
Mitochondrial Dysfunction Causes Cell Death in Patients Affected by Fragile-X-Associated Disorders
 , , , ,
, , , , 
 ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
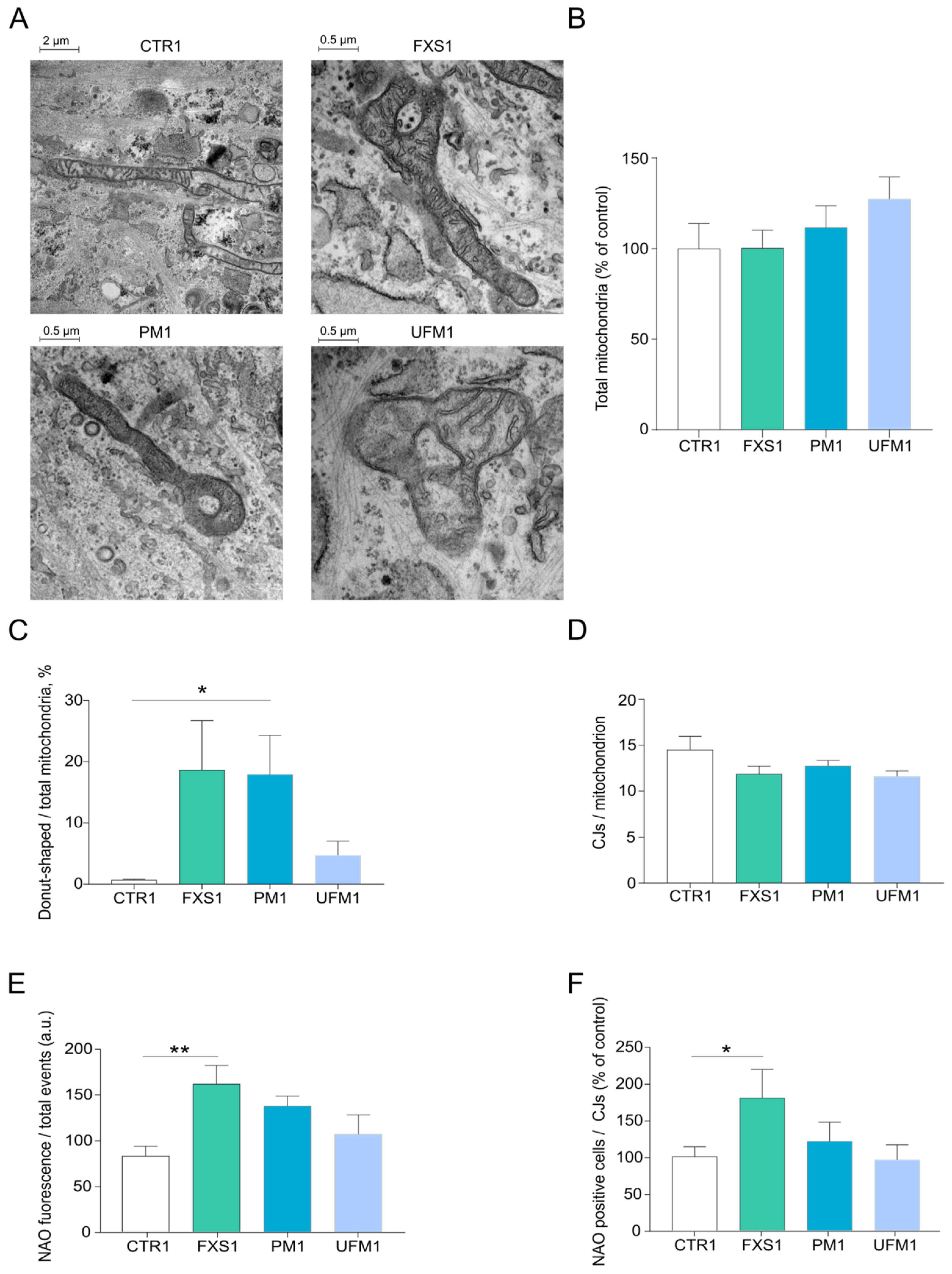
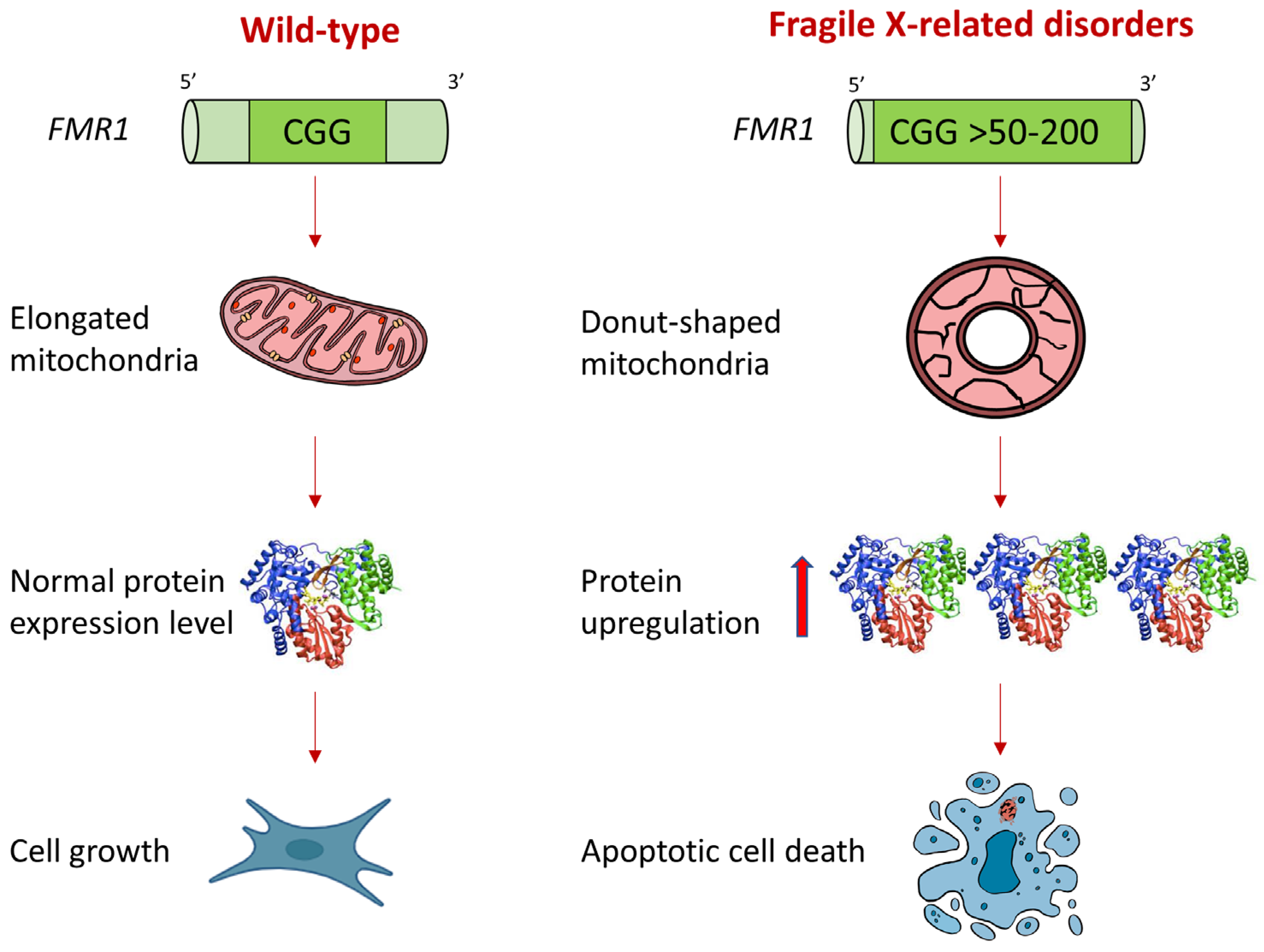
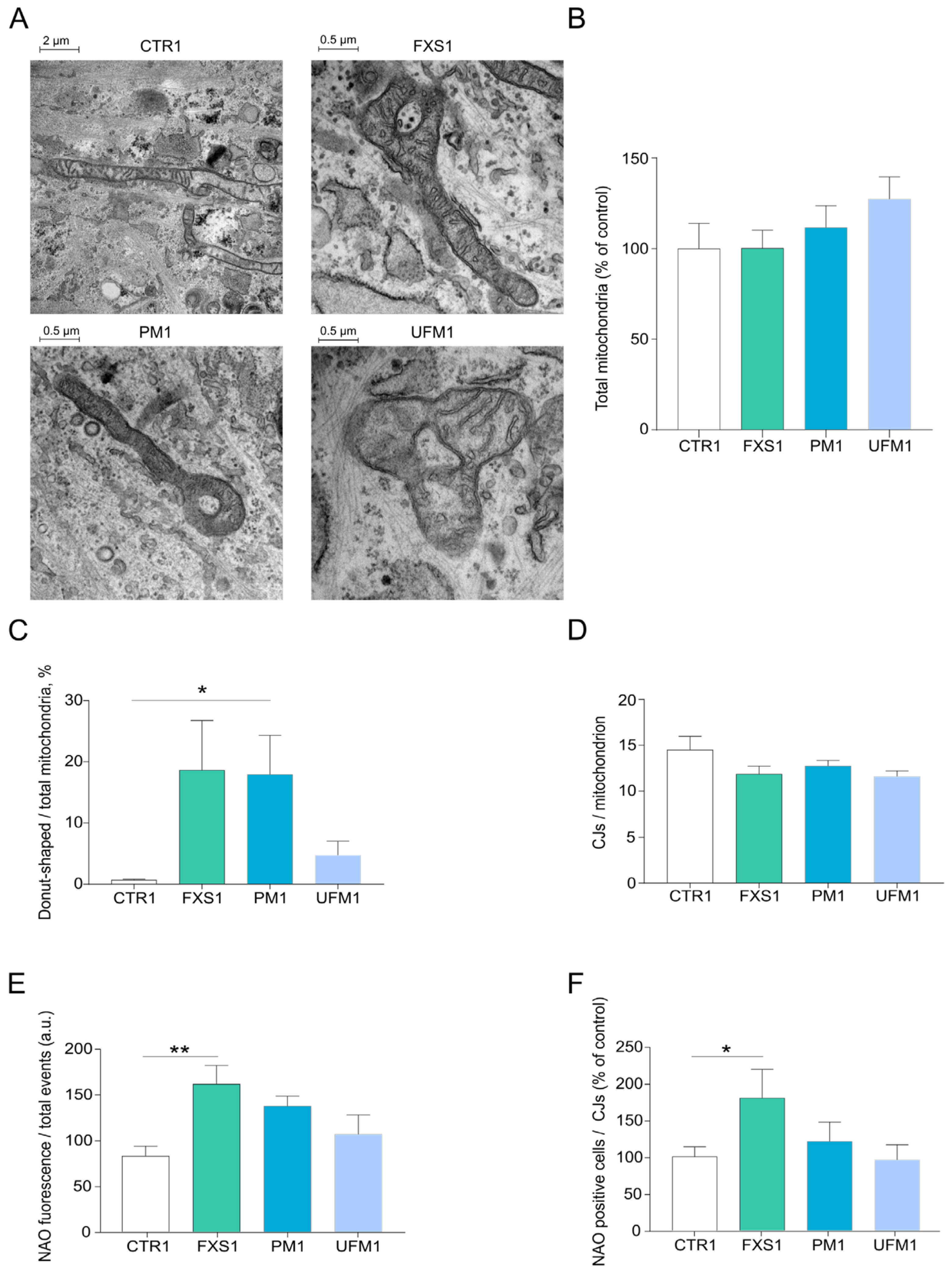
2.1. Mitochondria of FXD Patients Show Altered Morphology
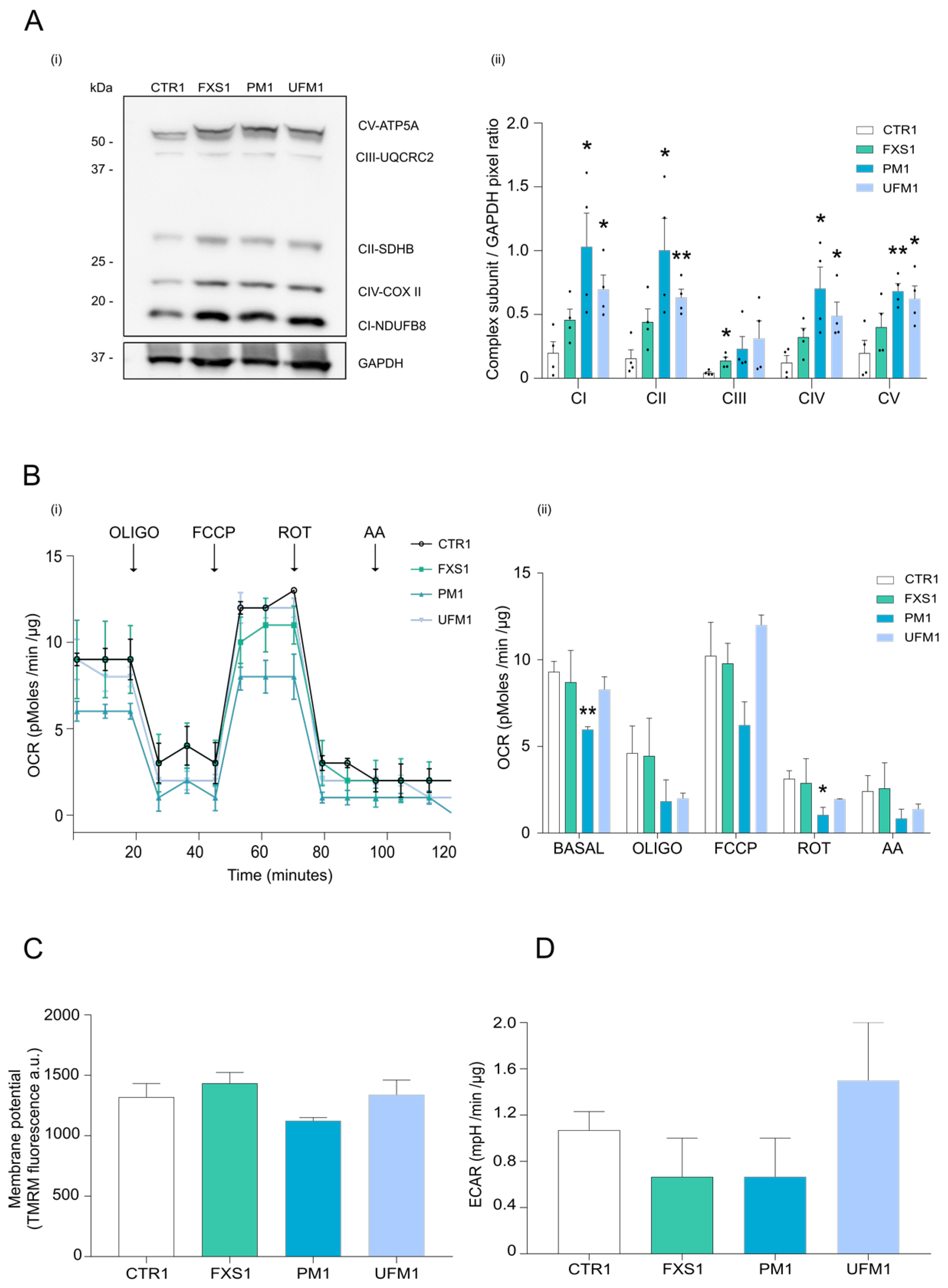
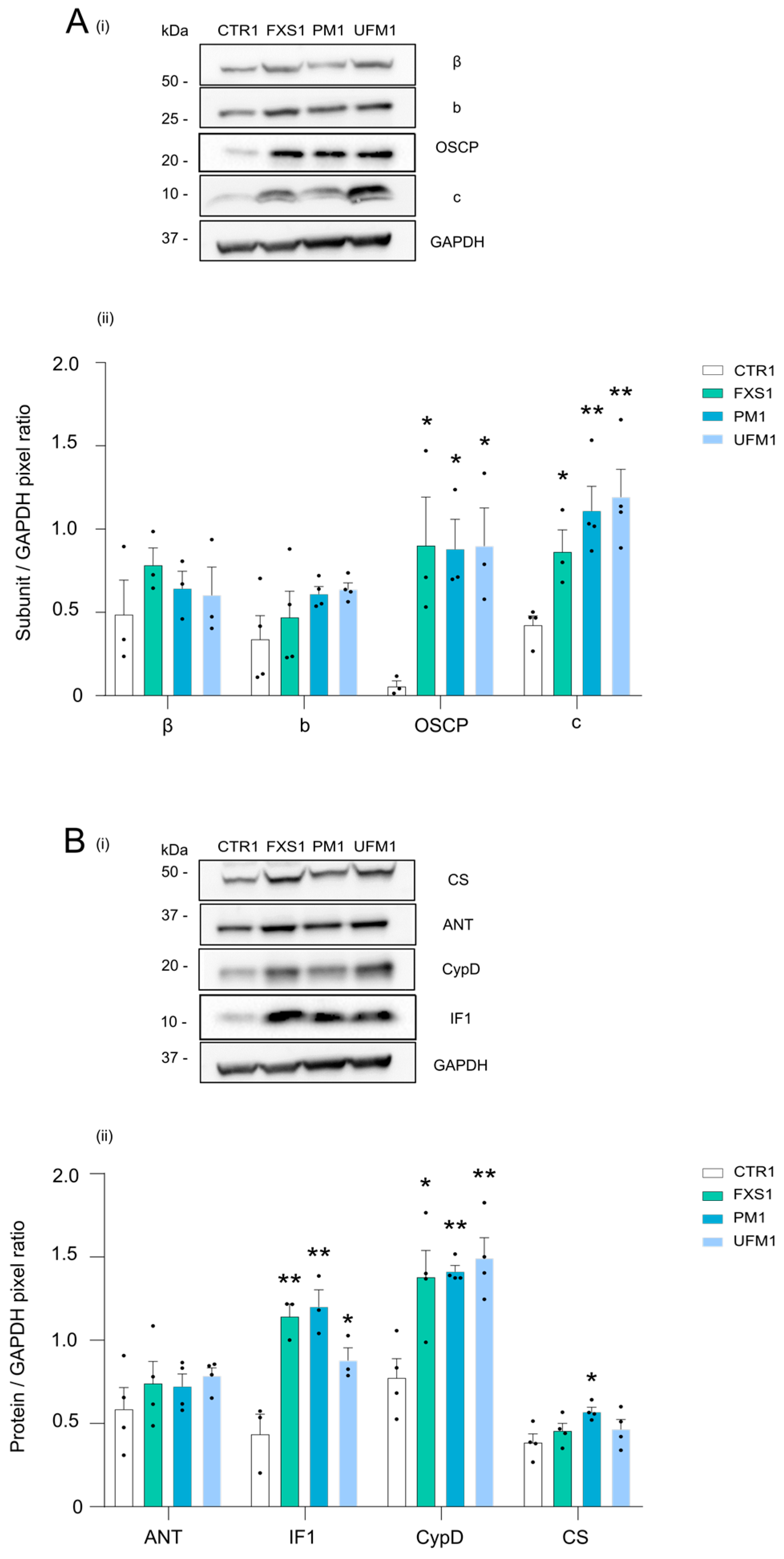
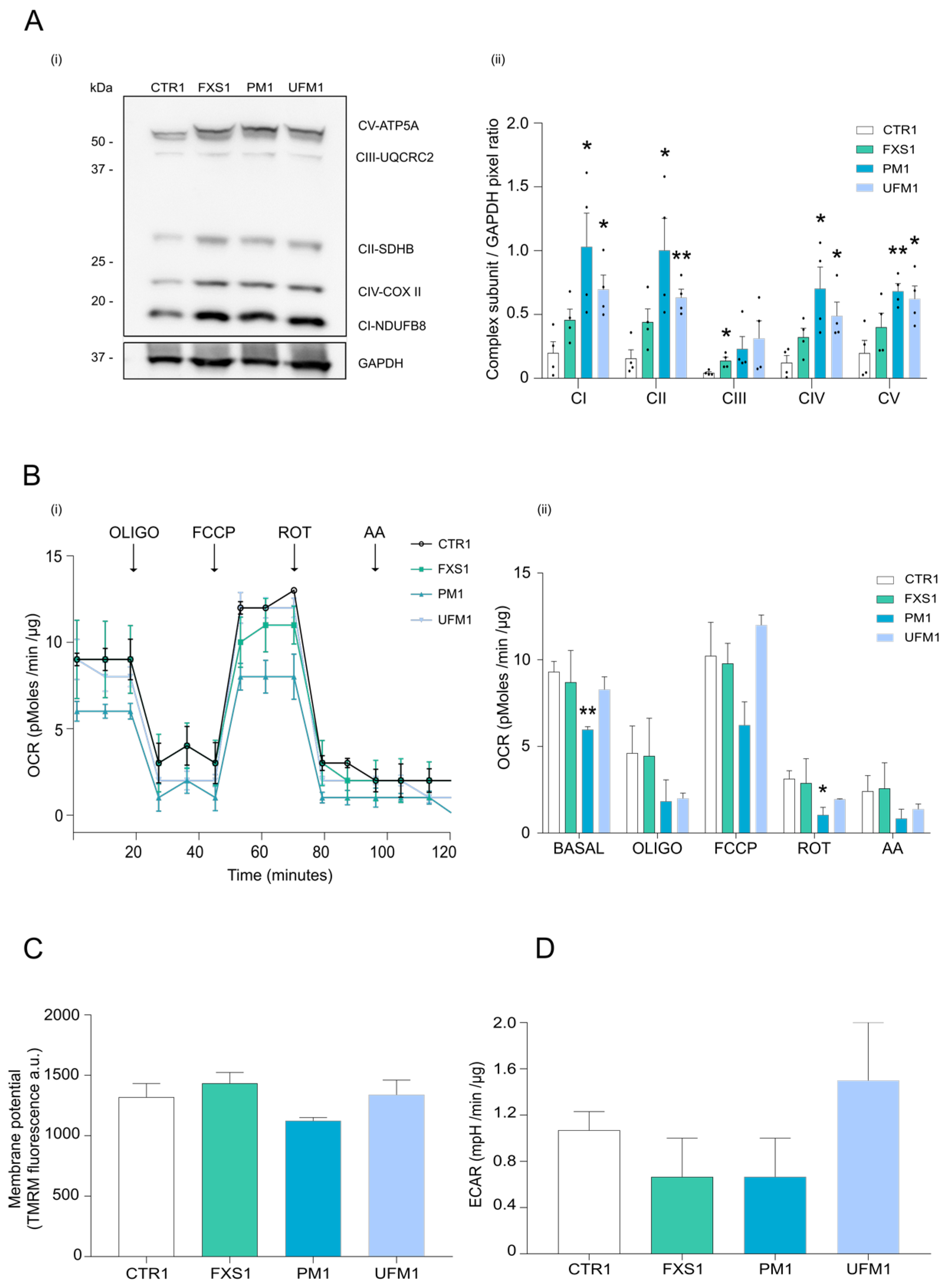
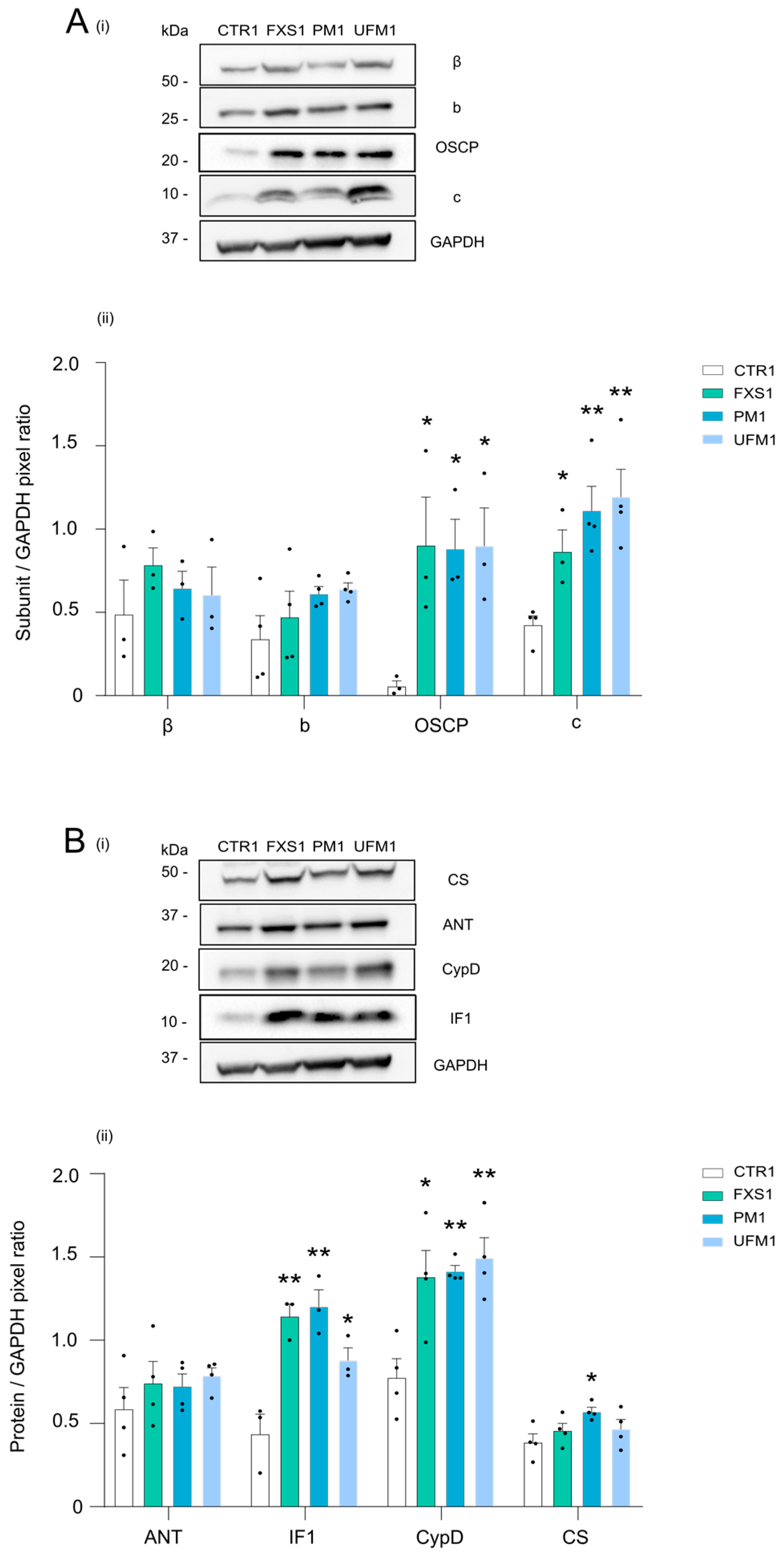
2.2. Mitochondrial OXPHOS Complexes and Other Proteins Are Upregulated in FXDs
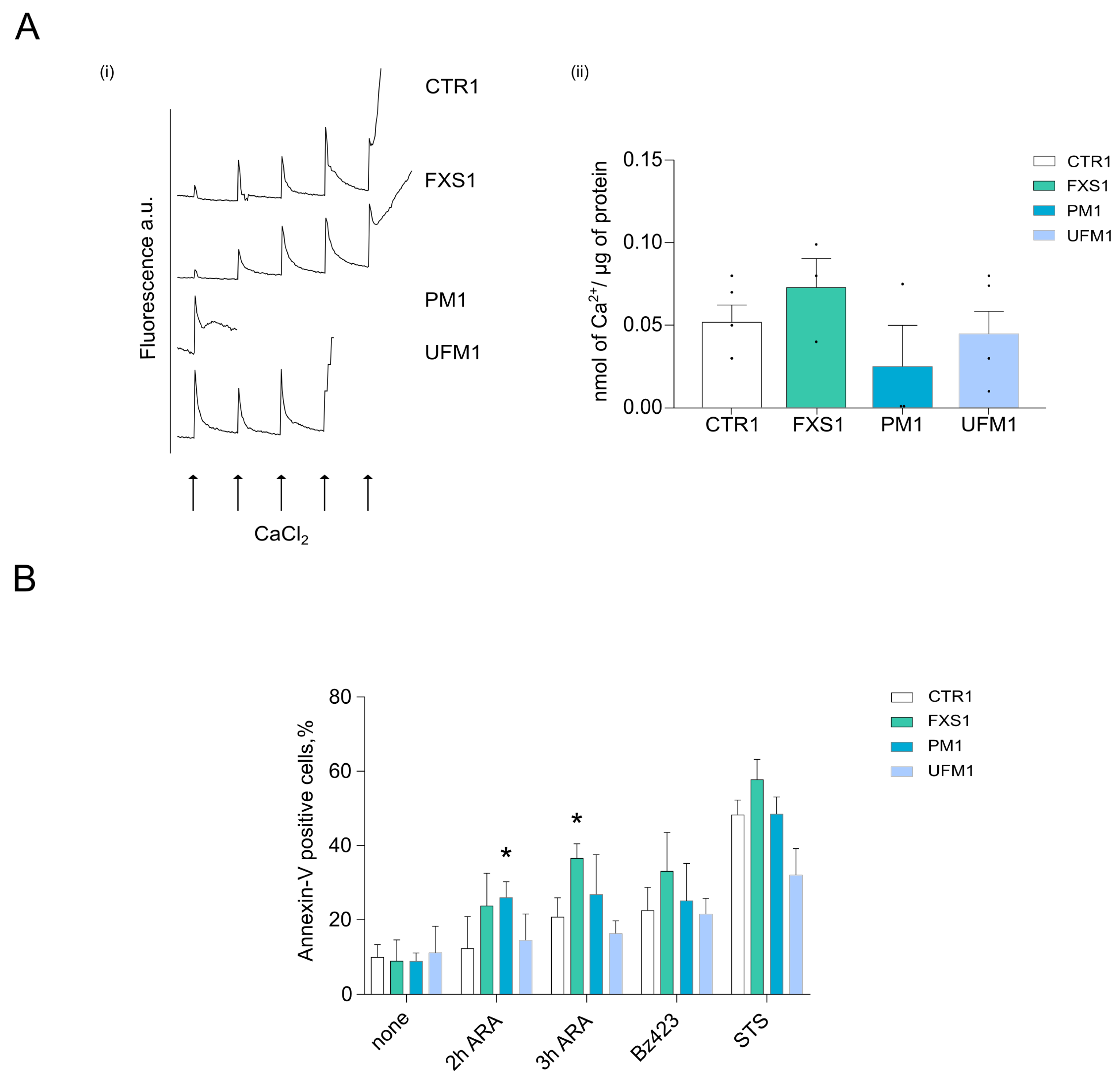
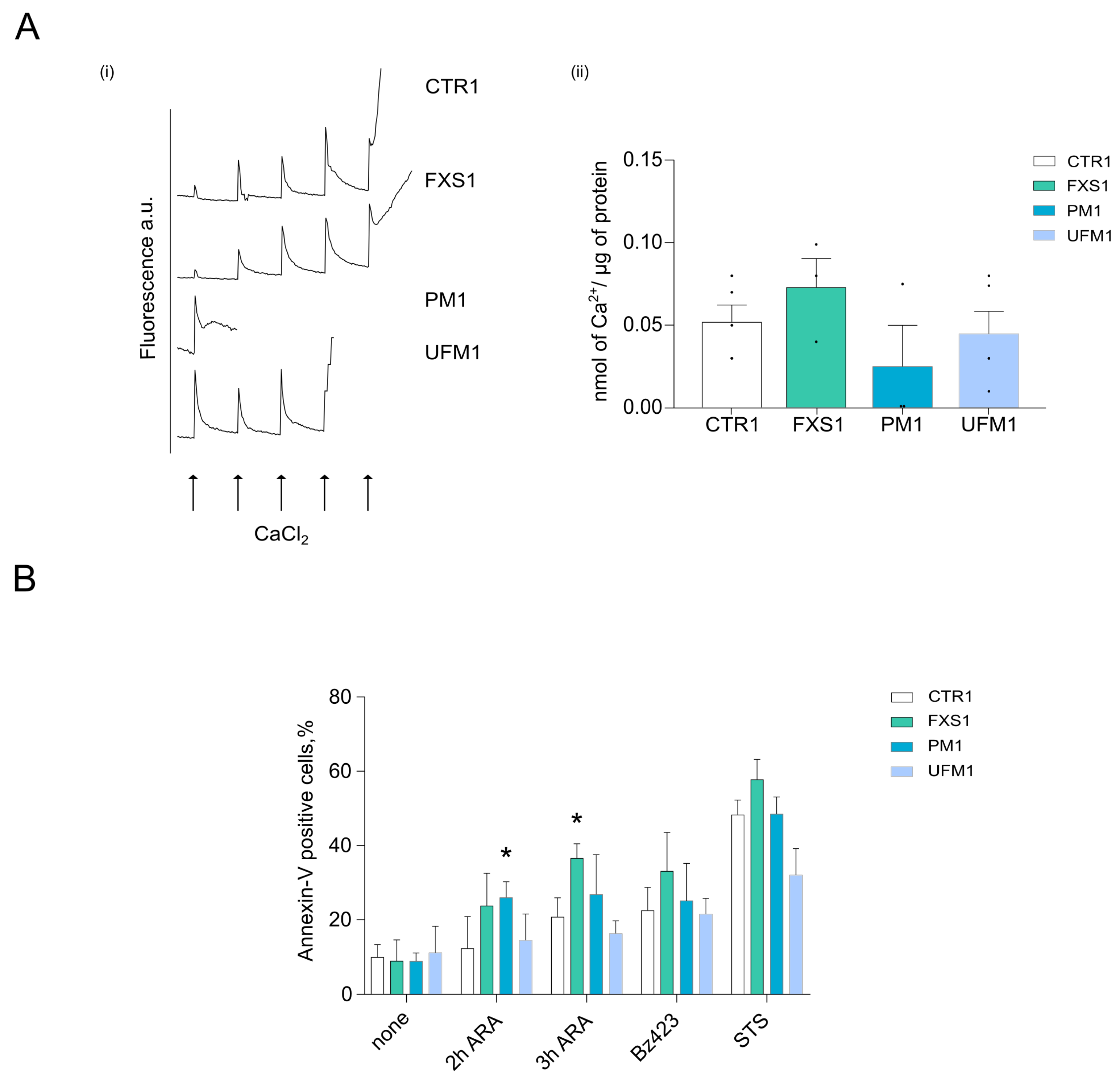
2.3. Cells from FXD Patients Are Sensitized to Apoptotic Stimuli
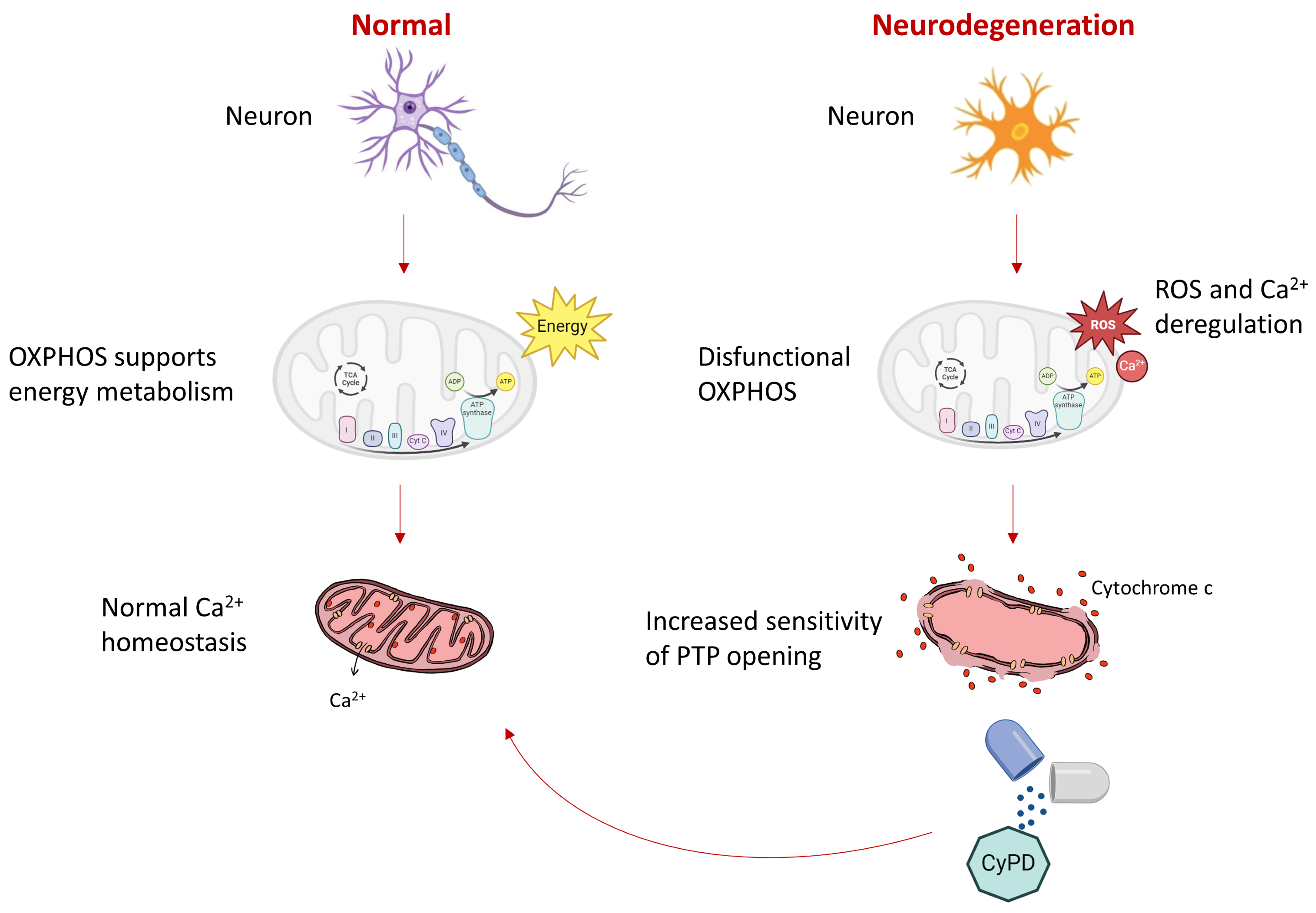
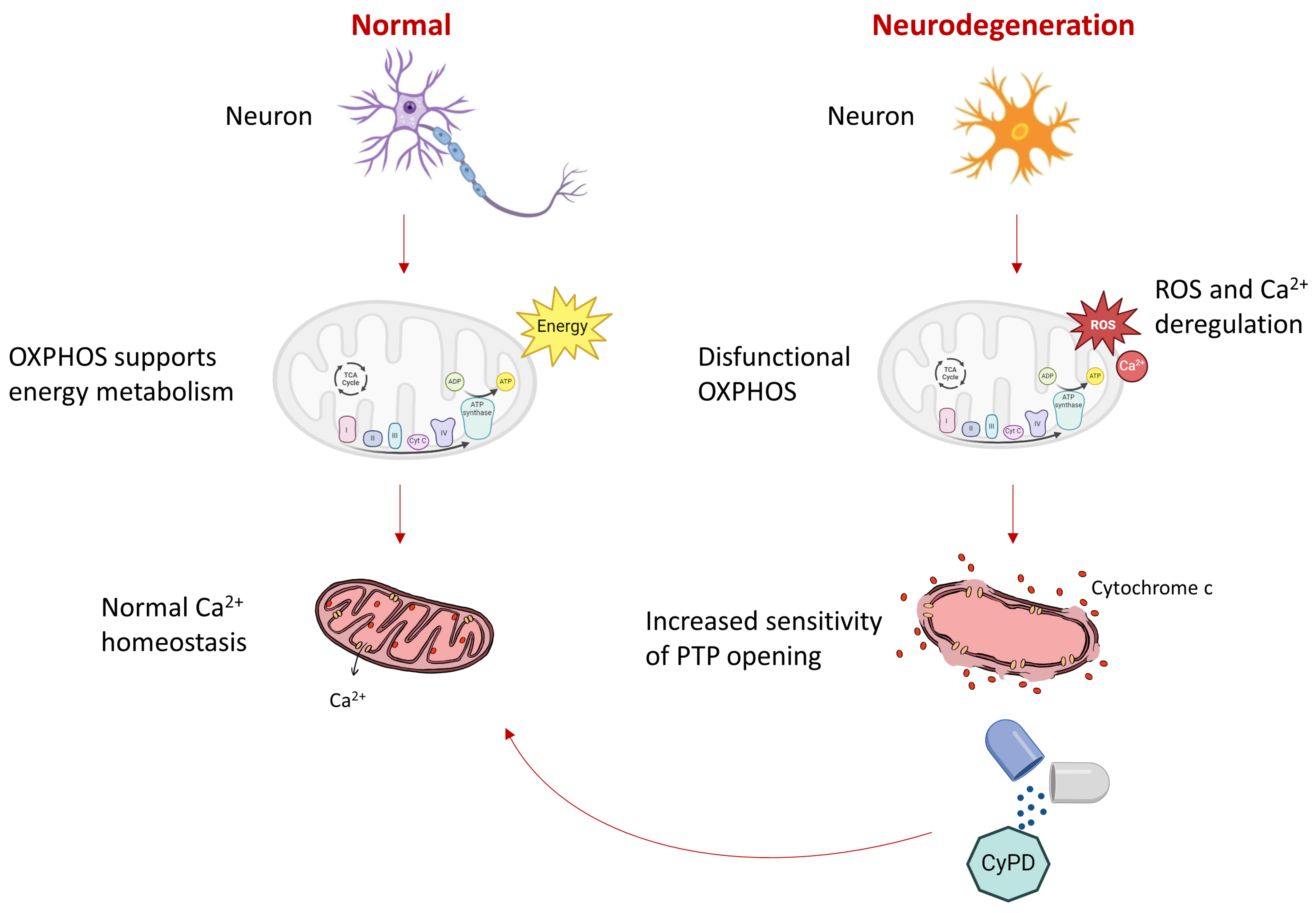
3. Discussion
4. Materials and Methods
4.1. Experimental Model
4.2. Electron Microscopy
4.3. Nonyl Acridine Orange Staining
4.4. Lysates, Gel Electrophoresis, Western Blotting
4.5. Oxygen Consumption Rate
4.6. Mitochondrial Membrane Potential
4.7. Calcium Retention Capacity
4.8. Cell Death
4.9. Quantification and Statistical Analysis
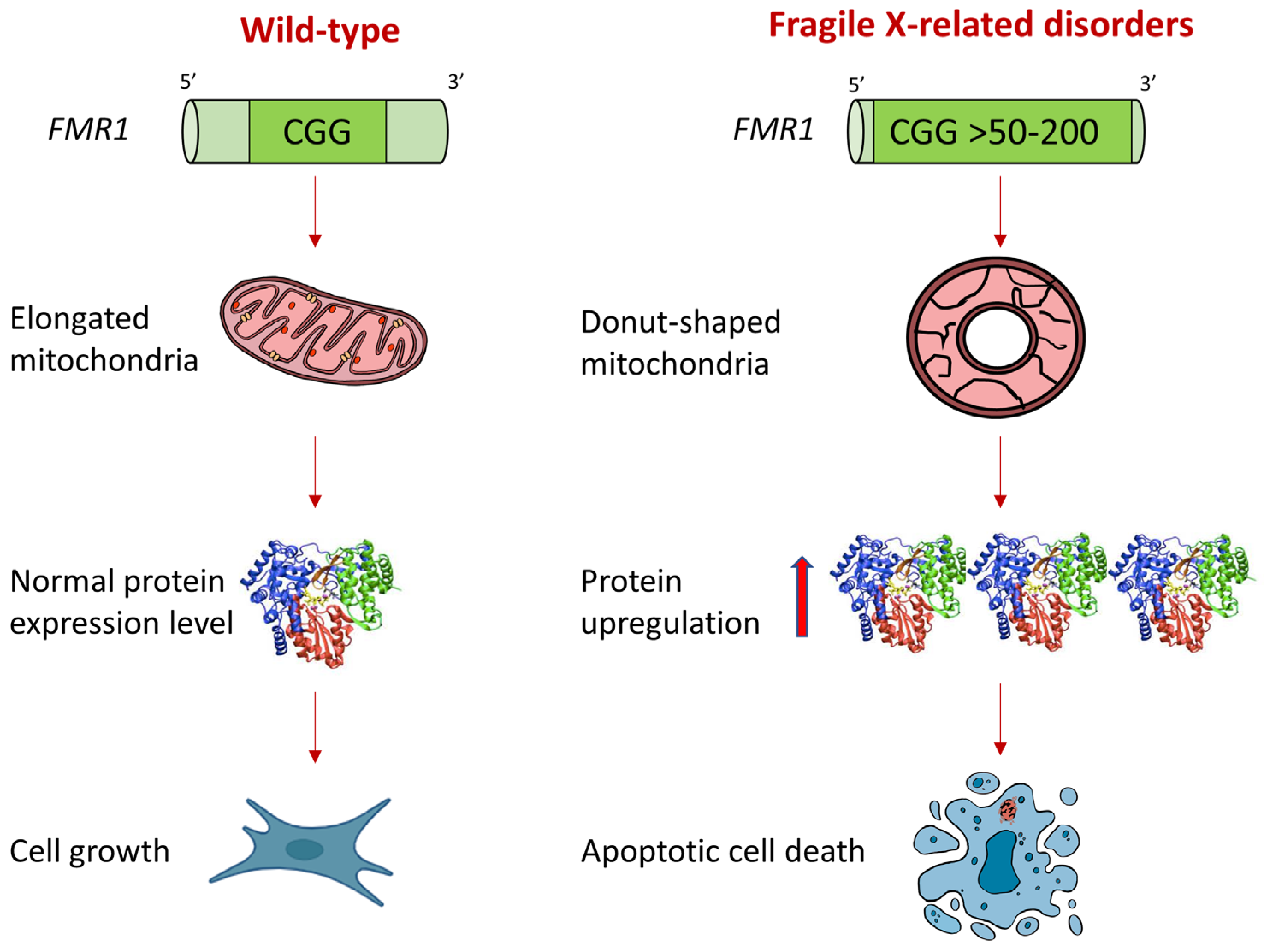
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bagni, C.; Tassone, F.; Neri, G.; Hagerman, R. Fragile X Syndrome: Causes, Diagnosis, Mechanisms, and Therapeutics. J. Clin. Investig. 2012, 122, 4314–4322. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X Syndrome. Nat. Rev. Dis. Prim. 2017, 3, 17065. [Google Scholar] [CrossRef]
- Verkerk, A.J.M.H.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.A.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a Gene (FMR-1) Containing a CGG Repeat Coincident with a Breakpoint Cluster Region Exhibiting Length Variation in Fragile X Syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Pieretti, M.; Zhang, F.; Fu, Y.-H.; Warren, S.T.; Oostra, B.A.; Caskey, C.T.; Nelson, D.L. Absence of Expression of the FMR-1 Gene in FXS. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Mills, J.B.; Harris, S.W.; Gane, L.W.; Hagerman, P. Clinical Involvement and Protein Expression in Individuals with the FMR1 Premutation. Am. J. Med. Genet. 2000, 91, 144–152. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Leehey, M.; Heinrichs, W.; Tassone, F.; Wilson, R.; Hills, J.; Grigsby, J.; Gage, B.; Hagerman, P.J. Intention Tremor, Parkinsonism, and Generalized Brain Atrophy in Male Carriers of Fragile X. Neurology 2001, 57, 127–130. [Google Scholar] [CrossRef]
- Greco, C.M.; Berman, R.F.; Martin, R.M.; Tassone, F.; Schwartz, P.H.; Chang, A.; Trapp, B.D.; Iwahashi, C.; Brunberg, J.; Grigsby, J.; et al. Neuropathology of Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS). Brain 2006, 129, 243–255. [Google Scholar] [CrossRef]
- Brykczynska, U.; Pecho-Vrieseling, E.; Thiemeyer, A.; Klein, J.; Fruh, I.; Doll, T.; Manneville, C.; Fuchs, S.; Iazeolla, M.; Beibel, M.; et al. CGG Repeat-Induced FMR1 Silencing Depends on the Expansion Size in Human IPSCs and Neurons Carrying Unmethylated Full Mutations. Stem Cell Reports 2016, 7, 1059–1071. [Google Scholar] [CrossRef]
- Smeets, H.J.M.; Smits, A.P.T.; Verheij, C.E.; Theelen, J.P.G.; Willemsen, R.; van de Burgt, I.; Hoogeveen, A.T.; Oosterwijk, J.C.; Oostra, B.A. Normal Phenotype in Two Brothers with a Full FMR1 Mutation. Hum. Mol. Genet. 1995, 4, 2103–2108. [Google Scholar] [CrossRef]
- Bagni, C.; Zukin, R.S. A Synaptic Perspective of Fragile X Syndrome and Autism Spectrum Disorders. Neuron 2019, 101, 1070–1088. [Google Scholar] [CrossRef]
- Davis, J.K.; Broadie, K. Multifarious Functions of the Fragile X Mental Retardation Protein. Trends Genet. 2017, 33, 703–714. [Google Scholar] [CrossRef]
- Pasciuto, E.; Bagni, C. SnapShot: FMRP MRNA Targets and Diseases. Cell 2014, 158, 1446.e1. [Google Scholar] [CrossRef]
- Pasciuto, E.; Bagni, C. SnapShot: FMRP Interacting Proteins. Cell 2014, 159, 218.e1. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular Mechanisms and Consequences of Mitochondrial Permeability Transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D Is a Component of Mitochondrial Permeability Transition and Mediates Neuronal Cell Death after Focal Cerebral Ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar] [CrossRef]
- Bonda, D.J.; Wang, X.; Gustaw-Rothenberg, K.A.; Perry, G.; Smith, M.A.; Zhu, X. Mitochondrial Drugs for Alzheimer Disease. Pharmaceuticals 2009, 2, 287–298. [Google Scholar] [CrossRef]
- Nobile, V.; Palumbo, F.; Lanni, S.; Ghisio, V.; Vitali, A.; Castagnola, M.; Marzano, V.; Maulucci, G.; De Angelis, C.; De Spirito, M.; et al. Altered Mitochondrial Function in Cells Carrying a Premutation or Unmethylated Full Mutation of the FMR1 Gene. Hum. Genet. 2020, 139, 227–245. [Google Scholar] [CrossRef]
- Napoli, E.; Ross-Inta, C.; Wong, S.; Omanska-Klusek, A.; Barrow, C.; Iwahashi, C.; Garcia-Arocena, D.; Sakaguchi, D.; Berry-Kravis, E.; Hagerman, R.; et al. Altered Zinc Transport Disrupts Mitochondrial Protein Processing/Import in Fragile X-Associated Tremor/Ataxia Syndrome. Hum. Mol. Genet. 2011, 20, 3079–3092. [Google Scholar] [CrossRef]
- Napoli, E.; Song, G.; Wong, S.; Hagerman, R.; Giulivi, C. Altered Bioenergetics in Primary Dermal Fibroblasts from Adult Carriers of the FMR1 Premutation before the Onset of the Neurodegenerative Disease Fragile X-Associated Tremor/Ataxia Syndrome. Cerebellum 2016, 15, 522–564. [Google Scholar] [CrossRef]
- Ross-Inta, C.; Omanska-Klusek, A.; Wong, S.; Barrow, C.; Garcia-Arocena, D.; Iwahashi, C.; Berry-Kravis, E.; Hagerman, R.J.; Hagerman, P.J.; Giulivi, C. Evidence of Mitochondrial Dysfunction in Fragile X-Associated Tremor/Ataxia Syndrome. Biochem. J. 2010, 429, 545–552. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Annesley, S.J.; Trost, N.; Bui, M.Q.; Lay, S.T.; Storey, E.; De Piazza, S.W.; Sanislav, O.; Francione, L.M.; Hammersley, E.M.; et al. Novel Blood Biomarkers Are Associated with White Matter Lesions in Fragile X- Associated Tremor/Ataxia Syndrome. Neurodegener. Dis. 2016, 17, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Licznerski, P.; Park, H.; Rolyan, H.; Chen, R.; Miranda, P.; Graham, M.; Wu, J.; Cruz-reyes, N.; Sohail, S.; Salcedo, J.; et al. ATP Synthase C-Subunit Leak Causes Aberrant Cellular Metabolismmetabolism in Fragile X Syndrome. Cell 2021, 182, 1170–1185. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; McLennan, Y.A.; Schneider, A.; Tassone, F.; Hagerman, R.J.; Giulivi, C. Characterization of the Metabolic, Clinical and Neuropsychological Phenotype of Female Carriers of the Premutation in the X-Linked FMR1 Gene. Front. Mol. Biosci. 2020, 7, 578640. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Kemp, B.E.; Bui, M.Q.; Fisher, P.R.; Allan, C.Y.; Sanislav, O.; Ngoei, K.R.W.; Atkinson, A.; Tassone, F.; Annesley, S.J.; et al. Cellular Bioenergetics and AMPK and TORC1 Signalling in Blood Lymphoblasts Are Biomarkers of Clinical Status in FMR1 Premutation Carriers. Front. Psychiatry 2021, 12, 747268. [Google Scholar] [CrossRef]
- Alvarez-Mora, M.I.; Rodriguez-Revenga, L.; Madrigal, I.; Guitart-Mampel, M.; Garrabou, G.; Milà, M. Impaired Mitochondrial Function and Dynamics in the Pathogenesis of FXTAS. Mol. Neurobiol. 2017, 54, 6896–6902. [Google Scholar] [CrossRef]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of Mitochondrial ATP Synthase Form the Permeability Transition Pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Jones, K.; Sartori, G.; Schiavone, M.; Antonucci, S.; Kucharczyk, R.; di Rago, J.P.; Franchin, C.; Arrigoni, G.; Forte, M.; et al. The Unique Cysteine of F-ATP Synthase OSCP Subunit Participates in Modulation of the Permeability Transition Pore. Cell Rep. 2020, 32, 108095. [Google Scholar] [CrossRef]
- Giorgio, V.; Fogolari, F.; Lippe, G.; Bernardi, P. OSCP Subunit of Mitochondrial ATP Synthase: Role in Regulation of Enzyme Function and of Its Transition to a Pore. Br. J. Pharmacol. 2019, 176, 4247–4257. [Google Scholar] [CrossRef]
- Gauba, E.; Guo, L.; Du, H. Cyclophilin D Promotes Brain Mitochondrial F1FO ATP Synthase Dysfunction in Aging Mice. J. Alzheimer’s Dis. 2017, 55, 1351–1362. [Google Scholar] [CrossRef]
- Gledhill, J.R.; Montgomery, M.G.; Leslie, A.G.W.; Walker, J.E. How the Regulatory Protein, IF1, Inhibits F1-ATPase from Bovine Mitochondria. Proc. Natl. Acad. Sci. USA 2007, 104, 15671–15676. [Google Scholar] [CrossRef]
- Bason, J.V.; Montgomery, M.G.; Leslie, A.G.W.; Walker, J.E. Pathway of Binding of the Intrinsically Disordered Mitochondrial Inhibitor Protein to F1-ATPase. Proc. Natl. Acad. Sci. USA 2014, 111, 11305–11310. [Google Scholar] [CrossRef]
- Galber, C.; Fabbian, S.; Gatto, C.; Grandi, M.; Carissimi, S.; Acosta, M.J.; Sgarbi, G.; Tiso, N.; Argenton, F.; Solaini, G.; et al. The Mitochondrial Inhibitor IF1 Binds to the ATP Synthase OSCP Subunit and Protects Cancer Cells from Apoptosis. Cell Death Dis. 2023, 14, 54. [Google Scholar] [CrossRef]
- Scorrano, L.; Penzo, D.; Petronilli, V.; Pagano, F.; Bernardi, P. Arachidonic Acid Causes Cell Death through the Mitochondrial Permeability Transition. Implications for Tumor Necrosis Factor-α Apoptotic Signaling. J. Biol. Chem. 2001, 276, 12035–12040. [Google Scholar] [CrossRef]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; Di Lisa, F. The Mitochondrial Permeability Transition, Release of Cytochrome c and Cell Death. Correlation with the Duration of Pore Openings in Situ. J. Biol. Chem. 2001, 276, 12030–12034. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Bott, L.C. The Ubiquitin-Proteasome System in Neurodegenerative Diseases: Precipitating Factor, yet Part of the Solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Primerano, B.; Tassone, F.; Hagerman, R.J.; Hagerman, P.; Amaldi, F.; Bagni, C. Reduced FMR1 MRNA Translation Efficiency in Fragile X Patients with Premutations. Rna 2002, 8, 1482–1488. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi, C.K.; Yasui, D.H.; An, H.J.; Greco, C.M.; Tassone, F.; Nannen, K.; Babineau, B.; Lebrilla, C.B.; Hagerman, R.J.; Hagerman, P.J. Protein Composition of the Intranuclear Inclusions of FXTAS. Brain 2006, 129, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. MITOCHONDRIAL FRAGMENTATION IN NEURODEGENERATION. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef]
- Maurya, S.K.; Gupta, S.; Bakshi, A.; Kaur, H.; Jain, A.; Senapati, S.; Baghel, M.S. Targeting Mitochondria in the Regulation of Neurodegenerative Diseases: A Comprehensive Review. J. Neurosci. Res. 2022, 100, 1845–1861. [Google Scholar] [CrossRef]
- Ariza, J.; Steward, C.; Rueckert, F.; Widdison, M.; Coffman, R.; Afjeia, A.; Noctor, S.; Hagerman, R.; Hagerman, P.; Martínez-Cerdeñoa, V. Dysregulated Iron Metabolism in the Choroid Plexus in Fragile X- Associated Tremor/Ataxia Syndrome. Brain Res. 2015, 1598, 88–96. [Google Scholar] [CrossRef]
- Ariza, J.; Rogers, H.; Hartvigsen, A.; Snell, M.; Judd, D.; Hagerman, P. Iron Accumulation and Dysregulation in the Putamen in Fragile X Associated Tremor/Ataxia Syndrome. Mov. Disord. 2018, 32, 585–591. [Google Scholar] [CrossRef]
- Giulivi, C.; Napoli, E.; Tassone, F.; Halmai, J.; Hagerman, R. Plasma Metabolic Profile Delineates Roles for Neurodegeneration, pro-Inflammatory Damage and Mitochondrial Dysfunction in the FMR1 Premutation. Biochem. J. 2016, 473, 3871–3888. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Wang, F.; Li, M.; Sah, N.; Stockton, M.E.; Tidei, J.J.; Gao, Y.; Korabelnikov, T.; Kannan, S.; Vevea, J.D.; et al. Reduced Mitochondrial Fusion and Huntingtin Levels Contribute to Impaired Dendritic Maturation and Behavioral Deficits in Fmr1-Mutant Mice. Nat. Neurosci. 2019, 22, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Hukema, R.K.; Buijsen, R.A.M.; Raske, C.; Severijnen, L.A.; Nieuwenhuizen-Bakker, I.; Minneboo, M.; Maas, A.; De Crom, R.; Kros, J.M.; Hagerman, P.J.; et al. Induced Expression of Expanded CGG RNA Causes Mitochondrial Dysfunction in Vivo. Cell Cycle 2014, 13, 2600–2608. [Google Scholar] [CrossRef] [PubMed]
- Gohel, D.; Sripada, L.; Prajapati, P.; Singh, K.; Roy, M.; Kotadia, D.; Tassone, F.; Charlet-Berguerand, N.; Singh, R. FMRpolyG Alters Mitochondrial Transcripts Level and Respiratory Chain Complex Assembly in Fragile X Associated Tremor/Ataxia Syndrome [FXTAS]. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Todd, P.K.; Oh, S.Y.; Krans, A.; He, F.; Sellier, C.; Frazer, M.; Renoux, A.J.; Chen, K.C.; Scaglione, K.M.; Basrur, V.; et al. CGG Repeat-Associated Translation Mediates Neurodegeneration in Fragile X Tremor Ataxia Syndrome. Neuron 2013, 78, 440–455. [Google Scholar] [CrossRef]
- Giorgio, V.; Guo, L.; Bassot, C.; Petronilli, V.; Bernardi, P. Calcium and Regulation of the Mitochondrial Permeability Transition. Cell Calcium 2018, 70, 56–63. [Google Scholar] [CrossRef]
- Rao, V.K.; Carlson, E.A.; Yan, S.S. Mitochondrial Permeability Transition Pore Is a Potential Drug Target for Neurodegeneration. Biochim Biophys Acta 2014, 1842, 1267–1272. [Google Scholar] [CrossRef]
- Biosa, A.; Arduini, I.; Soriano, M.E.; Giorgio, V.; Bernardi, P.; Bisaglia, M.; Bubacco, L. Dopamine Oxidation Products as Mitochondrial Endotoxins, a Potential Molecular Mechanism for Preferential Neurodegeneration in Parkinson’s Disease. ACS Chem. Neurosci. 2018, 9, 2849–2858. [Google Scholar] [CrossRef]
- Long, Q.; Zhao, D.; Fan, W.; Yang, L.; Zhou, Y.; Qi, J.; Wang, X.; Liu, X. Modeling of Mitochondrial Donut Formation. Biophys. J. 2015, 109, 892–899. [Google Scholar] [CrossRef]
- Liu, X.; Hajnóczky, G. Altered Fusion Dynamics Underlie Unique Morphological Changes in Mitochondria during Hypoxia-Reoxygenation Stress. Cell Death Differ. 2011, 18, 1561–1572. [Google Scholar] [CrossRef]
- Galber, C.; Minervini, G.; Cannino, G.; Boldrin, F.; Petronilli, V.; Tosatto, S.; Lippe, G.; Giorgio, V. The f Subunit of Human ATP Synthase Is Essential for Normal Mitochondrial Morphology and Permeability Transition. Cell Rep. 2021, 35, 109111. [Google Scholar] [CrossRef]
- Song, G.; Napoli, E.; Wong, S.; Hagerman, R.; Liu, S.; Tassone, F.; Giulivi, C. Altered Redox Mitochondrial Biology in the Neurodegenerative Disorder Fragile X-Tremor/Ataxia Syndrome: Use of Antioxidants in Precision Medicine. Mol. Med. 2016, 22, 548–559. [Google Scholar] [CrossRef]
- Napoli, E.; Schneider, A.; Wang, J.Y.; Trivedi, A.; Carrillo, N.R.; Tassone, F.; Rogawski, M.; Hagerman, R.J.; Giulivi, C. Allopregnanolone Treatment Improves Plasma Metabolomic Profile Associated with GABA Metabolism in Fragile X-Associated Tremor/Ataxia Syndrome: A Pilot Study. Mol. Neurobiol. 2019, 56, 3702–3713. [Google Scholar] [CrossRef]
- Napoli, E.; Flores, A.; Mansuri, Y.; Hagerman, R.J.; Giulivi, C. Sulforaphane Improves Mitochondrial Metabolism in Fibroblasts from Patients with Fragile X-Associated Tremor and Ataxia Syndrome. Neurobiol. Dis. 2021, 157, 105427. [Google Scholar] [CrossRef]
- Santos, E.; Clark, C.; Biag, H.M.B.; Tang, S.J.; Kim, K.; Ponzini, M.D.; Schneider, A.; Giulivi, C.; Montanaro, F.A.M.; Gipe, J.T.E.; et al. Open-Label Sulforaphane Trial in FMR1 Premutation Carriers with Fragile-X-Associated Tremor and Ataxia Syndrome (FXTAS). Cells 2023, 12, 2773. [Google Scholar] [CrossRef] [PubMed]
- Šileikytė, J.; Devereaux, J.; de Jong, J.; Schiavone, M.; Jones, K.; Nilsen, A.; Bernardi, P.; Forte, M.; Cohen, M.S. Second-Generation Inhibitors of the Mitochondrial Permeability Transition Pore with Improved Plasma Stability. ChemMedChem 2019, 14, 1771–1782. [Google Scholar] [CrossRef]
- Naryzhnaya, N.V.; Maslov, L.N.; Oeltgen, P.R. Pharmacology of Mitochondrial Permeability Transition Pore Inhibitors. Drug Dev. Res. 2019, 80, 1013–1030. [Google Scholar] [CrossRef]
- Warne, J.; Pryce, G.; Hill, J.M.; Shi, X.; Lennerås, F.; Puentes, F.; Kip, M.; Hilditch, L.; Walker, P.; Simone, M.I.; et al. Selective Inhibition of the Mitochondrial Permeability Transition Pore Protects against Neurodegeneration in Experimental Multiple Sclerosis. J. Biol. Chem. 2016, 291, 4356–4373. [Google Scholar] [CrossRef]
- Roy, S.; Šileikyte, J.; Neuenswander, B.; Hedrick, M.P.; Chung, T.D.Y.; Aubé, J.; Schoenen, F.J.; Forte, M.A.; Bernardi, P. N-Phenylbenzamides as Potent Inhibitors of the Mitochondrial Permeability Transition Pore. ChemMedChem 2016, 11, 283–288. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grandi, M.; Galber, C.; Gatto, C.; Nobile, V.; Pucci, C.; Schaldemose Nielsen, I.; Boldrin, F.; Neri, G.; Chiurazzi, P.; Solaini, G.; et al. Mitochondrial Dysfunction Causes Cell Death in Patients Affected by Fragile-X-Associated Disorders. Int. J. Mol. Sci. 2024, 25, 3421. https://doi.org/10.3390/ijms25063421
Grandi M, Galber C, Gatto C, Nobile V, Pucci C, Schaldemose Nielsen I, Boldrin F, Neri G, Chiurazzi P, Solaini G, et al. Mitochondrial Dysfunction Causes Cell Death in Patients Affected by Fragile-X-Associated Disorders. International Journal of Molecular Sciences. 2024; 25(6):3421. https://doi.org/10.3390/ijms25063421
Chicago/Turabian StyleGrandi, Martina, Chiara Galber, Cristina Gatto, Veronica Nobile, Cecilia Pucci, Ida Schaldemose Nielsen, Francesco Boldrin, Giovanni Neri, Pietro Chiurazzi, Giancarlo Solaini, and et al. 2024. "Mitochondrial Dysfunction Causes Cell Death in Patients Affected by Fragile-X-Associated Disorders" International Journal of Molecular Sciences 25, no. 6: 3421. https://doi.org/10.3390/ijms25063421
APA StyleGrandi, M., Galber, C., Gatto, C., Nobile, V., Pucci, C., Schaldemose Nielsen, I., Boldrin, F., Neri, G., Chiurazzi, P., Solaini, G., Baracca, A., Giorgio, V., & Tabolacci, E. (2024). Mitochondrial Dysfunction Causes Cell Death in Patients Affected by Fragile-X-Associated Disorders. International Journal of Molecular Sciences, 25(6), 3421. https://doi.org/10.3390/ijms25063421