Radic. Biol. Med. 2000, 29, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Gan, T.-Y.; Zhang, R.-C.; Zhang, Y.-H. miR-23a Regulates Cardiomyocyte Apoptosis by Targeting Manganese Superoxide Dismutase. Mol. Cells 2017, 40, 542–549. [Google Scholar] [CrossRef]
- Simmons, T.W.; Jamall, I.S. Relative importance of intracellular glutathione peroxidase and catalase in vivo for prevention of peroxidation to the heart. Cardiovasc. Res. 1989, 23, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belardinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation 2015, 131, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zheng, H.; Nilcham, P.; Bucur, O.; Vogt, F.; Slabu, I.; Liehn, E.A.; Rusu, M. Vitamin C Regulates the Profibrotic Activity of Fibroblasts in In Vitro Replica Settings of Myocardial Infarction. Int. J. Mol. Sci. 2023, 24, 8379. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef]
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell Cardiol. 2014, 73, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Johar, S.; Cave, A.C.; Narayanapanicker, A.; Grieve, D.J.; Shah, A.M.; Johar, S.; Cave, A.C.; Narayanapanicker, A.; Grieve, D.J.; Shah, A.M. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. 2006, 20, 1546–1548. [Google Scholar] [CrossRef]
- Cau, S.B.; Guimaraes, D.A.; Rizzi, E.; Ceron, C.S.; Gerlach, R.F.; Tanus-Santos, J.E. The Nuclear Factor kappaB Inhibitor Pyrrolidine Dithiocarbamate Prevents Cardiac Remodelling and Matrix Metalloproteinase-2 Up-Regulation in Renovascular Hypertension. Basic Clin. Pharmacol. Toxicol. 2015, 117, 234–241. [Google Scholar] [CrossRef]
- Ryu, H.M.; Kim, Y.J.; Oh, E.J.; Oh, S.H.; Choi, J.Y.; Cho, J.H.; Kim, C.D.; Park, S.H.; Kim, Y.L. Hypoxanthine induces cholesterol accumulation and incites atherosclerosis in apolipoprotein E-deficient mice and cells. J. Cell. Mol. Med. 2016, 20, 2160–2172. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, J.; Hu, S.; Zhu, H.; Toanc, S.; Ren, J. BI1 alleviates cardiac microvascular ischemia-reperfusion injury via modifying mitochondrial fission and inhibiting XO/ROS/F-actin pathways. J. Cell. Physiol. 2019, 234, 5056–5069. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Zhang, M.; Prosser, B.L.; Bamboye, M.A.; Gondim, A.N.; Santos, C.X.; Martin, D.; Ghigo, A.; Perino, A.; Brewer, A.C.; Ward, C.W. Contractile function during angiotensin-II activation: Increased Nox2 activity modulates cardiac calcium handling via phospholamban phosphorylation. J. Am. Coll. Cardiol. 2015, 66, 261–272. [Google Scholar] [CrossRef]
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH oxidases in heart failure: Poachers or gamekeepers? Antioxid. Redox Signal. 2013, 18, 1024–1041. [Google Scholar] [CrossRef]
- Zhang, M.; Brewer, A.C.; Schröder, K.; Santos, C.X.; Grieve, D.J.; Wang, M.; Anilkumar, N.; Yu, B.; Dong, X.; Walker, S.J. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18121–18126. [Google Scholar] [CrossRef]
- Smyrnias, I.; Zhang, X.; Zhang, M.; Murray, T.V.; Brandes, R.P.; Schröder, K.; Brewer, A.C.; Shah, A.M. Nicotinamide adenine dinucleotide phosphate oxidase-4–dependent upregulation of nuclear factor erythroid–derived 2-like 2 protects the heart during chronic pressure overload. Hypertension 2015, 65, 547–553. [Google Scholar] [CrossRef]
- Brewer, A.C.; Murray, T.V.; Arno, M.; Zhang, M.; Anilkumar, N.P.; Mann, G.E.; Shah, A.M. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic. Biol. Med. 2011, 51, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Volpe, M.; Sadoshima, J. NOX4 regulates autophagy during energy deprivation. Autophagy 2014, 10, 699–701. [Google Scholar] [CrossRef] [PubMed]
- Nomura, J.; Busso, N.; Ives, A.; Matsui, C.; Tsujimoto, S.; Shirakura, T.; Tamura, M.; Kobayashi, T.; So, A.; Yamanaka, Y. Xanthine oxidase inhibition by febuxostat attenuates experimental atherosclerosis in mice. Sci. Rep. 2014, 4, 4554. [Google Scholar] [CrossRef]
- Takimoto, E.; Champion, H.C.; Li, M.; Ren, S.; Rodriguez, E.R.; Tavazzi, B.; Lazzarino, G.; Paolocci, N.; Gabrielson, K.L.; Wang, Y.; et al. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J. Clin. Investig. 2005, 115, 1221–1231. [Google Scholar] [CrossRef]
- Okazaki, T.; Otani, H.; Shimazu, T.; Yoshioka, K.; Fujita, M.; Katano, T.; Ito, S.; Iwasaka, T. Reversal of inducible nitric oxide synthase uncoupling unmasks tolerance to ischemia/reperfusion injury in the diabetic rat heart. J. Mol. Cell. Cardiol. 2011, 50, 534–544. [Google Scholar] [PubMed]
- Gottlieb, R.A. Cytochrome P450: Major player in reperfusion injury. Arch. Biochem. Biophys. 2003, 420, 262–267. [Google Scholar] [PubMed]
- Granville, D.; Tashakkor, B.; Takeuchi, C.; Gustafsson, A.B.; Huang, C.; Sayen, M.R.; Wentworth, P., Jr.; Yeager, M.; Gottlieb, R.A. Reduction of ischemia and reperfusion-induced myocardial damage by cytochrome P450 inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 1321–1326. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Rupp, H.; Dhalla, K.S.; Dhalla, N.S. Mechanisms of cardiac cell damage due to catecholamines: Significance of drugs regulating central sympathetic outflow. J. Cardiovasc. Pharmacol. 1994, 24, S16–S24. [Google Scholar] [CrossRef]
- Zhang, G.-X.; Kimura, S.; Nishiyama, A.; Shokoji, T.; Rahman, M.; Yao, L.; Nagai, Y.; Fujisawa, Y.; Miyatake, A.; Abe, Y. Cardiac oxidative stress in acute and chronic isoproterenol-infused rats. Cardiovasc. Res. 2005, 65, 230–238. [Google Scholar] [CrossRef]
- Oknińska, M.; Zambrowska, Z.; Zajda, K.; Paterek, A.; Brodaczewska, K.; Mackiewicz, U.; Szczylik, C.; Torbicki, A.; Kieda, C.; Mączewski, M. Right ventricular myocardial oxygen tension is reduced in monocrotaline-induced pulmonary hypertension in the rat and restored by myo-inositol trispyrophosphate. Sci. Rep. 2021, 11, 18002. [Google Scholar] [CrossRef]
- Balachander, K.; Priyadharsini, J.V.; Paramasivam, A. Role of exosomal mitochondria in cardiovascular diseases. Hypertens. Res. 2023, 46, 812–813. [Google Scholar] [CrossRef]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetić, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell Dev. Biol. 2020, 8, 624216. [Google Scholar] [CrossRef]
- Wagner, M.; Bertero, E.; Nickel, A.; Kohlhaas, M.; Gibson, G.E.; Heggermont, W.; Heymans, S.; Maack, C. Selective NADH communication from α-ketoglutarate dehydrogenase to mitochondrial transhydrogenase prevents reactive oxygen species formation under reducing conditions in the heart. Basic Res. Cardiol. 2020, 115, 53. [Google Scholar] [CrossRef]
- Edalat, A.; Schulte-Mecklenbeck, P.; Bauer, C.; Undank, S.; Krippeit-Drews, P.; Drews, G.; Düfer, M. Mitochondrial succinate dehydrogenase is involved in stimulus-secretion coupling and endogenous ROS formation in murine beta cells. Diabetologia 2015, 58, 1532–1541. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Gerencser, A.A.; Treberg, J.R.; Brand, M.D. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J. Biol. Chem. 2011, 286, 31361–31372. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Perevoschikova, I.V.; Goncalves, R.L.S.; Hey-Mogensen, M.; Brand, M.D. The determination and analysis of site-specific rates of mitochondrial reactive oxygen species production. Methods Enzym. 2013, 526, 189–217. [Google Scholar]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Cardona, L.R.; Kong, H.; Vasan, K.; McElroy, G.S.; Werner, M.; Kihshen, H.; Reczek, C.R.; Weinberg, S.E.; Gao, P.; et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 2020, 585, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.D. The Four Elements and the Recovery of Referentiality in Ecocriticism, Introduction. In Ecocritical Exploration in Literary and Cultural Studies: Fences, Boundaries, and Fields; Rowman & Littlefield: New York, NY, USA, 2009; pp. 1–13. [Google Scholar]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH: Ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [PubMed]
- Smeitink, J.; van den Heuvel, L.; DiMauro, S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2001, 2, 342–352. [Google Scholar] [CrossRef]
- Ochi, R.; Dhagia, V.; Lakhkar, A.; Patel, D.; Wolin, M.S.; Gupte, S.A. Rotenone-stimulated superoxide release from mitochondrial complex I acutely augments L-type Ca2+ current in A7r5 aortic smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1118–H1128. [Google Scholar] [CrossRef]
- Dikalov, S.; Itani, H.; Richmond, B.; Vergeade, A.; Rahman, S.M.J.; Boutaud, O.; Blackwell, T.; Massion, P.P.; Harrison, D.G.; Dikalova, A. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H639–H646. [Google Scholar] [CrossRef]
- Vignais, P.; Colbeau, A. Molecular biology of microbial hydrogenases. Curr. Issues Mol. Biol. 2004, 6, 159–188. [Google Scholar]
- Sag, C.M.; Wagner, S.; Maier, L.S. Role of oxidants on calcium and sodium movement in healthy and diseased cardiac myocytes. Free Radic. Biol. Med. 2013, 63, 338–349. [Google Scholar] [CrossRef]
- Stepanova, A.; Sosunov, S.; Niatsetskaya, Z.; Konrad, C.; Starkov, A.A.; Manfredi, G.; Wittig, I.; Ten, V.; Galkin, A. Redox-dependent loss of flavin by mitochondrial complex I in brain ischemia/reperfusion injury. Antioxid. Redox Signal. 2019, 31, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Brandt, U. Energy converting NADH: Quinone oxidoreductase (complex I). Annu. Rev. Biochem. 2006, 75, 69–92. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.; Smith, A.C. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.C.; Chen, C.; Wu, Y.N.; Ahmed, M.; Kitmitto, A.; Greenstein, A.S.; Kim, S.J.; Shao, Y.F.; Zhang, Y.H. Cardiac complex II activity is enhanced by fat and mediates greater mitochondrial oxygen consumption following hypoxic re-oxygenation. Pflügers Arch.-Eur. J. Physiol. 2020, 472, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Markevich, N.I.; Galimova, M.H.; Markevich, L.N. Hysteresis and bistability in the succinate-CoQ reductase activity and reactive oxygen species production in the mitochondrial respiratory complex II. Redox Biol. 2020, 37, 101630. [Google Scholar] [CrossRef]
- Korge, P.; Calmettes, G.; John, S.A.; Weiss, J.N. Reactive oxygen species production induced by pore opening in cardiac mitochondria: The role of complex III. J. Biol. Chem. 2017, 292, 9882–9895. [Google Scholar] [CrossRef]
- Dona, M.; Lamers, M.; Rohde, S.; Gorissen, M.; Timmers, H.J. Targeting the Redox Balance Pathway Using Ascorbic Acid in sdhb Zebrafish Mutant Larvae. Cancers 2021, 13, 5124. [Google Scholar] [CrossRef]
- Valls-Lacalle, L.; Barba, I.; Miró-Casas, E.; Alburquerque-Béjar, J.J.; Ruiz-Meana, M.; Fuertes-Agudo, M.; Rodríguez-Sinovas, A.; García-Dorado, D. Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc. Res. 2016, 109, 374–384. [Google Scholar] [CrossRef]
- Di Trani, J.M.; Liu, Z.; Whitesell, L.; Brzezinski, P.; Cowen, L.E.; Rubinstein, J.L. Rieske head domain dynamics and indazole-derivative inhibition of Candida albicans complex III. Structure 2022, 30, 129–138.e4. [Google Scholar] [CrossRef] [PubMed]
- Vennam, P.R.; Fisher, N.; Krzyaniak, M.D.; Kramer, D.M.; Bowman, M.K. A caged, destabilized, free radical intermediate in the q-cycle. Chembiochem 2013, 14, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial complex III Rieske Fe-S protein processing and assembly. Cell Cycle 2018, 17, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Gudz, T.I.; Moghaddas, S.; Migita, C.T.; Ikeda-Saito, M.; Turkaly, P.J.; Hoppel, C.L. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J. Mol. Cell. Cardiol. 2001, 33, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.E.; Trumpower, B.L. Subunit 6 regulates half-of-the-sites reactivity of the dimeric cytochrome bc1 complex in Saccharomyces cerevisiae. J. Biol. Chem. 1990, 265, 17005–17011. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Kang, P.T.; Zhang, L.; Xiao, K.; Zweier, J.L.; Chilian, W.M.; Chen, Y.-R. Reperfusion mediates heme impairment with increased protein cysteine sulfonation of mitochondrial complex III in the post-ischemic heart. J. Mol. Cell. Cardiol. 2021, 161, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-R.; Zweier, J.L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef]
- Lai, B.; Zhang, L.; Dong, L.-Y.; Zhu, Y.-H.; Sun, F.-Y.; Zheng, P. Inhibition of Qi site of mitochondrial complex III with antimycin A decreases persistent and transient sodium currents via reactive oxygen species and protein kinase C in rat hippocampal CA1 cells. Exp. Neurol. 2005, 194, 484–494. [Google Scholar] [CrossRef]
- Lai, B.; Zhang, L.; Dong, L.-Y.; Zhu, Y.-H.; Sun, F.-Y.; Zheng, P. Impact of inhibition of Qo site of mitochondrial complex III with myxothiazol on persistent sodium currents via superoxide and protein kinase C in rat hippocampal CA1 cells. Neurobiol. Dis. 2006, 21, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Baudry, N.; Laemmel, E.; Vicaut, E. In vivo reactive oxygen species production induced by ischemia in muscle arterioles of mice: Involvement of xanthine oxidase and mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H821–H828. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, Q.; Su, H.; Qi, B.; Wang, Y.; Yan, K.; Wang, X.; Li, X.; Zhao, D. A SIRT1 activator, ginsenoside Rc, promotes energy metabolism in cardiomyocytes and neurons. J. Am. Chem. Soc. 2021, 143, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Svahn, E.; Swanson, J.M.J.; Lepp, H.; Voth, G.A.; Brzezinski, P.; Gennis, R.B. Intricate role of water in proton transport through cytochrome c oxidase. J. Am. Chem. Soc. 2010, 132, 16225–16239. [Google Scholar] [CrossRef] [PubMed]
- Kolbe, F.; Safarian, S.; Piórek, Ż.; Welsch, S.; Müller, H.; Michel, H. Cryo-EM structures of intermediates suggest an alternative catalytic reaction cycle for cytochrome c oxidase. Nat. Commun. 2021, 12, 6903. [Google Scholar] [CrossRef] [PubMed]
- Bouillaud, F.; Blachier, F. Mitochondria and sulfide: A very old story of poisoning, feeding, and signaling? Antioxid. Redox Signal. 2011, 15, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Timkovich, R.; Thrasher, J.S. Carbon monoxide oxygenase activity of cytochrome cd1. Biochemistry 1988, 27, 5383–5388. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.E. Nitric oxide and cytochrome oxidase: Substrate, inhibitor or effector? Trends Biochem. Sci. 2002, 27, 33–39. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, H.; Shi, M.; Chen, Y.; Yang, T.; Fan, H. Toxic effects of hydrogen sulfide donor NaHS induced liver apoptosis is regulated by complex IV subunits and reactive oxygen species generation in rats. Environ. Toxicol. 2020, 35, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Prabu, S.K.; Anandatheerthavarada, H.K.; Raza, H.; Srinivasan, S.; Spear, J.F.; Avadhani, N.G. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J. Biol. Chem. 2006, 281, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-L.; Chen, C.-L.; Yeh, S.T.; Zweier, J.L.; Chen, Y.-R. Biphasic modulation of the mitochondrial electron transport chain in myocardial ischemia and reperfusion. Am. J. Physiol.-Heart Circ. Physiol. 2012, 302, H1410–H1422. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.-I.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef]
- Rosca, M.; Minkler, P.; Hoppel, C.L. Cardiac mitochondria in heart failure: Normal cardiolipin profile and increased threonine phosphorylation of complex IV. Biochim. Biophys. Acta (BBA)-Bioenergy 2011, 1807, 1373–1382. [Google Scholar] [CrossRef]
- Fang, J.-K.; Prabu, S.K.; Sepuri, N.B.; Raza, H.; Anandatheerthavarada, H.K.; Galati, D.; Spear, J.; Avadhani, N.G. Site specific phosphorylation of cytochrome c oxidase subunits I, IVi1 and Vb in rabbit hearts subjected to ischemia/reperfusion. FEBS Lett. 2007, 581, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Antoniel, M.; Jones, K.; Antonucci, S.; Spolaore, B.; Fogolari, F.; Petronilli, V.; Giorgio, V.; Carraro, M.; Di Lisa, F.; Forte, M.; et al. The unique histidine in OSCP subunit of F-ATP synthase mediates inhibition of the permeability transition pore by acidic pH. EMBO Rep. 2018, 19, 257–268. [Google Scholar] [CrossRef]
- Naoi, M.; Wu, Y.; Shamoto-Nagai, M.; Maruyama, W. Mitochondria in neuroprotection by phytochemicals: Bioactive polyphenols modulate mitochondrial apoptosis system, function and structure. Int. J. Mol. Sci. 2019, 20, 2451. [Google Scholar] [CrossRef]
- Azarashvili, T.; Odinokova, I.; Bakunts, A.; Ternovsky, V.; Krestinina, O.; Tyynelä, J.; Saris, N.-E.L. Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 2014, 55, 69–77. [Google Scholar]
- Hausenloy, D.J.; Yellon, D.M. The mitochondrial permeability transition pore: Its fundamental role in mediating cell death during ischaemia and reperfusion. J. Mol. Cell. Cardiol. 2003, 35, 339–341. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Gupta, R.C.; Kohli, S.; Wang, M.; Hachem, S.; Zhang, K. Chronic Therapy with Elamipretide (MTP-131), a Novel Mitochondria-Targeting Peptide, Improves Left Ventricular and Mitochondrial Function in Dogs With Advanced Heart Failure. Circ. Heart Fail. 2016, 9, e002206. [Google Scholar] [CrossRef] [PubMed]
- Efremov, R.G.; Sazanov, L.A. Structure of the membrane domain of respiratory complex I. Nature 2011, 476, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.-I.; Byun, J.; Huang, C.-Y.; Imai, N.; Ralda, G.; Zhai, P.; Xu, X.; Kashyap, S.; Warren, J.S.; Maschek, J.A.; et al. Nampt Potentiates Antioxidant Defense in Diabetic Cardiomyopathy. Circ. Res. 2021, 129, 114–130. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Moghaddas, S.; Hassan, M.O.; Tandler, B.; Hoppel, C.L. Blockade of electron transport during ischemia protects cardiac mitochondria. J. Biol. Chem. 2004, 279, 47961–47967. [Google Scholar] [CrossRef]
- Wu, Z.; Jankowski, V.; Jankowski, J. Irreversible post-translational modifications—Emerging cardiovascular risk factors. Mol. Asp. Med. 2022, 86, 101010. [Google Scholar] [CrossRef]
- Nanni, E.J., Jr.; Stallings, M.D.; Sawyer, D.T. Does superoxide ion oxidize catechol,. alpha.-tocopherol, and ascorbic acid by direct electron transfer? J. Am. Chem. Soc. 1980, 102, 4481–4485. [Google Scholar] [CrossRef]
- Sawyer, D.T.; Calderwood, T.S.; Johlman, C.L.; Wilkins, C.L. Oxidation by superoxide ion of catechols, ascorbic acid, dihydrophenazine, and reduced flavins to their respective anion radicals. A common mechanism via a combined proton-hydrogen atom transfer. J. Org. Chem. 1985, 50, 1409–1412. [Google Scholar] [CrossRef]
- Sawyer, D.T.; Chiericato, G., Jr.; Tsuchiya, T. Oxidation of ascorbic acid and dehydroascorbic acid by superoxide ion in aprotic media. J. Am. Chem. Soc. 1982, 104, 6273–6278. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, X.; Gao, M.; Xu, L.; Qi, Y.; Wang, J.; Yin, L. Dioscin alleviates myocardial infarction injury via regulating BMP4/NOX1-mediated oxidative stress and inflammation. Phytomedicine 2022, 103, 154222. [Google Scholar] [CrossRef]
- Imran, M.; Hassan, M.Q.; Akhtar, M.S.; Rahman, O.; Akhtar, M.; Najmi, A.K. Sacubitril and valsartan protect from experimental myocardial infarction by ameliorating oxidative damage in Wistar rats. Clin. Exp. Hypertens. 2019, 41, 62–69. [Google Scholar] [CrossRef]
- Chen, X.; Touyz, R.M.; Park, J.B.; Schiffrin, E.L. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR. Hypertension 2001, 38, 606–611. [Google Scholar] [CrossRef]
- Valdecantos, M.P.; Pérez-Matute, P.; Quintero, P.; Martínez, J.A. Vitamin C, resveratrol and lipoic acid actions on isolated rat liver mitochondria: All antioxidants but different. Redox Rep. 2010, 15, 207–216. [Google Scholar] [CrossRef]
- Sadi, G.; Yılmaz, Ö.; Güray, T. Effect of vitamin C and lipoic acid on streptozotocin-induced diabetes gene expression: mRNA and protein expressions of Cu–Zn SOD and catalase. Mol. Cell. Biochem. 2008, 309, 109–116. [Google Scholar] [CrossRef]
- Song, Y.; Wang, B.; Zhu, X.; Hu, J.; Sun, J.; Xuan, J.; Ge, Z. Human umbilical cord blood-derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biol. Toxicol. 2021, 37, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wan, W.; Ran, Q.; Ye, T.; Sun, Y.; Liu, Z.; Liu, X.; Shi, S.; Qu, C.; Zhang, C.; et al. Pinocembrin mediates antiarrhythmic e ff ects in rats with isoproterenol-induced cardiac remodeling. Eur. J. Pharmacol. 2022, 920, 174799. [Google Scholar] [CrossRef]
- Ting, H.H.; Timimi, F.K.; Haley, E.A.; Roddy, M.-A.; Ganz, P.; Creager, M.A. Vitamin C improves endothelium-dependent vasodilation in forearm resistance vessels of humans with hypercholesterolemia. Circulation 1997, 95, 2617–2622. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, A.K.; May, J.M. α-Lipoic acid and ascorbate prevent LDL oxidation and oxidant stress in endothelial cells. Mol. Cell. Biochem. 2008, 309, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudabadi, M.M.S.; Rahbar, A.R. Effect of EPA and vitamin C on superoxide dismutase, glutathione peroxidase, total antioxidant capacity and malondialdehyde in type 2 diabetic patients. Oman Med. J. 2014, 29, 39. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Berdun, F.; Bartoli, C.; Steelheart, C.; Alegre, M.; Bayir, H.; Tyurina, Y.Y.; Kagan, V.E.; Salerno, G.; Pagnussat, G.; et al. C-ferroptosis is an iron-dependent form of regulated cell death in cyanobacteria. J. Cell Biol. 2022, 221, e201911005. [Google Scholar] [CrossRef]
- Valgimigli, L. Lipid Peroxidation and Antioxidant Protection. Biomolecules 2023, 13, 1291. [Google Scholar] [CrossRef]
- Vu, T.D.; Pal, S.N.; Ti, L.-K.; Martinez, E.C.; Rufaihah, A.J.; Ling, L.H.; Lee, C.-N.; Richards, A.M.; Kofidis, T. An autologous platelet-rich plasma hydrogel compound restores left ventricular structure, function and ameliorates adverse remodeling in a minimally invasive large animal myocardial restoration model: A translational approach: Vu and Pal “Myocardial Repair: PRP, Hydrogel and Supplements”. Biomaterials 2015, 45, 27–35. [Google Scholar]
- Jing, L.; Wang, Y.; Zhao, X.-M.; Zhao, B.; Han, J.-J.; Qin, S.-C.; Sun, X.-J. Cardioprotective Effect of Hydrogen-rich Saline on Isoproterenol-induced Myocardial Infarction in Rats. Heart Lung Circ. 2015, 24, 602–610. [Google Scholar] [CrossRef]
- Patel, P.; Parikh, M.; Shah, H.; Gandhi, T. Inhibition of RhoA/Rho kinase by ibuprofen exerts cardioprotective effect on isoproterenol induced myocardial infarction in rats. Eur. J. Pharmacol. 2016, 791, 91–98. [Google Scholar] [CrossRef]
- Nabzdyk, C.S.; Bittner, E.A. Vitamin C in the critically ill—Indications and controversies. World J. Crit. Care Med. 2018, 7, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Kaidow, A.; Ishii, N.; Suzuki, S.; Shiina, T.; Kasahara, H. Vitamin C Maintenance against Cell Growth Arrest and Reactive Oxygen Species Accumulation in the Presence of Redox Molecular Chaperone hslO Gene. Int. J. Mol. Sci. 2022, 23, 12786. [Google Scholar] [CrossRef]
- Tosato, M.; Calvani, R.; Picca, A.; Ciciarello, F.; Galluzzo, V.; Coelho-Júnior, H.J.; Di Giorgio, A.; Di Mario, C.; Gervasoni, J.; Gremese, E.; et al. Effects of l-Arginine Plus Vitamin C Supplementation on Physical Performance, Endothelial Function, and Persistent Fatigue in Adults with Long COVID: A Single-Blind Randomized Controlled Trial. Nutrients 2022, 14, 4984. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.S.; Fisher, B.J.; McCarter, J.; Sweeney, C.; Martin, E.J.; Middleton, P.; Ellenberg, M.; Fowler, E.; Brophy, D.F.; Fowler, A.A.; et al. Interventional vitamin C: A strategy for attenuation of coagulopathy and inflammation in a swine multiple injuries model. J. Trauma Acute Care Surg. 2018, 85, S57–S67. [Google Scholar] [CrossRef] [PubMed]
- Szyller, J.; Jagielski, D.; Bil-Lula, I. Antioxidants in Arrhythmia Treatment-Still a Controversy? A Review of Selected Clinical and Laboratory Research. Antioxidants 2022, 11, 1109. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Gambardella, J.; Castellanos, V.; Trimarco, V.; Santulli, G. Vitamin C and Cardiovascular Disease: An Update. Antioxidants 2020, 9, 1227. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Folsom, A.R.; Harnack, L.; Halliwell, B.; Jacobs, D.R. Does supplemental vitamin C increase cardiovascular disease risk in women with diabetes? Am. J. Clin. Nutr. 2004, 80, 1194–1200. [Google Scholar] [CrossRef]
- O’Connor, E.A.; Evans, C.V.; Ivlev, I.; Rushkin, M.C.; Thomas, R.G.; Martin, A.; Lin, J.S. Vitamin and Mineral Supplements for the Primary Prevention of Cardiovascular Disease and Cancer: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2022, 327, 2334–2347. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Feng, M.; Chen, Q.; Long, X.; Park, K.-Y.; Zhao, X. The Effect of Lactobacillus plantarum CQPC02 on Fatigue and Biochemical Oxidation Levels in a Mouse Model of Physical Exhaustion. Front. Nutr. 2021, 8, 641544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, L.; Zhang, L.; Chen, Q.; Tan, F.; Zhao, X. Effect of Lactobacillus fermentum HFY03 on the Antifatigue and Antioxidation Ability of Running Exhausted Mice. Oxid. Med. Cell. Longev. 2021, 2021, 8013681. [Google Scholar] [CrossRef] [PubMed]
- Martín-Calvo, N.; Martínez-González, M.Á. Vitamin C Intake is Inversely Associated with Cardiovascular Mortality in a Cohort of Spanish Graduates: The SUN Project. Nutrients 2017, 9, 954. [Google Scholar] [CrossRef] [PubMed]
- Basili, S.; Tanzilli, G.; Mangieri, E.; Raparelli, V.; Di Santo, S.; Pignatelli, P.; Violi, F. Intravenous ascorbic acid infusion improves myocardial perfusion grade during elective percutaneous coronary intervention: Relationship with oxidative stress markers. JACC Cardiovasc. Interv. 2010, 3, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Arad, Y.; Spadaro, L.A.; Roth, M.; Newstein, D.; Guerci, A.D. Treatment of asymptomatic adults with elevated coronary calcium scores with atorvastatin, vitamin C, and vitamin E: The St. Francis Heart Study randomized clinical trial. J. Am. Coll. Cardiol. 2005, 46, 166–172. [Google Scholar] [CrossRef]
- Jaxa-Chamiec, T.; Bednarz, B.; Drozdowska, D.; Gessek, J.; Gniot, J.; Janik, K.; Kawka-Urbanek, T.; Maciejewski, P.; Ogórek, M.; Szpajer, M. Antioxidant effects of combined vitamins C and E in acute myocardial infarction. The randomized, double-blind, placebo controlled, multicenter pilot Myocardial Infarction and VITamins (MIVIT) trial. Kardiol. Pol. 2005, 62, 344–350. [Google Scholar]
- Sesso, H.D.; Buring, J.E.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Glynn, R.J.; Gaziano, J.M. Vitamins E and C in the prevention of cardiovascular disease in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2008, 300, 2123–2133. [Google Scholar] [CrossRef]
- Laskowski, H.; Minczykowski, A.; Wysocki, H. Mortality and clinical course of patients with acute myocardial infarction treated with streptokinase and antioxidants: Mannitol and ascorbic acid. Int. J. Cardiol. 1995, 48, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Bhakuni, P.; Chandra, M.; Misra, M.K. Effect of ascorbic acid supplementation on certain oxidative stress parameters in the post reperfusion patients of myocardial infarction. Mol. Cell. Biochem. 2006, 290, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Fukuma, N.; Kimura-Kato, Y.; Aisu, N.; Tuchida, T.; Mabuchi, K.; Takano, T. Improvement of sympathetic response to exercise by oral administration of ascorbic acid in patients after myocardial infarction. Int. J. Cardiol. 2006, 111, 240–246. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, H.; Xu, Y.; Liehn, E.A.; Rusu, M. Vitamin C as Scavenger of Reactive Oxygen Species during Healing after Myocardial Infarction. Int. J. Mol. Sci. 2024, 25, 3114. https://doi.org/10.3390/ijms25063114
Zheng H, Xu Y, Liehn EA, Rusu M. Vitamin C as Scavenger of Reactive Oxygen Species during Healing after Myocardial Infarction. International Journal of Molecular Sciences. 2024; 25(6):3114. https://doi.org/10.3390/ijms25063114
Chicago/Turabian StyleZheng, Huabo, Yichen Xu, Elisa A. Liehn, and Mihaela Rusu. 2024. "Vitamin C as Scavenger of Reactive Oxygen Species during Healing after Myocardial Infarction" International Journal of Molecular Sciences 25, no. 6: 3114. https://doi.org/10.3390/ijms25063114
APA StyleZheng, H., Xu, Y., Liehn, E. A., & Rusu, M. (2024). Vitamin C as Scavenger of Reactive Oxygen Species during Healing after Myocardial Infarction. International Journal of Molecular Sciences, 25(6), 3114. https://doi.org/10.3390/ijms25063114