
Downregulation of Mitochondrial Fusion Protein Expression Affords Protection from Canonical Necroptosis in H9c2 Cardiomyoblasts
 , , ,
, , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
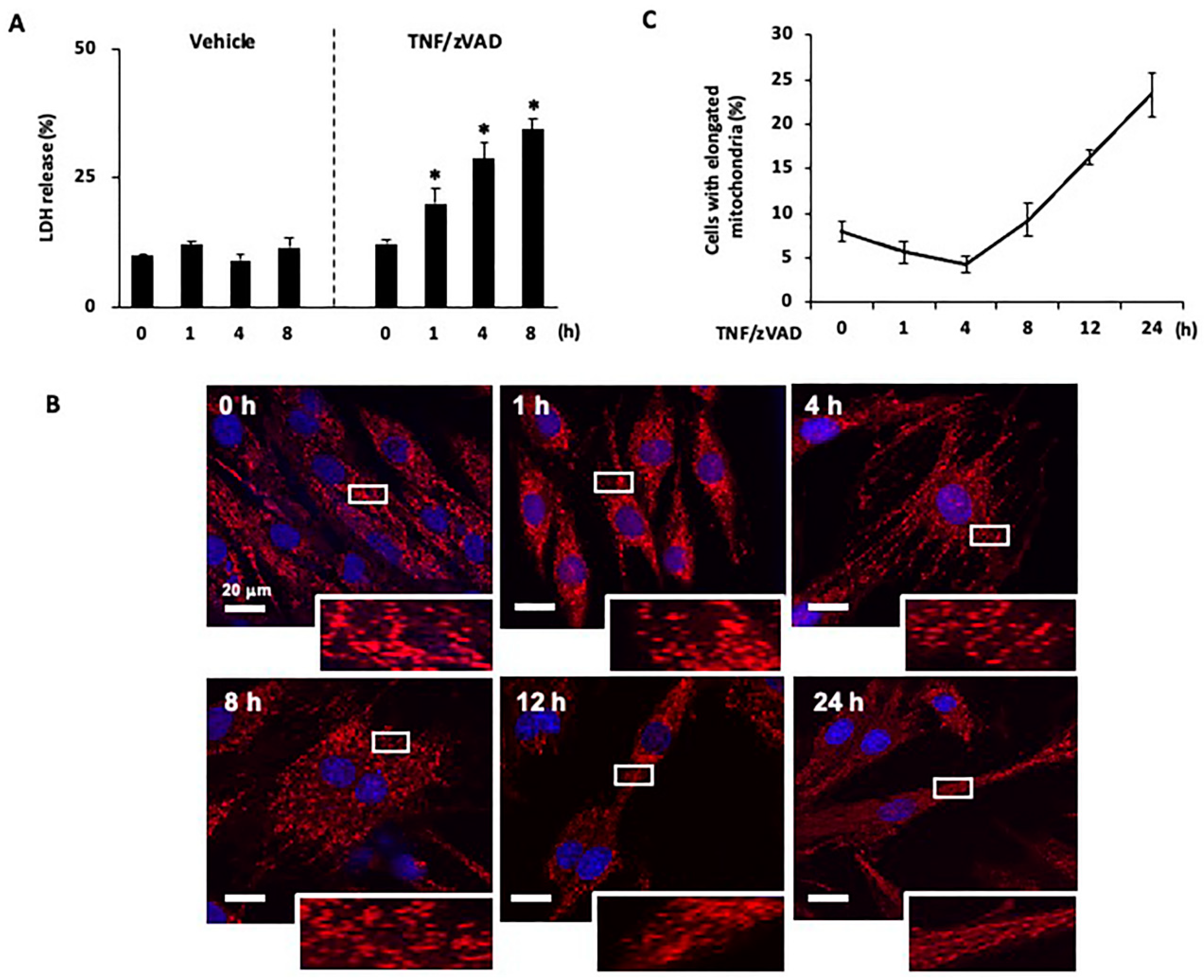
2.1. Time-Dependent Changes in Mitochondrial Morphology after the Induction of Necroptosis
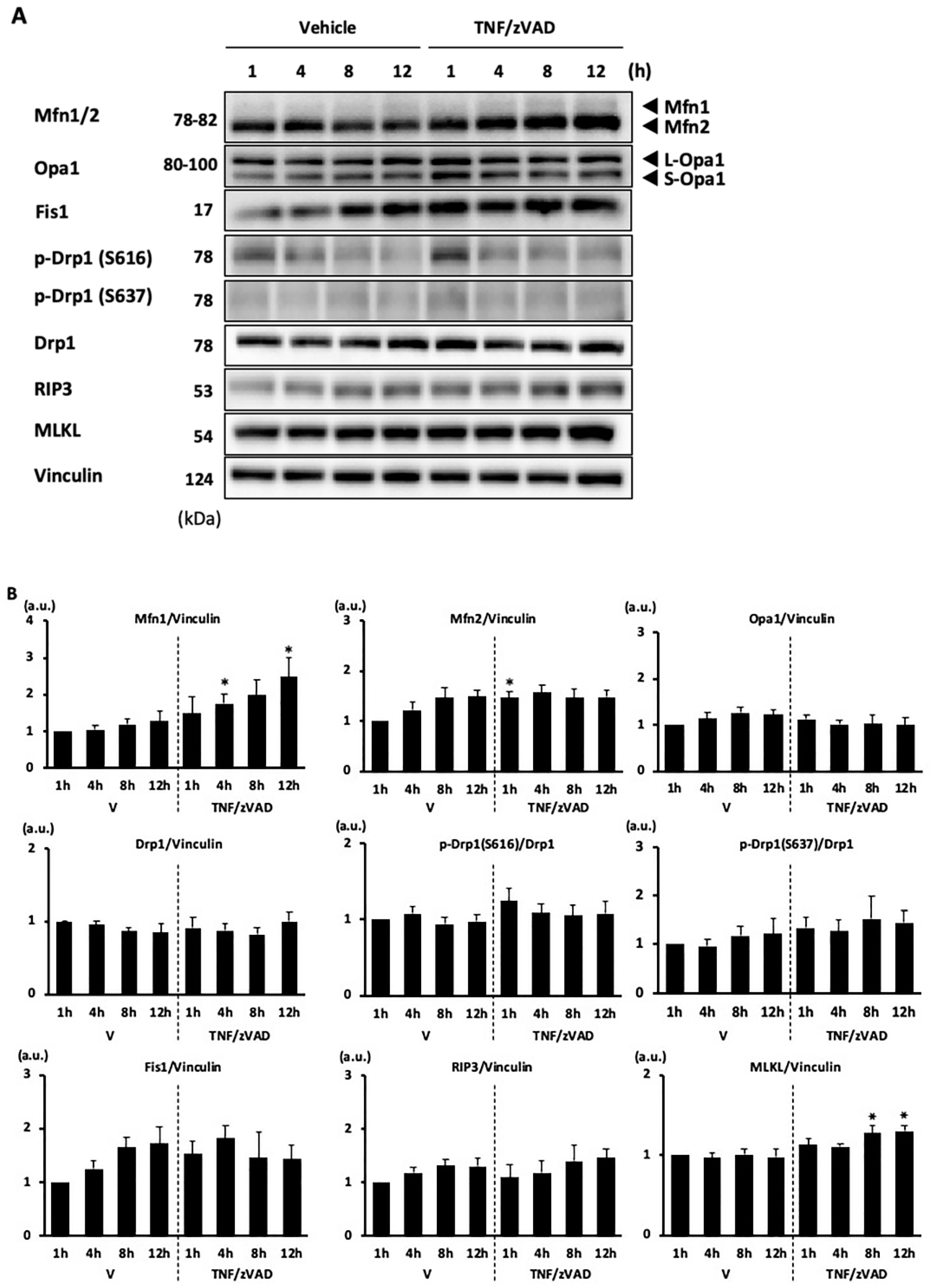
2.2. Changes in Mitochondrial Fusion/Fission Proteins following the Addition of TNF/zVAD
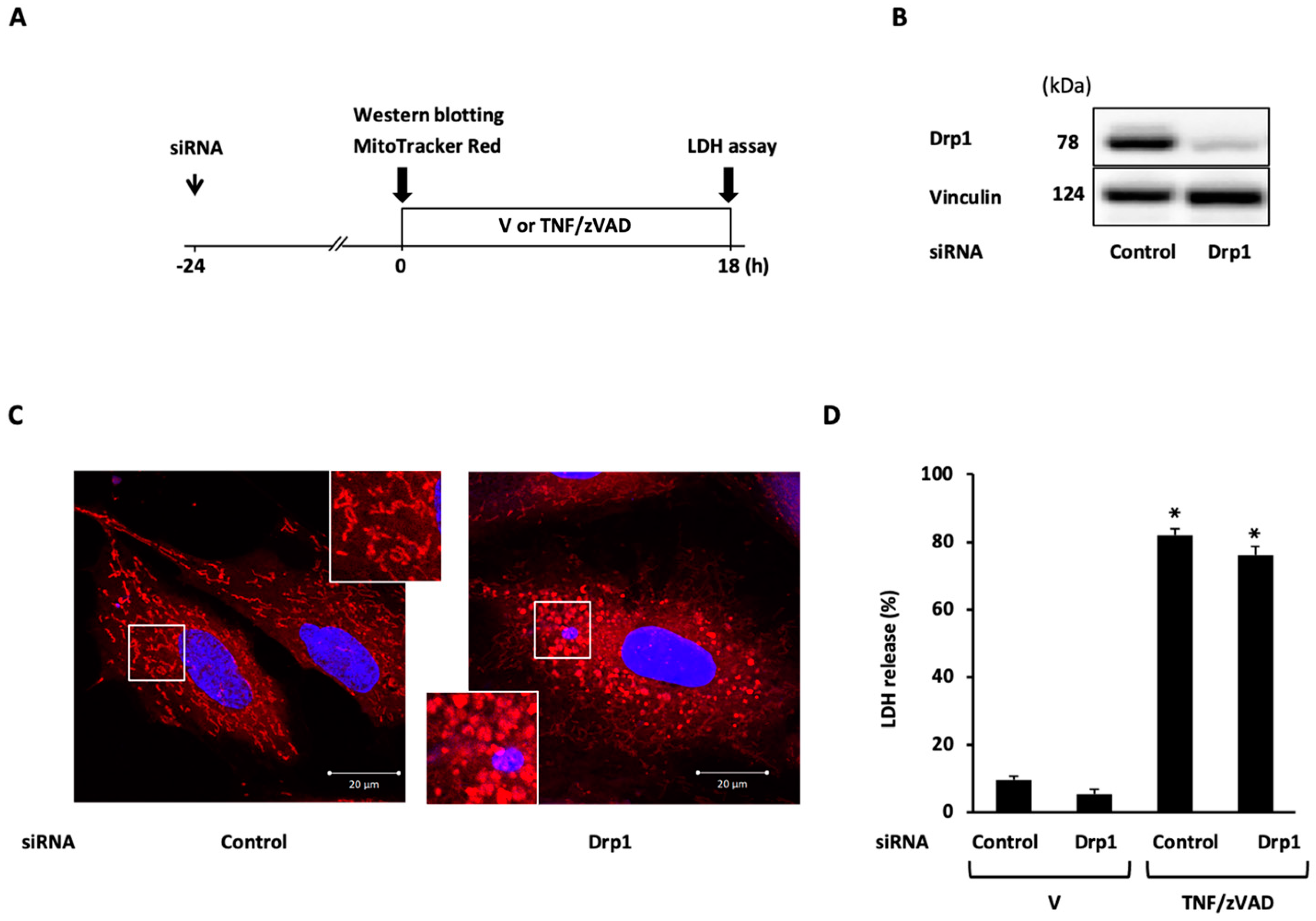
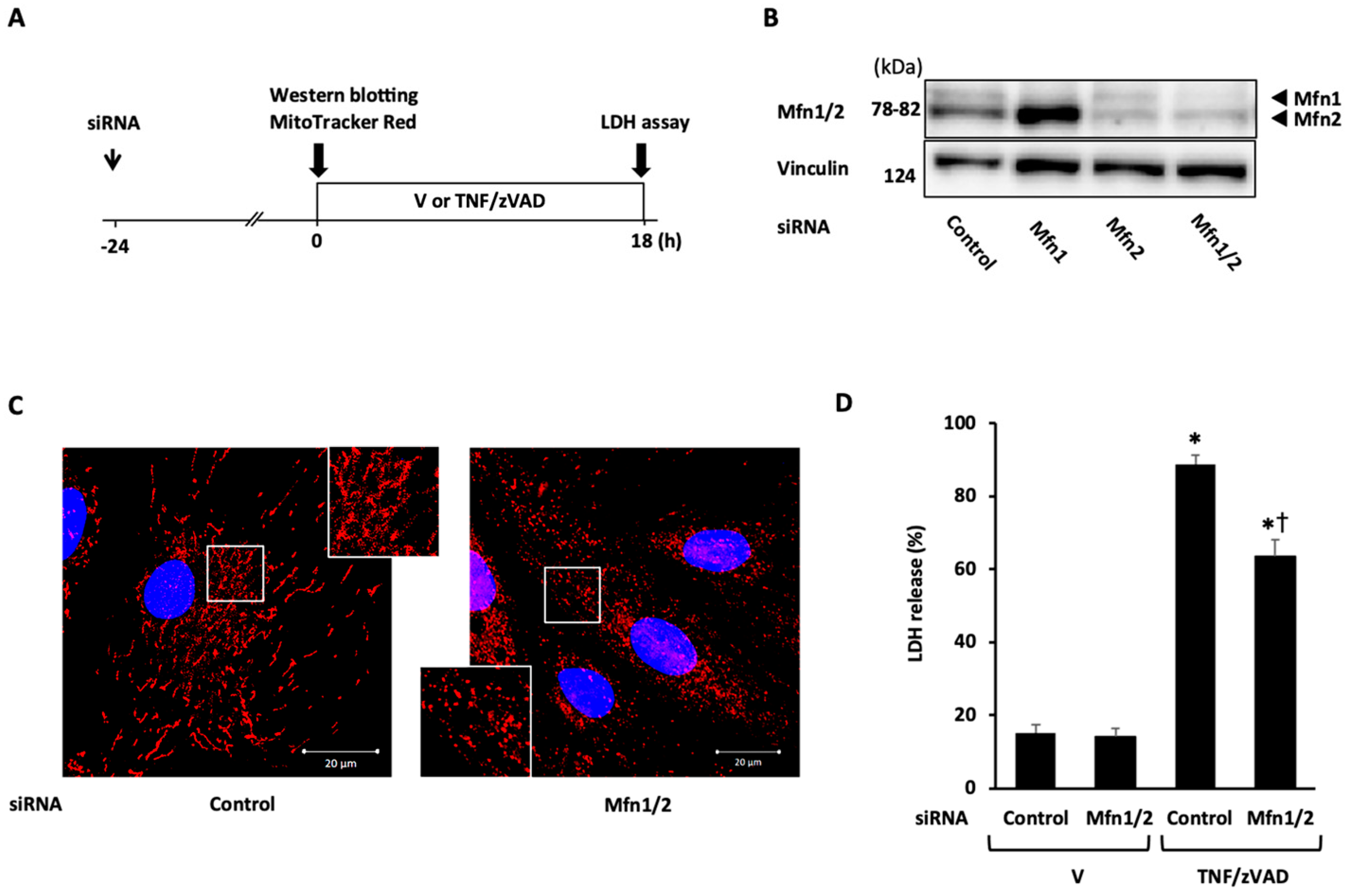
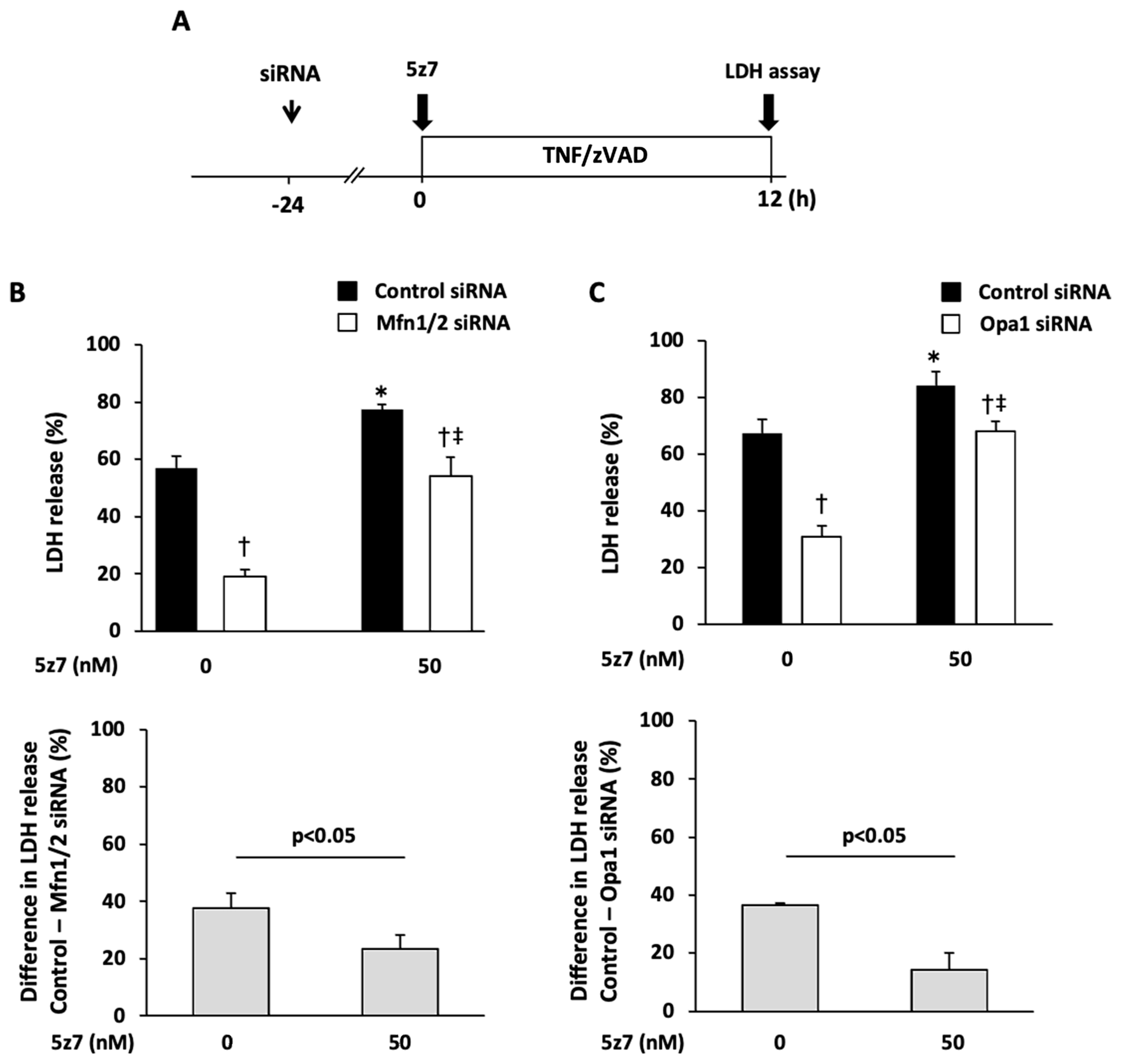
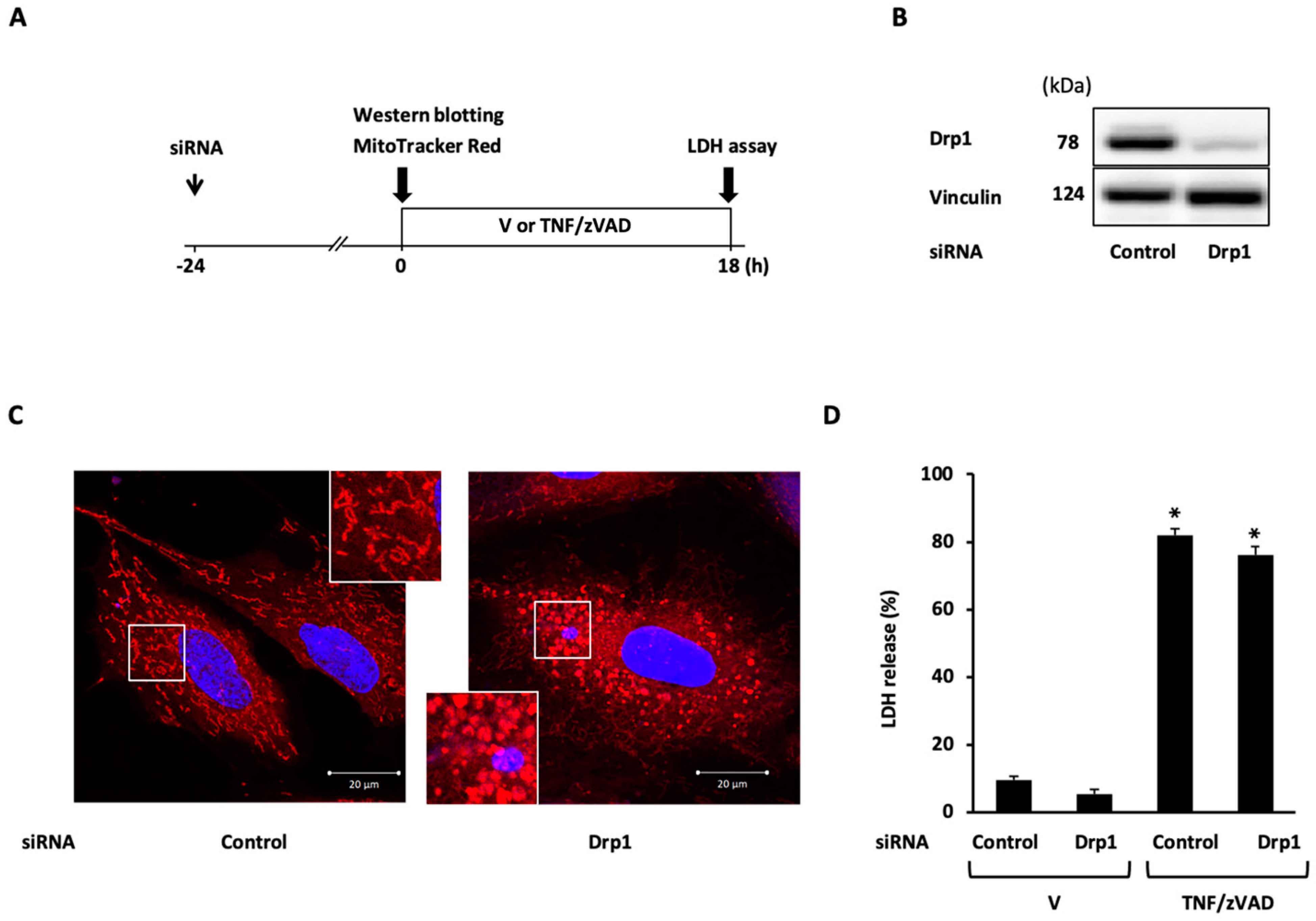
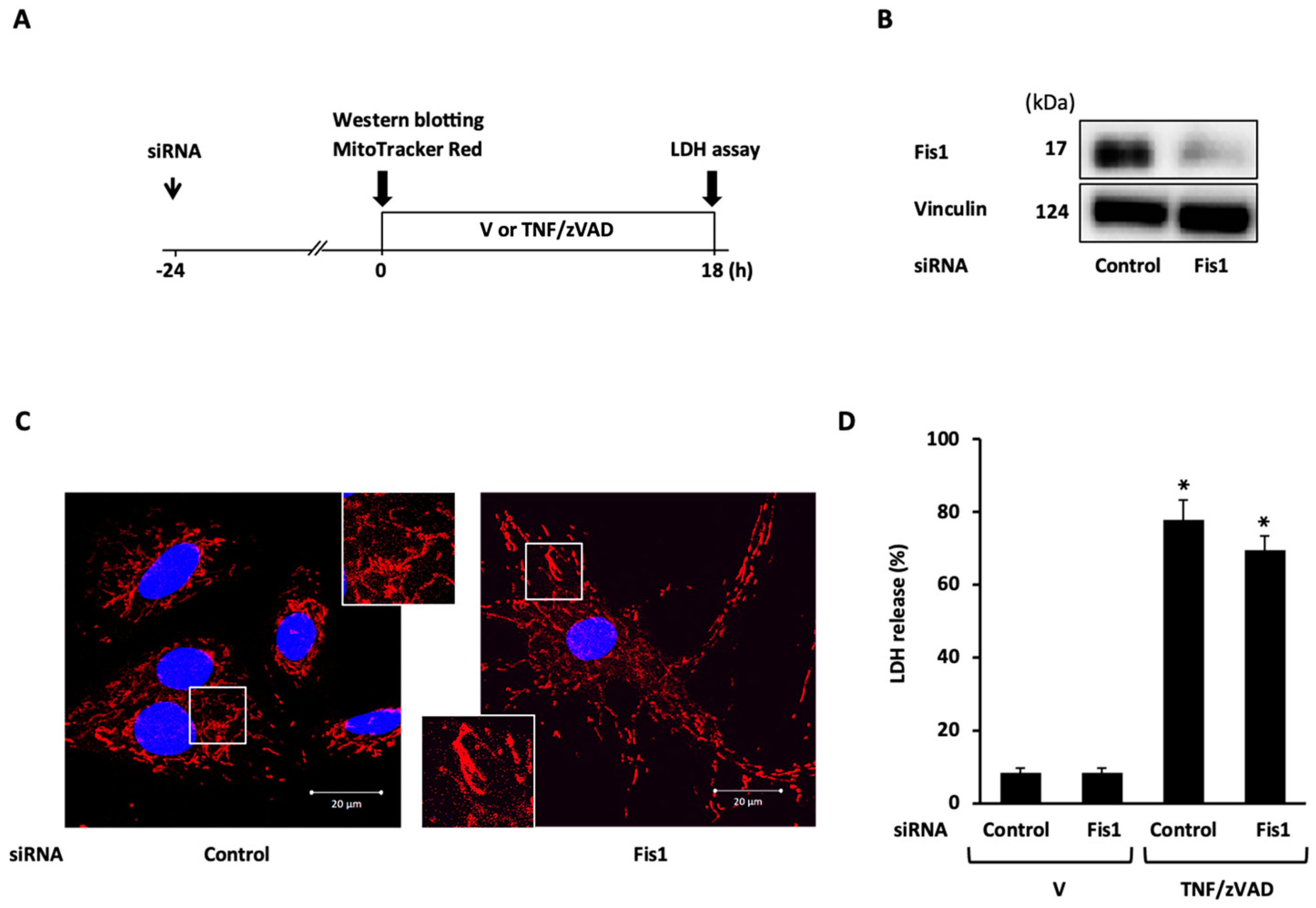
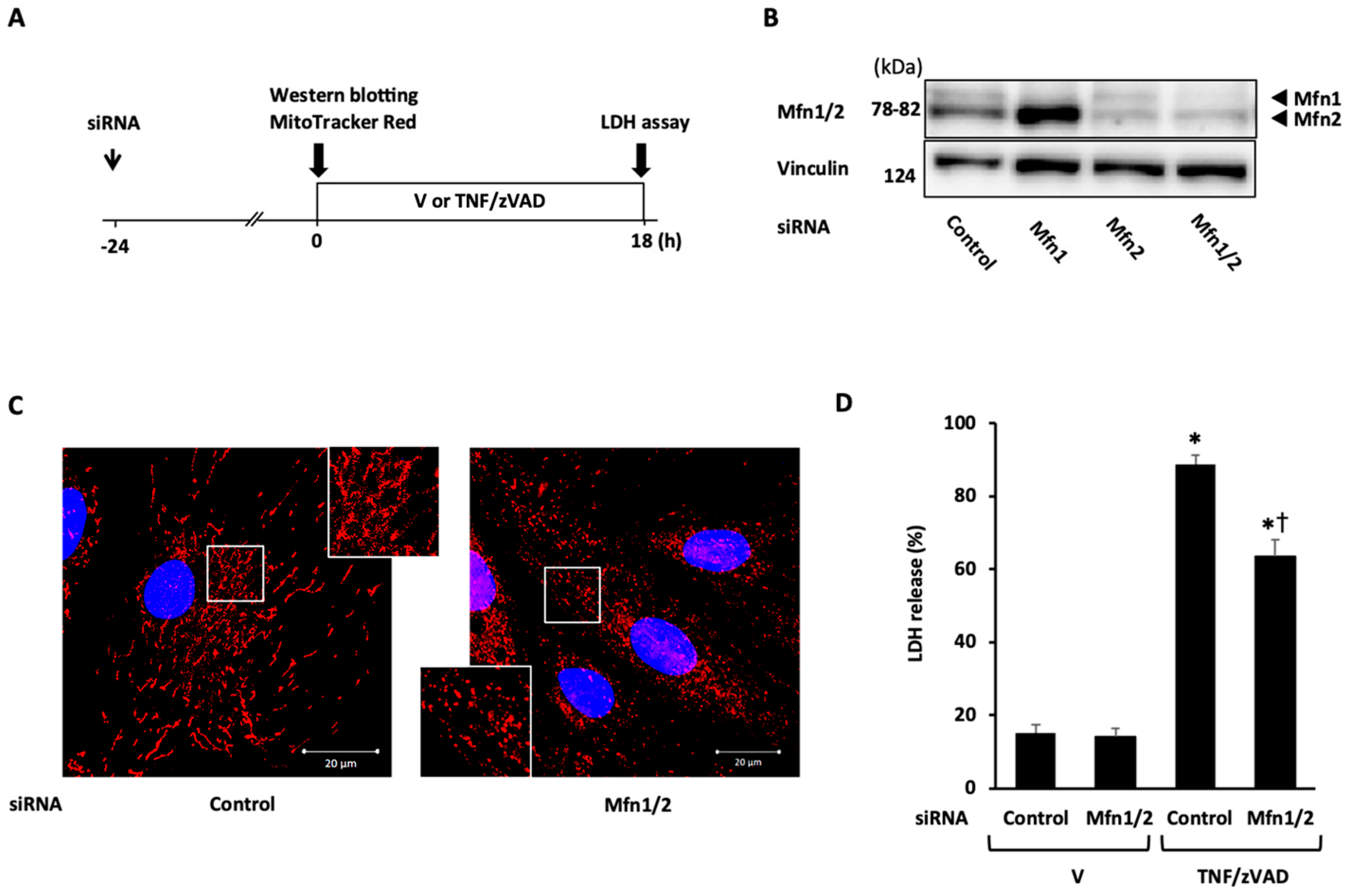
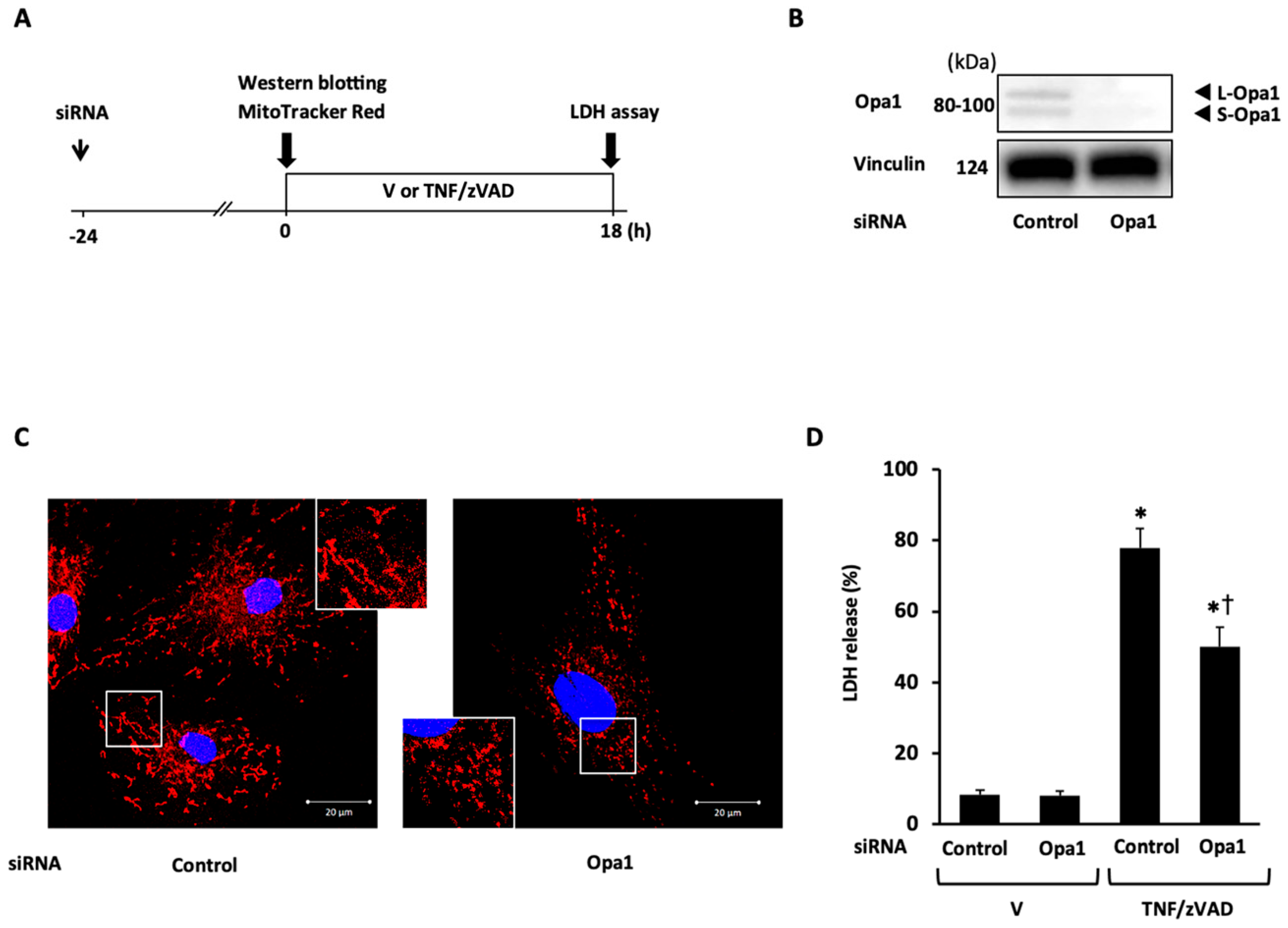
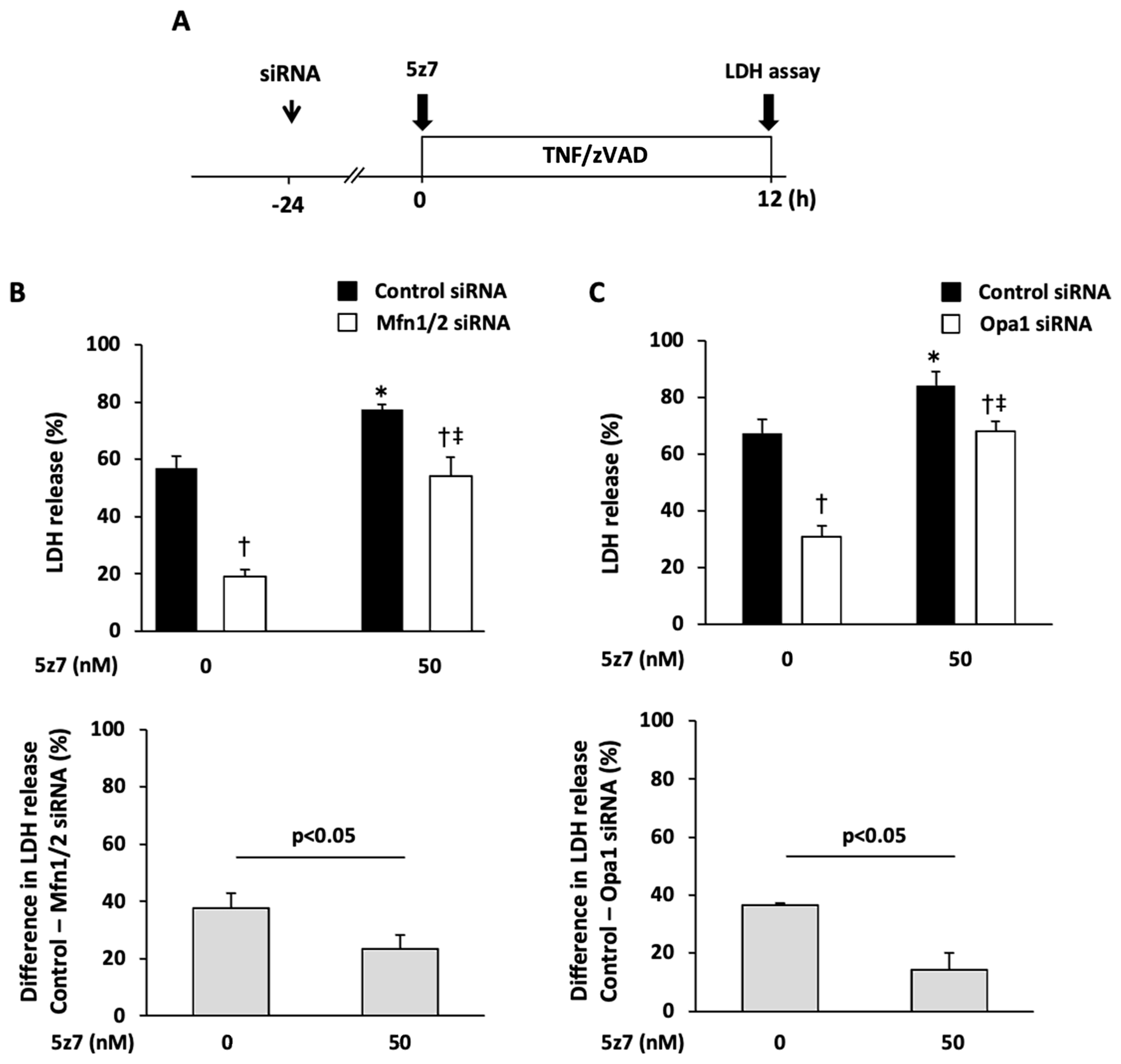
2.3. Role of Mitochondrial Fusion/Fission Proteins in Necroptosis
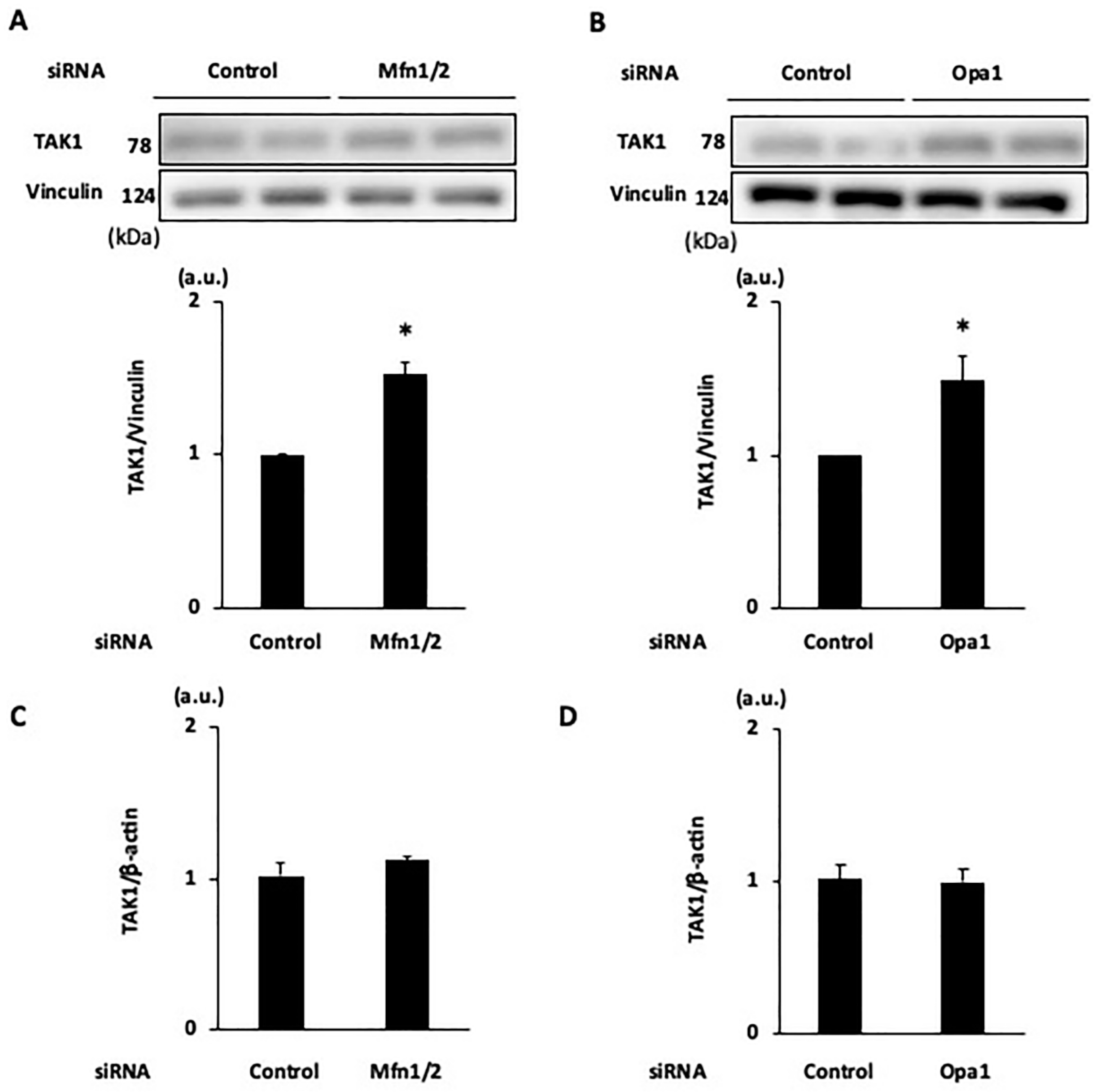
2.4. Effects of Knockdown of Mitochondrial Fusion Proteins on Necroptotic Signaling
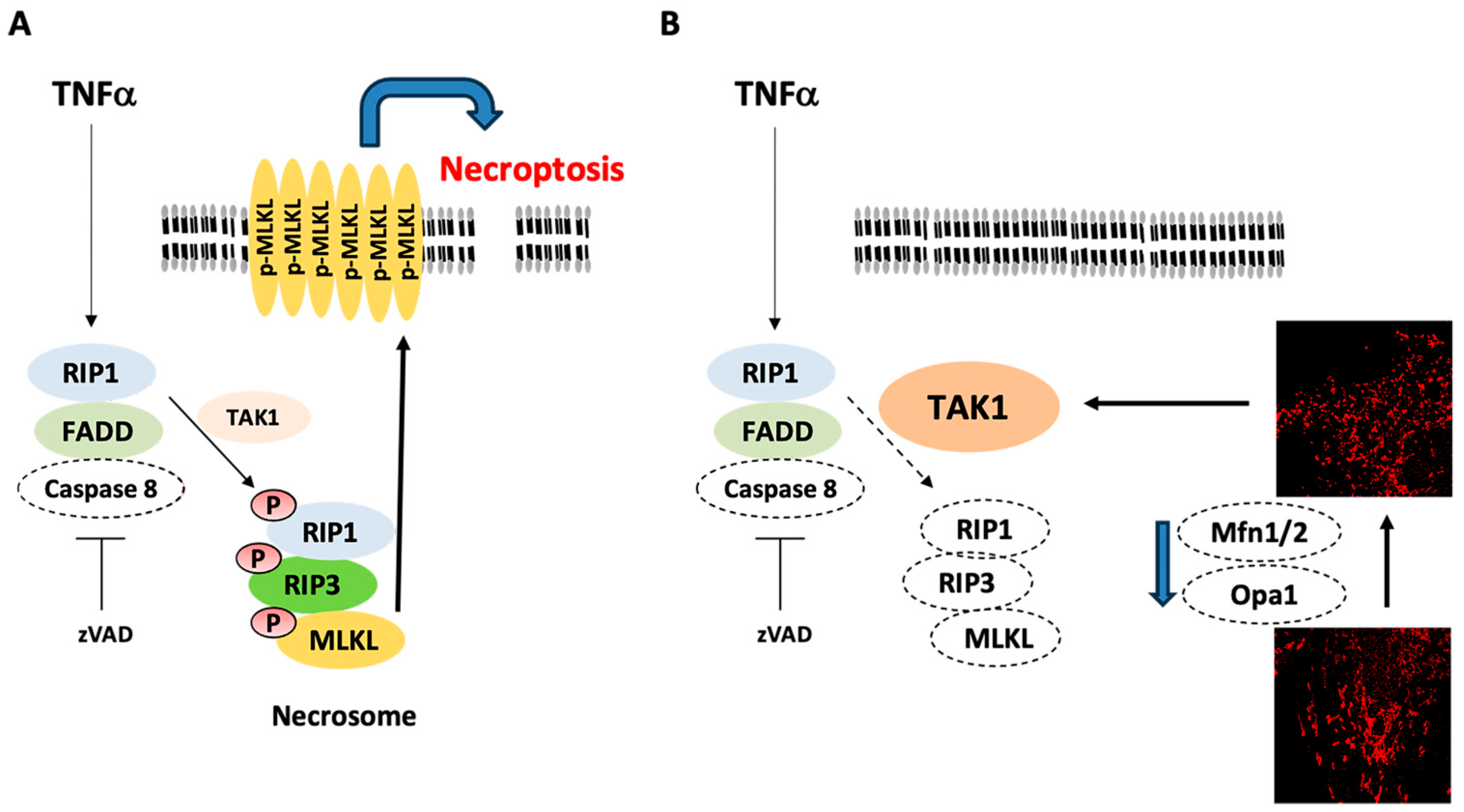
3. Discussion
4. Materials and Methods
4.1. Chemical Compounds
4.2. Cell Culture and Transfection
4.3. Experimental Protocols and Cell Death Assay
4.4. Western Blotting
4.5. Fluorescence Microscopy Analysis
4.6. mRNA Quantification
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arya, R.; White, K. Cell death in development: Signaling pathways and core mechanisms. Semin. Cell Dev. Biol. 2015, 39, 12–19. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and Execution Mechanisms of Necroptosis: An Overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Rall, G.F. Benefits and Perils of Necroptosis in Influenza Virus Infection. J. Virol. 2020, 94, e01101-19. [Google Scholar] [CrossRef]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef]
- Khoury, M.K.; Gupta, K.; Franco, S.R.; Liu, B. Necroptosis in the Pathophysiology of Disease. Am. J. Pathol. 2020, 190, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Huang, M.; Yang, X.; Hou, J. MLKL: Functions beyond serving as the Executioner of Necroptosis. Theranostics 2021, 11, 4759–4769. [Google Scholar] [CrossRef]
- Abe, K.; Yano, T.; Tanno, M.; Miki, T.; Kuno, A.; Sato, T.; Kouzu, H.; Nakata, K.; Ohwada, W.; Kimura, Y.; et al. mTORC1 inhibition attenuates necroptosis through RIP1 inhibition-mediated TFEB activation. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165552. [Google Scholar] [CrossRef]
- Li, L.; Tong, A.; Zhang, Q.; Wei, Y.; Wei, X. The molecular mechanisms of MLKL-dependent and MLKL-independent necrosis. J. Mol. Cell Biol. 2021, 13, 3–14. [Google Scholar] [CrossRef]
- Yoon, S.; Bogdanov, K.; Kovalenko, A.; Wallach, D. Necroptosis Is Preceded by Nuclear Translocation of the Signaling Proteins That Induce It. Cell Death Differ. 2016, 23, 253–260. [Google Scholar] [CrossRef]
- Weber, K.; Roelandt, R.; Bruggeman, I.; Estornes, Y.; Vandenabeele, P. Nuclear RIPK3 and MLKL Contribute to Cytosolic Necrosome Formation and Necroptosis. Commun. Biol. 2018, 1, 6. [Google Scholar] [CrossRef]
- Ino, S.; Yano, T.; Kuno, A.; Tanno, M.; Kouzu, H.; Sato, T.; Yamashita, T.; Ohwada, W.; Osanami, A.; Ogawa, T.; et al. Nuclear translocation of MLKL enhances necroptosis by a RIP1/RIP3-independent mechanism in H9c2 cardiomyoblasts. J. Pharmacol. Sci. 2023, 151, 134–143. [Google Scholar] [CrossRef]
- Chen, W.; Zhou, Z.; Li, L.; Zhong, C.Q.; Zheng, X.; Wu, X.; Zhang, Y.; Ma, H.; Huang, D.; Li, W.; et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J. Biol. Chem. 2013, 288, 16247–16261. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Ong, S.B.; Kalkhoran, S.B.; Hernández-Reséndiz, S.; Samangouei, P.; Ong, S.G.; Hausenloy, D.J. Mitochondrial-Shaping Proteins in Cardiac Health and Disease—The Long and the Short of It! Cardiovasc. Drugs Ther. 2017, 31, 87–107. [Google Scholar] [CrossRef]
- Delmotte, P.; Marin Mathieu, N.; Sieck, G.C. TNFα induces mitochondrial fragmentation and biogenesis in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L137–L151. [Google Scholar] [CrossRef]
- Shen, Y.L.; Shi, Y.Z.; Chen, G.G.; Wang, L.L.; Zheng, M.Z.; Jin, H.F.; Chen, Y.Y. TNF-α induces Drp1-mediated mitochondrial fragmentation during inflammatory cardiomyocyte injury. Int. J. Mol. Med. 2018, 41, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Preston, K.J.; Cooper, H.A.; Boyer, M.J.; Escoto, K.M.; Poltronetti, A.J.; Elliott, K.J.; Kuroda, R.; Miyao, M.; Sesaki, H.; et al. Mitochondrial Fission Mediates Endothelial Inflammation. Hypertension 2020, 76, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Nan, J.; Hu, H.; Sun, Y.; Zhu, L.; Wang, Y.; Zhong, Z.; Zhao, J.; Zhang, N.; Wang, Y.; Wang, Y.; et al. TNFR2 Stimulation Promotes Mitochondrial Fusion via Stat3- and NF-kB-Dependent Activation of OPA1 Expression. Circ. Res. 2017, 121, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, M.; Yano, T.; Tanno, M.; Abe, K.; Ishikawa, S.; Miki, T.; Kuno, A.; Tobisawa, T.; Muratsubaki, S.; Ohno, K.; et al. Suppression of autophagic flux contributes to cardiomyocyte death by activation of necroptotic pathways. J. Mol. Cell. Cardiol. 2017, 108, 203–213. [Google Scholar] [CrossRef]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Ishikita, A.; Matoba, T.; Ikeda, G.; Koga, J.; Mao, Y.; Nakano, K.; Takeuchi, O.; Sadoshima, J.; Egashira, K. Nanoparticle-Mediated Delivery of Mitochondrial Division Inhibitor 1 to the Myocardium Protects the Heart From Ischemia-Reperfusion Injury Through Inhibition of Mitochondria Outer Membrane Permeabilization: A New Therapeutic Modality for Acute Myocardial Infarction. J. Am. Heart Assoc. 2016, 5, e003872. [Google Scholar]
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn, G.W., 2nd. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015, 21, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Li, A.; Liu, B.; Jiang, W.; Gao, M.; Tian, X.; Gong, G. Mitochondrial fusion mediated by fusion promotion and fission inhibition directs adult mouse heart function toward a different direction. FASEB J. 2019, 34, 663–675. [Google Scholar] [CrossRef]
- Marshall, K.D.; Baines, C.P. Necroptosis: Is there a role for mitochondria? Front. Physiol. 2014, 5, 323. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The Mitochondrial Phosphatase PGAM5 Functions at the Convergence Point of Multiple Necrotic Death Pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef]
- Remijsen, Q.; Goossens, V.; Grootjans, S.; Van den Haute, C.; Vanlangenakker, N.; Dondelinger, Y.; Roelandt, R.; Bruggeman, I.; Gonçalves, A.; Bertrand, M.J.M.; et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 2014, 5, e1004. [Google Scholar] [CrossRef]
- Moujalled, D.M.; Cook, W.D.; Murphy, J.M.; Vaux, D.L. Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis. 2014, 5, e1086. [Google Scholar] [CrossRef]
- Hildebrand, J.M.; Tanzer, M.C.; Lucet, I.S.; Young, S.N.; Spall, S.K.; Sharma, P.; Pierotti, C.; Garnier, J.-M.; Dobson, R.C.; Webb, A.I.; et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 15072–15077. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Davies, K.A.; Fitzgibbon, C.; Young, S.N.; Garnish, S.E.; Horne, C.R.; Luo, C.; Garnier, J.-M.; Liang, L.-Y.; Cowan, A.D.; et al. Human RIPK3 maintains MLKL in an inactive conformation prior to cell death by necroptosis. Nat. Commun. 2021, 12, 6783. [Google Scholar] [CrossRef]
- Tait, S.W.; Oberst, A.; Quarato, G.; Milasta, S.; Haller, M.; Wang, R.; Karvela, M.; Ichim, G.; Yatim, N.; Albert, M.L.; et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013, 5, 878–885. [Google Scholar] [CrossRef]
- Sidarala, V.; Zhu, J.; Levi-D’Ancona, E.; Pearson, G.L.; Reck, E.C.; Walker, E.M.; Kaufman, B.A.; Soleimanpour, S.A. Mitofusin 1 and 2 regulation of mitochondrial DNA content is a critical determinant of glucose homeostasis. Nat. Commun. 2022, 13, 2340. [Google Scholar] [CrossRef]
- Kawalec, M.; Boratyńska-Jasińska, A.; Beręsewicz, M.; Dymkowska, D.; Zabłocki, K.; Zabłocka, B. Mitofusin 2 Deficiency Affects Energy Metabolism and Mitochondrial Biogenesis in MEF Cells. PLoS ONE 2015, 10, e0134162. [Google Scholar] [CrossRef]
- Li, L.; Chen, Y.; Doan, J.; Murray, J.; Molkentin, J.D.; Liu, Q. Transforming Growth Factor β–Activated Kinase 1 Signaling Pathway Critically Regulates Myocardial Survival and Remodeling. Circulation 2014, 130, 2162–2172. [Google Scholar] [CrossRef] [PubMed]
- Hindi, S.M.; Sato, S.; Xiong, G.; Bohnert, K.R.; Gibb, A.A.; Gallot, Y.S.; McMillan, J.D.; Hill, B.G.; Uchida, S.; Kumar, A. TAK1 regulates skeletal muscle mass and mitochondrial function. J. Clin. Investig. 2018, 3, e98441. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Sharma, A.K.; Nellore, K.; A Narkar, V.; Kumar, A. TAK1 preserves skeletal muscle mass and mitochondrial function through redox homeostasis. FASEB BioAdv. 2020, 2, 538–553. [Google Scholar] [CrossRef]
- Deng, L.; Yi, S.; Yin, X.; Li, Y.; Luan, Q. MFN2 knockdown promotes osteogenic differentiation of iPSC-MSCs through aerobic glycolysis mediated by the Wnt/β-catenin signaling pathway. Stem. Cell Res. Ther. 2022, 13, 162. [Google Scholar] [CrossRef]
- Son, J.M.; Sarsour, E.H.; Balaraju, A.K.; Fussell, J.; Kalen, A.L.; Wagner, B.A.; Buettner, G.R.; Goswami, P.C. Mitofusin 1 and optic atrophy 1 shift metabolism to mitochondrial respiration during aging. Aging Cell 2017, 16, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Chen, K.-H.; Guo, Y.; Liao, H.; Tang, J.; Xiao, R.-P. Mitofusin 2 Triggers Vascular Smooth Muscle Cell Apoptosis via Mitochondrial Death Pathway. Circ. Res. 2007, 101, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Chen, G.; Li, X.; Wu, X.; Chang, Z.; Xu, J.; Zhu, Y.; Yin, P.; Liang, X.; Dong, L. MFN2 suppresses cancer progression through inhibition of mTORC2/Akt signaling. Sci Rep. 2017, 7, 41718. [Google Scholar] [CrossRef]
- Zhang, D.; Gaussin, V.; Taffet, G.E.; Belaguli, N.S.; Yamada, M.; Schwartz, R.J.; Michael, L.H.; Overbeek, P.A.; Schneider, M.D. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat. Med. 2000, 6, 556–563. [Google Scholar] [CrossRef]
- Koitabashi, N.; Danner, T.; Zaiman, A.L.; Pinto, Y.M.; Rowell, J.; Mankowski, J.; Zhang, D.; Nakamura, T.; Takimoto, E.; Kass, D.A. Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload. J. Clin. Investig. 2011, 121, 2301–2312. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toda, Y.; Ong, S.-B.; Yano, T.; Kuno, A.; Kouzu, H.; Sato, T.; Ohwada, W.; Tatekoshi, Y.; Ogawa, T.; Shimizu, M.; et al. Downregulation of Mitochondrial Fusion Protein Expression Affords Protection from Canonical Necroptosis in H9c2 Cardiomyoblasts. Int. J. Mol. Sci. 2024, 25, 2905. https://doi.org/10.3390/ijms25052905
Toda Y, Ong S-B, Yano T, Kuno A, Kouzu H, Sato T, Ohwada W, Tatekoshi Y, Ogawa T, Shimizu M, et al. Downregulation of Mitochondrial Fusion Protein Expression Affords Protection from Canonical Necroptosis in H9c2 Cardiomyoblasts. International Journal of Molecular Sciences. 2024; 25(5):2905. https://doi.org/10.3390/ijms25052905
Chicago/Turabian StyleToda, Yuki, Sang-Bing Ong, Toshiyuki Yano, Atsushi Kuno, Hidemichi Kouzu, Tatsuya Sato, Wataru Ohwada, Yuki Tatekoshi, Toshifumi Ogawa, Masaki Shimizu, and et al. 2024. "Downregulation of Mitochondrial Fusion Protein Expression Affords Protection from Canonical Necroptosis in H9c2 Cardiomyoblasts" International Journal of Molecular Sciences 25, no. 5: 2905. https://doi.org/10.3390/ijms25052905
APA StyleToda, Y., Ong, S.-B., Yano, T., Kuno, A., Kouzu, H., Sato, T., Ohwada, W., Tatekoshi, Y., Ogawa, T., Shimizu, M., Tanno, M., & Furuhashi, M. (2024). Downregulation of Mitochondrial Fusion Protein Expression Affords Protection from Canonical Necroptosis in H9c2 Cardiomyoblasts. International Journal of Molecular Sciences, 25(5), 2905. https://doi.org/10.3390/ijms25052905