

Gemcitabine Modulates HLA-I Regulation to Improve Tumor Antigen Presentation by Pancreatic Cancer Cells
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
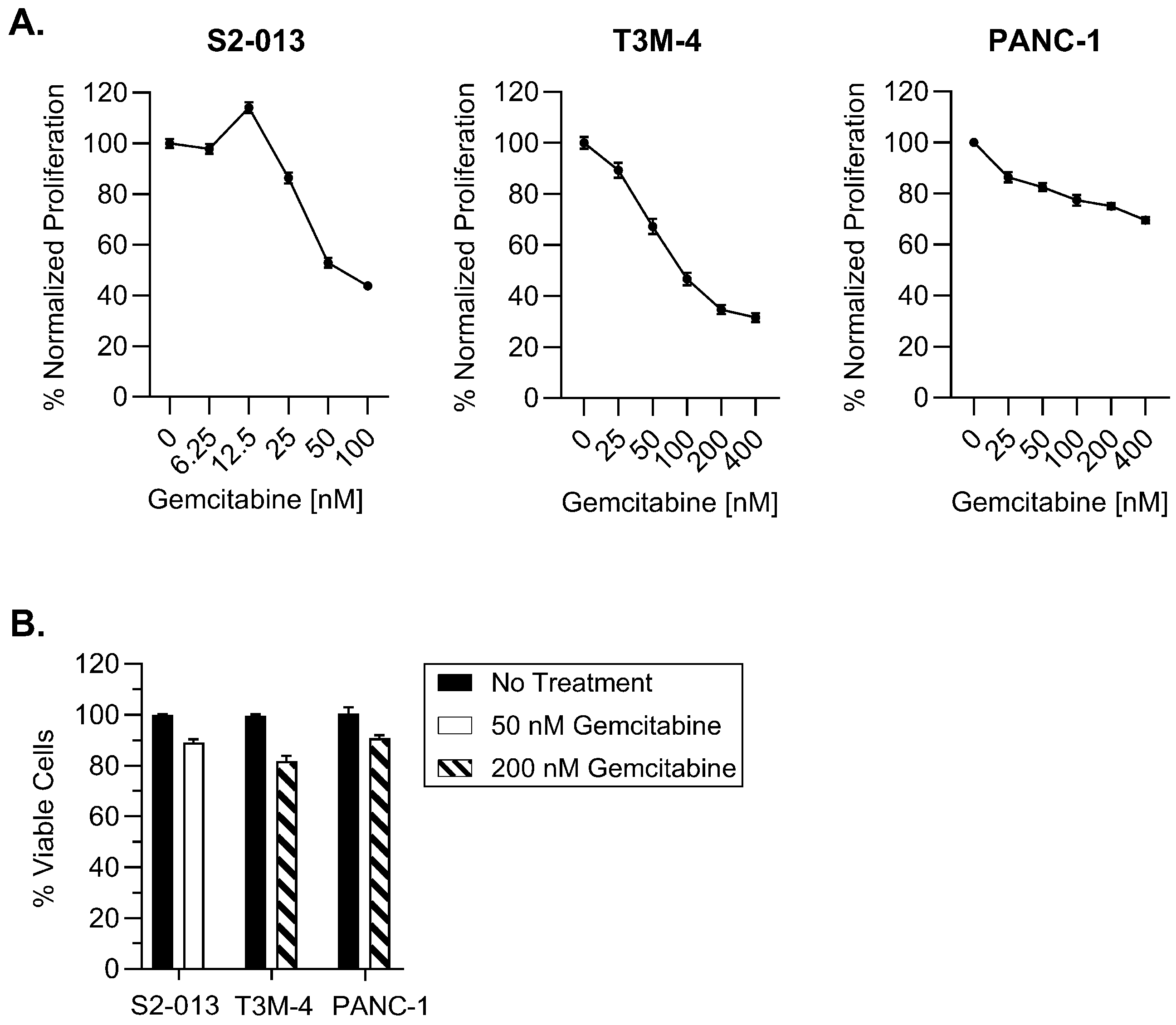
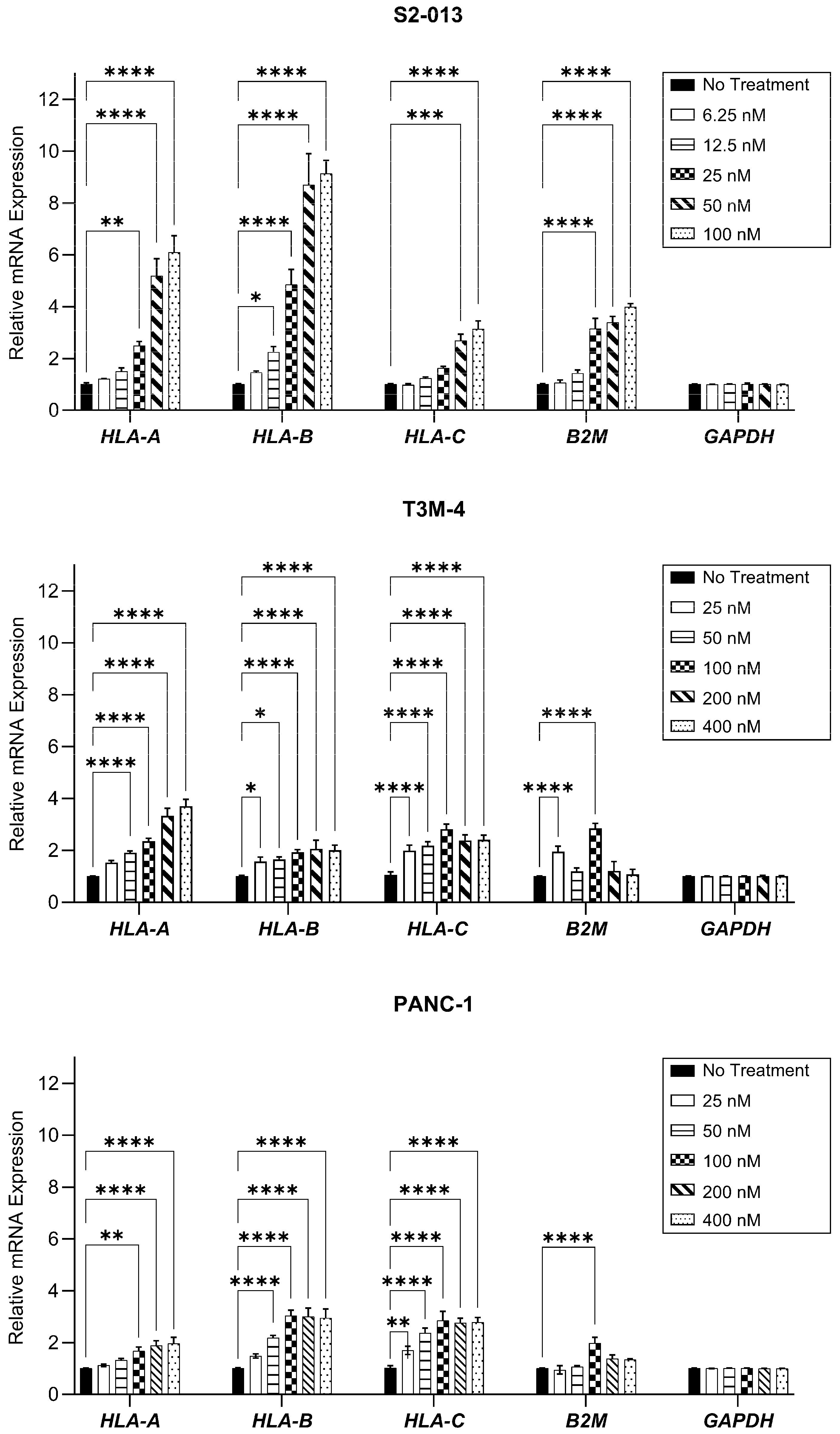
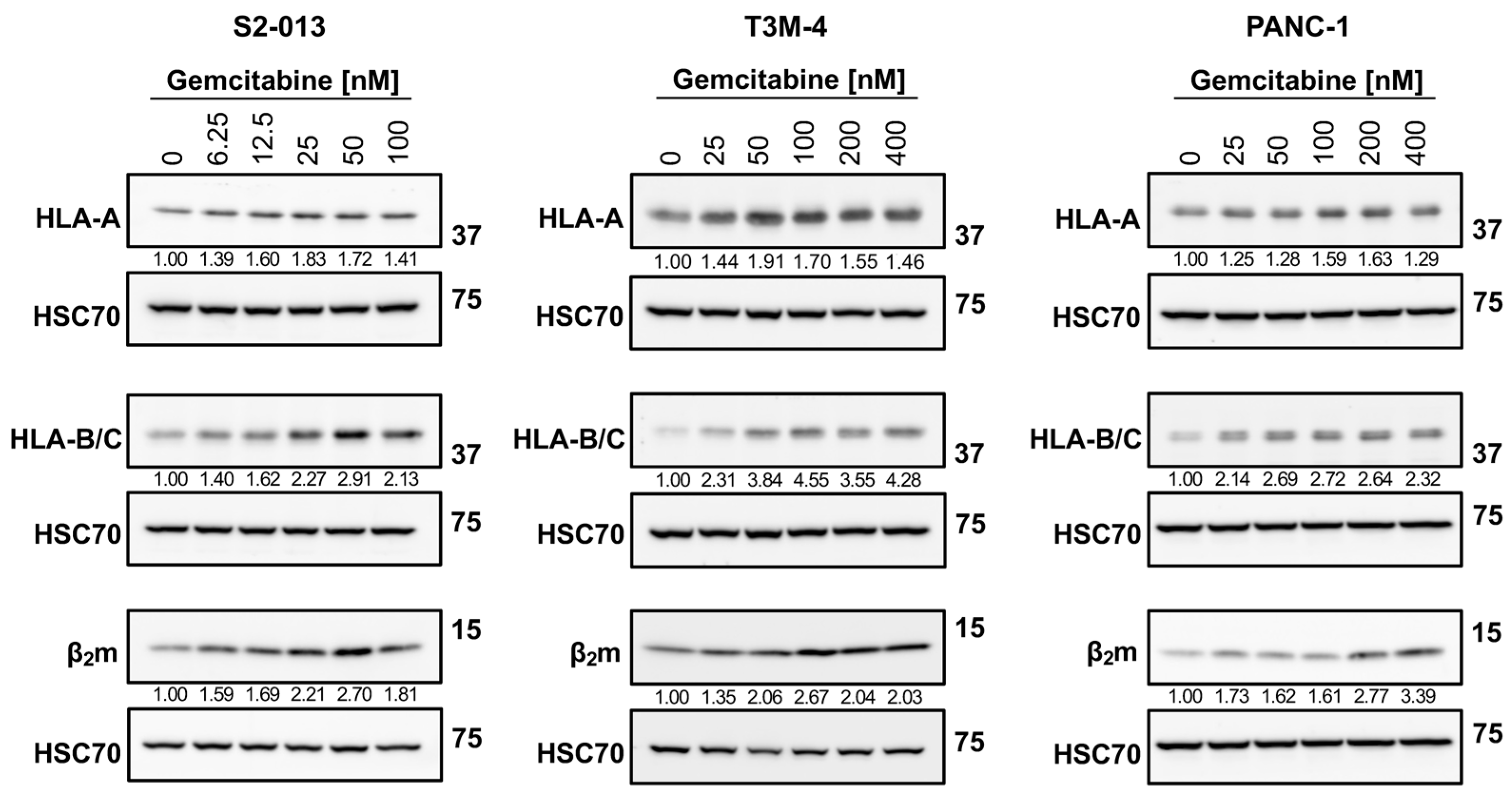
2.1. Gemcitabine Increases HLA-I Expression by Pancreatic Cancer Cells
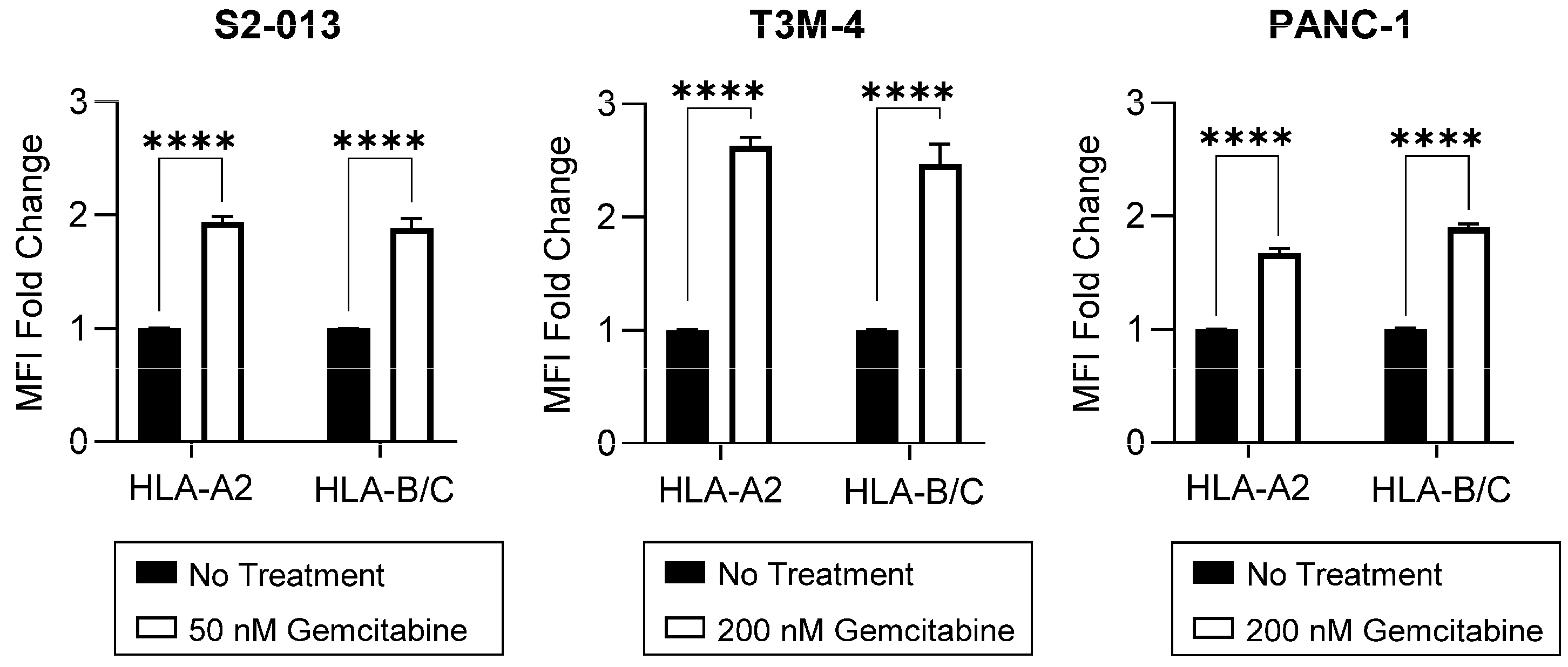
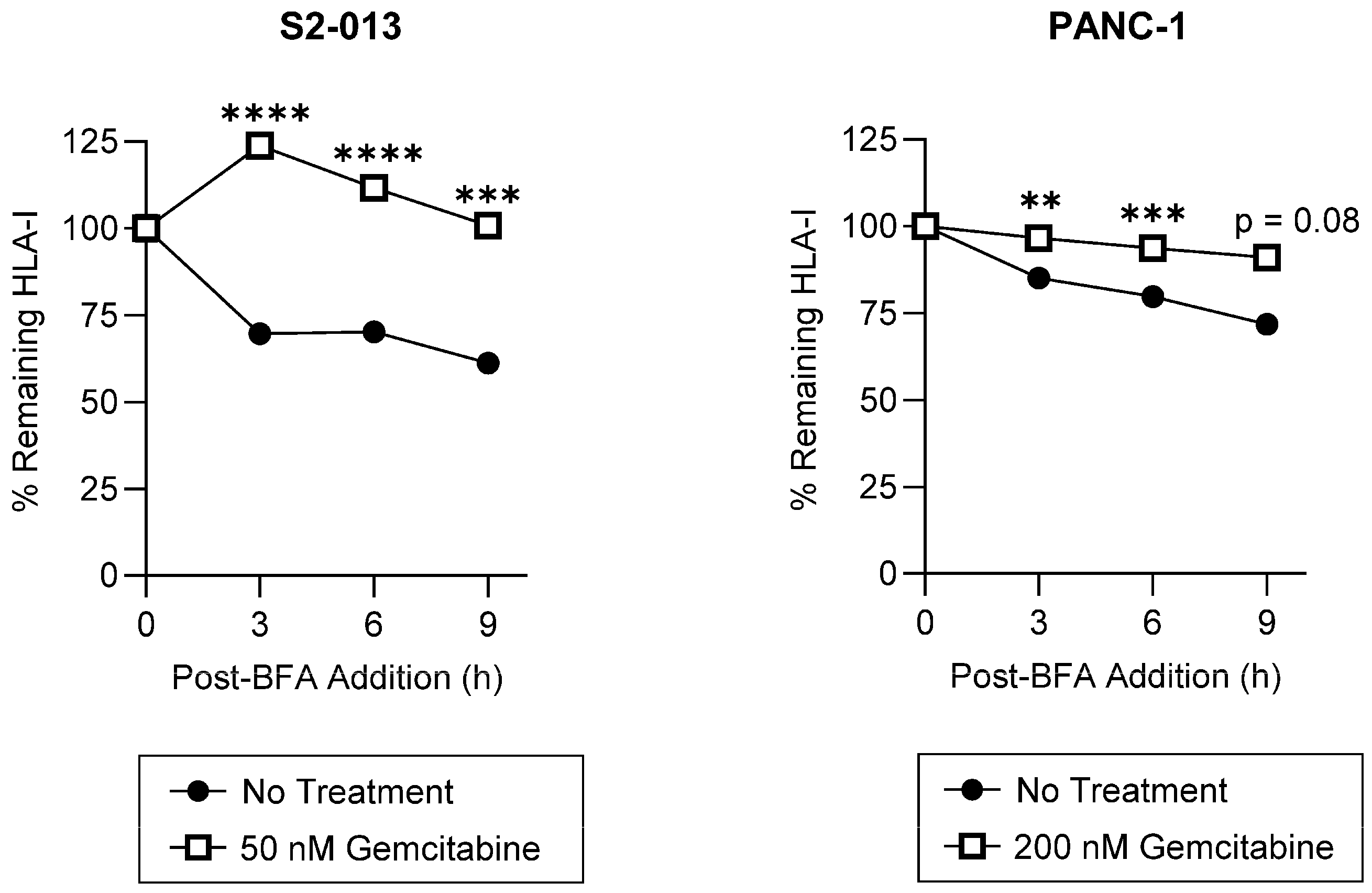
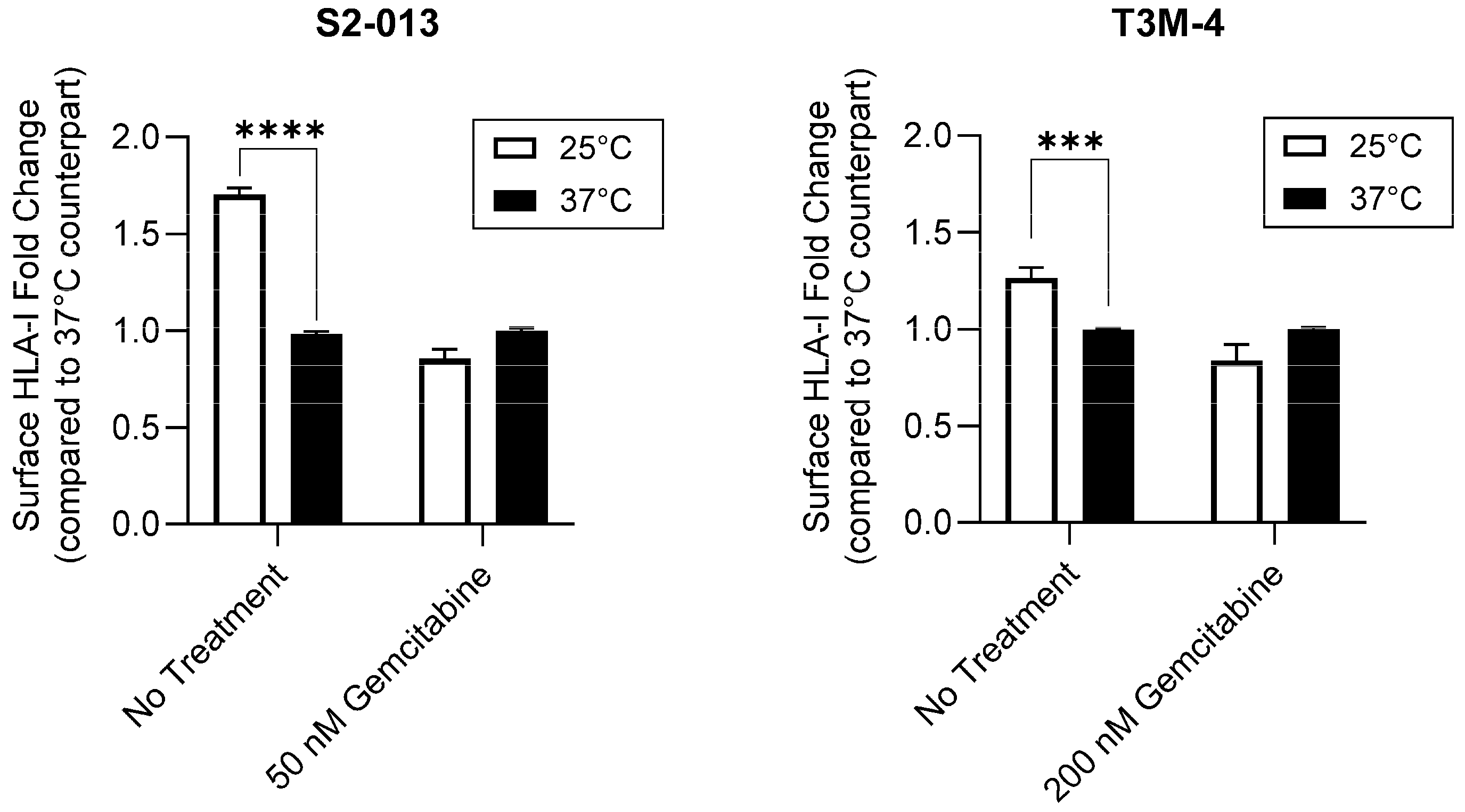
2.2. Gemcitabine Enhances Surface Stability of HLA-I on Pancreatic Cancer Cells
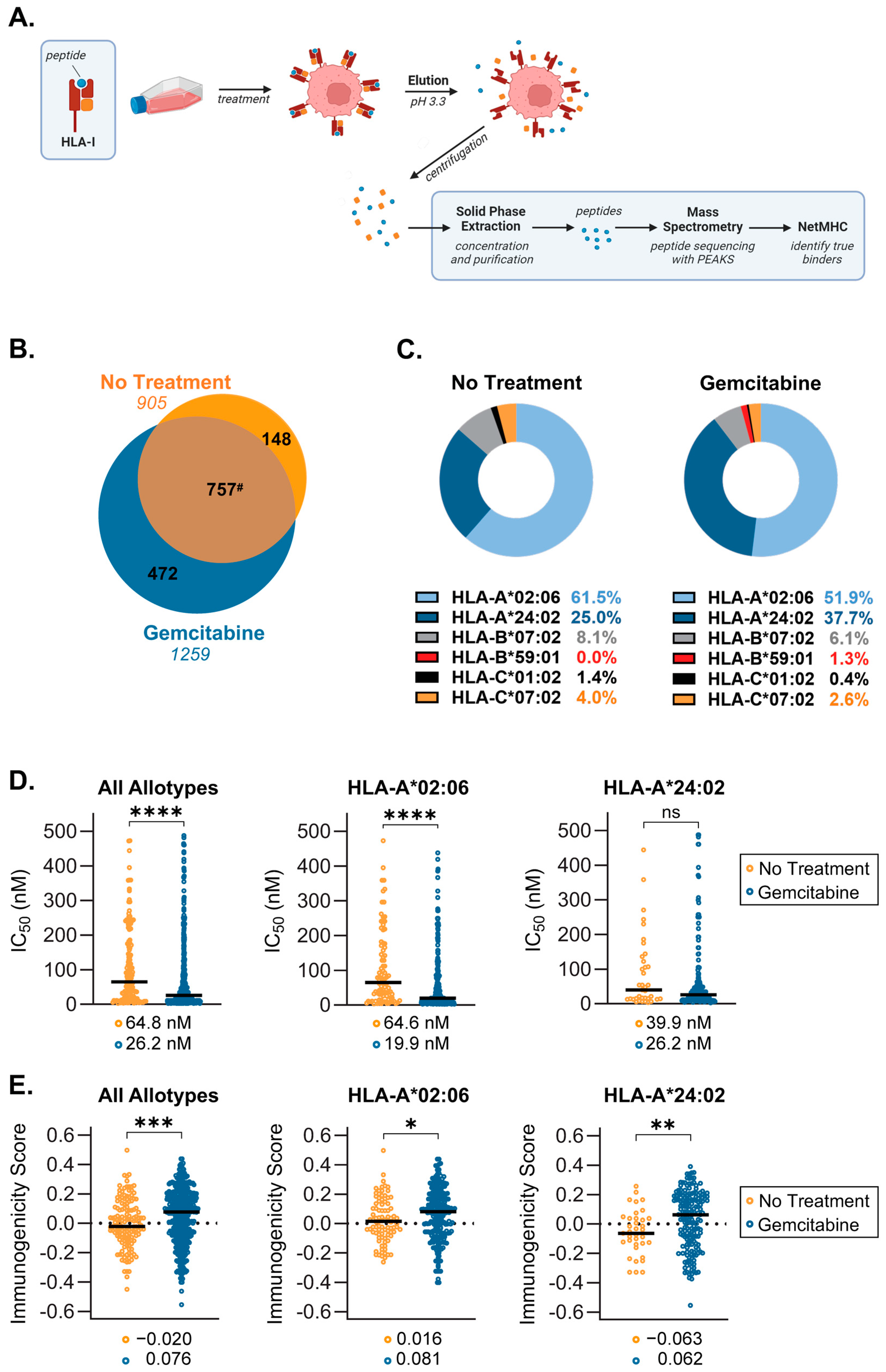
2.3. Gemcitabine Induces Presentation of Unique, Higher Quality Peptides by S2-013 Cells
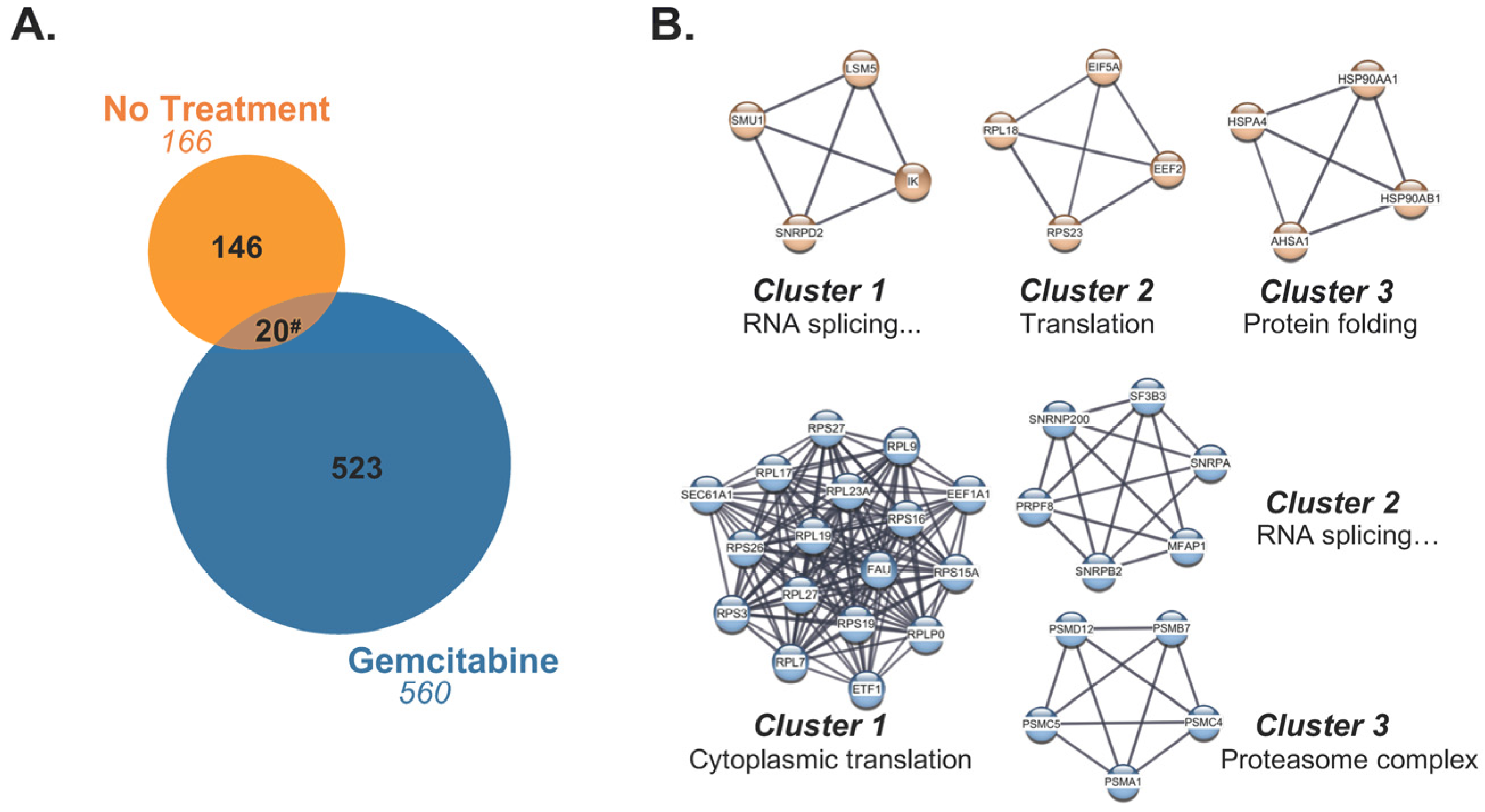
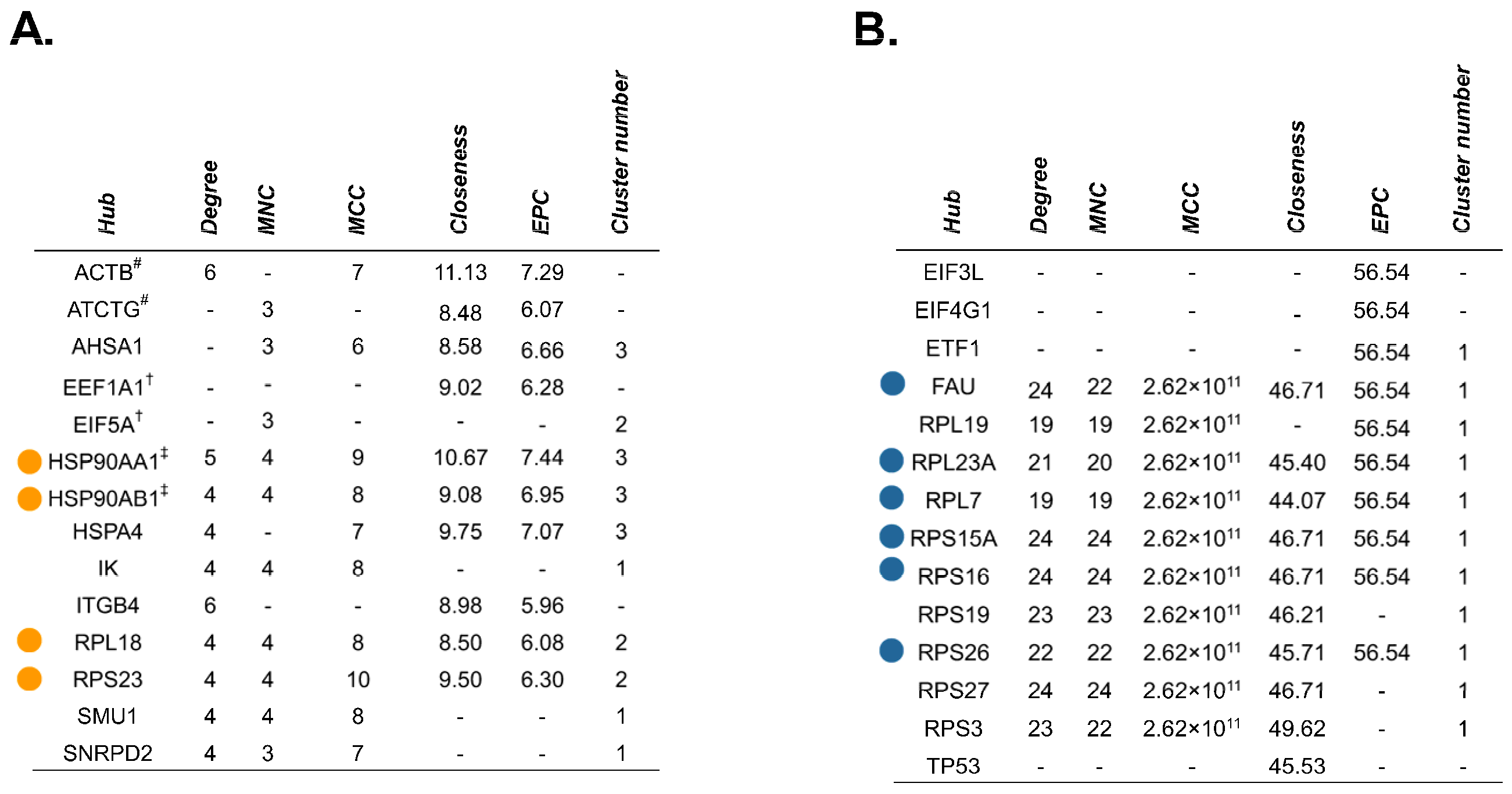
2.4. Source Proteins which Generated HLA-I-Bound Peptides Differ after Gemcitabine Exposure in S2-013 Cells
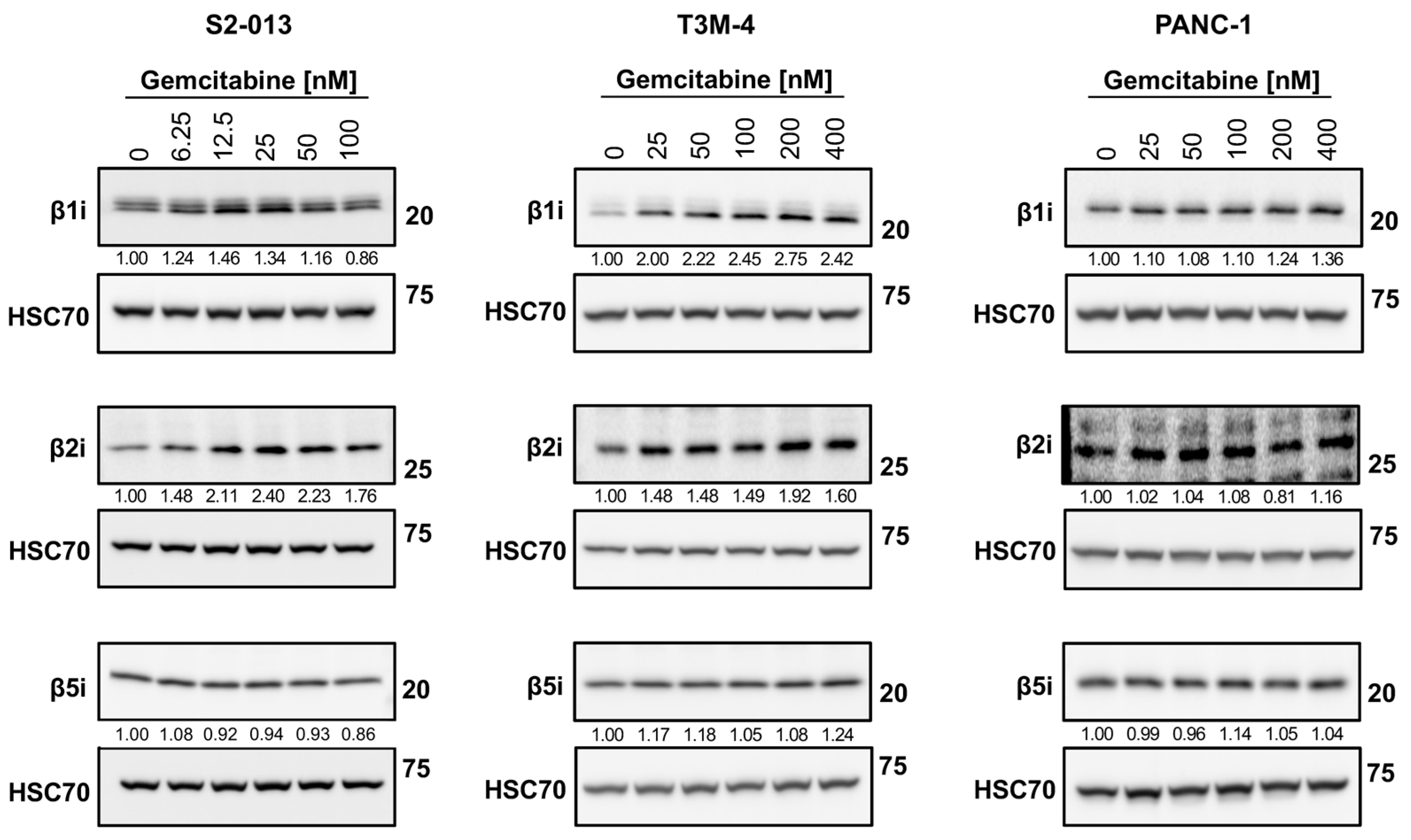
2.5. Gemcitabine Alters Immunoproteasome Catalytic Subunit Expression in Pancreatic Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Antibodies and Drugs
4.3. Proliferation and Viability Assays
4.3.1. MTT
4.3.2. Trypan Blue
4.4. Flow Cytometry
4.4.1. Brefeldin A (BFA) Assay
4.4.2. Temperature-Dependence Assay
4.5. Western Blotting
4.6. RNA Extraction and Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.7. HLA-I Peptide Elution, Mass Spectrometry, and Peptide Analysis
4.7.1. Mild Acid Elution of HLA-Bound Peptides
4.7.2. Mass Spectrometry Data Acquisition
4.7.3. Identification of Putative Binders and Assessment of Peptides’ Features
4.7.4. Evaluation of Source Proteins
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic Cancer: A Review of Clinical Diagnosis, Epidemiology, Treatment and Outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.S.; Kennedy, E.B.; Cinar, P.; Conroy, T.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Lau, M.W.; Johnson, T.; Krishnamurthi, S.; et al. Metastatic Pancreatic Cancer: ASCO Guideline Update. J. Clin. Oncol. 2020, 38, 3217–3230. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Oh, D.-Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.-C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Rong, Y.; Qin, X.; Jin, D.; Lou, W.; Wu, L.; Wang, D.; Wu, W.; Ni, X.; Mao, Z.; Kuang, T.; et al. A Phase I Pilot Trial of MUC1-Peptide-Pulsed Dendritic Cells in the Treatment of Advanced Pancreatic Cancer. Clin. Exp. Med. 2012, 12, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Bear, A.S.; Vonderheide, R.H.; O’Hara, M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yanagimoto, H.; Satoi, S.; Toyokawa, H.; Yamao, J.; Kim, S.; Terakawa, N.; Takahashi, K.; Kwon, A.-H. Circulating Myeloid Dendritic Cells as Prognostic Factors in Patients with Pancreatic Cancer Who Have Undergone Surgical Resection. J. Surg. Res. 2012, 173, 299–308. [Google Scholar] [CrossRef]
- Fukunaga, A.; Miyamoto, M.; Cho, Y.; Murakami, S.; Kawarada, Y.; Oshikiri, T.; Kato, K.; Kurokawa, T.; Suzuoki, M.; Nakakubo, Y.; et al. CD8+ Tumor-Infiltrating Lymphocytes Together with CD4+ Tumor-Infiltrating Lymphocytes and Dendritic Cells Improve the Prognosis of Patients with Pancreatic Adenocarcinoma. Pancreas 2004, 28, e26–e31. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Rajamanickam, V.; Bui, C.; Bernard, B.; Pucilowska, J.; Ballesteros-Merino, C.; Schmidt, M.; McCarty, K.; Philips, M.; Piening, B.; et al. Germinal center reactions in tertiary lymphoid structures associate with neoantigen burden, humoral immunity and long-term survivorship in pancreatic cancer. Oncoimmunology 2021, 10, 1900635. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhao, G.; Wu, W.; Rong, Y.; Jin, D.; Wang, D.; Lou, W.; Qin, X. Low Intratumoral Regulatory T Cells and High Peritumoral CD8(+) T Cells Relate to Long-Term Survival in Patients with Pancreatic Ductal Adenocarcinoma after Pancreatectomy. Cancer Immunol. Immunother. 2016, 65, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Kamath, S.D.; Korc, M.; Munshi, H.G. The Immune Modifying Effects of Chemotherapy and Advances in Chemo-Immunotherapy. Pharmacol. Ther. 2022, 236, 108111. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, E.; Wenthe, J.; Irenaeus, S.; Loskog, A.; Ullenhag, G. Gemcitabine Reduces MDSCs, Tregs and TGFβ-1 While Restoring the Teff/Treg Ratio in Patients with Pancreatic Cancer. J. Transl. Med. 2016, 14, 282. [Google Scholar] [CrossRef] [PubMed]
- Gürlevik, E.; Fleischmann-Mundt, B.; Brooks, J.; Demir, I.E.; Steiger, K.; Ribback, S.; Yevsa, T.; Woller, N.; Kloos, A.; Ostroumov, D.; et al. Administration of Gemcitabine After Pancreatic Tumor Resection in Mice Induces an Antitumor Immune Response Mediated by Natural Killer Cells. Gastroenterology 2016, 151, 338–350.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, D.; Li, Z.; Jiao, D.; Jin, L.; Cong, J.; Zheng, X.; Xu, L. Low-Dose Gemcitabine Treatment Enhances Immunogenicity and Natural Killer Cell-Driven Tumor Immunity in Lung Cancer. Front. Immunol. 2020, 11, 331. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.K.; Lake, R.A.; Marzo, A.L.; Scott, B.; Heath, W.R.; Collins, E.J.; Frelinger, J.A.; Robinson, B.W.S. Induction of Tumor Cell Apoptosis in Vivo Increases Tumor Antigen Cross-Presentation, Cross-Priming Rather than Cross-Tolerizing Host Tumor-Specific CD8 T Cells. J. Immunol. 2003, 170, 4905–4913. [Google Scholar] [CrossRef]
- Principe, D.R.; Narbutis, M.; Kumar, S.; Park, A.; Viswakarma, N.; Dorman, M.J.; Kamath, S.D.; Grippo, P.J.; Fishel, M.L.; Hwang, R.F.; et al. Long-Term Gemcitabine Treatment Reshapes the Pancreatic Tumor Microenvironment and Sensitizes Murine Carcinoma to Combination Immunotherapy. Cancer Res. 2020, 80, 3101–3115. [Google Scholar] [CrossRef]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef]
- Hurley, C.K. Naming HLA Diversity: A Review of HLA Nomenclature. Hum. Immunol. 2021, 82, 457–465. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: Structure, Function, and Antigen Presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [CrossRef]
- Schwarz, K.; van Den Broek, M.; Kostka, S.; Kraft, R.; Soza, A.; Schmidtke, G.; Kloetzel, P.M.; Groettrup, M. Overexpression of the Proteasome Subunits LMP2, LMP7, and MECL-1, but Not PA28 Alpha/Beta, Enhances the Presentation of an Immunodominant Lymphocytic Choriomeningitis Virus T Cell Epitope. J. Immunol. 2000, 165, 768–778. [Google Scholar] [CrossRef]
- Sijts, A.J.; Standera, S.; Toes, R.E.; Ruppert, T.; Beekman, N.J.; van Veelen, P.A.; Ossendorp, F.A.; Melief, C.J.; Kloetzel, P.M. MHC Class I Antigen Processing of an Adenovirus CTL Epitope Is Linked to the Levels of Immunoproteasomes in Infected Cells. J. Immunol. 2000, 164, 4500–4506. [Google Scholar] [CrossRef] [PubMed]
- Kalaora, S.; Lee, J.S.; Barnea, E.; Levy, R.; Greenberg, P.; Alon, M.; Yagel, G.; Bar Eli, G.; Oren, R.; Peri, A.; et al. Immunoproteasome Expression Is Associated with Better Prognosis and Response to Checkpoint Therapies in Melanoma. Nat. Commun. 2020, 11, 896. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, N.; Ino, Y.; Hori, S.; Yamazaki-Itoh, R.; Naito, C.; Shimasaki, M.; Esaki, M.; Nara, S.; Kishi, Y.; Shimada, K.; et al. Expression of Classical Human Leukocyte Antigen Class I Antigens, HLA-E and HLA-G, Is Adversely Prognostic in Pancreatic Cancer Patients. Cancer Sci. 2020, 111, 3057–3070. [Google Scholar] [CrossRef] [PubMed]
- Filip, I.; Wang, A.; Kravets, O.; Orenbuch, R.; Zhao, J.; Perea-Chamblee, T.E.; Manji, G.A.; López de Maturana, E.; Malats, N.; Olive, K.P.; et al. Pervasiveness of HLA Allele-Specific Expression Loss across Tumor Types. Genome Med. 2023, 15, 8. [Google Scholar] [CrossRef]
- Imai, D.; Yoshizumi, T.; Okano, S.; Uchiyama, H.; Ikegami, T.; Harimoto, N.; Itoh, S.; Soejima, Y.; Aishima, S.; Oda, Y.; et al. The Prognostic Impact of Programmed Cell Death Ligand 1 and Human Leukocyte Antigen Class I in Pancreatic Cancer. Cancer Med. 2017, 6, 1614–1626. [Google Scholar] [CrossRef]
- Ryschich, E.; Nötzel, T.; Hinz, U.; Autschbach, F.; Ferguson, J.; Simon, I.; Weitz, J.; Fröhlich, B.; Klar, E.; Büchler, M.W.; et al. Control of T-Cell-Mediated Immune Response by HLA Class I in Human Pancreatic Carcinoma. Clin. Cancer Res. 2005, 11, 498–504. [Google Scholar] [CrossRef]
- Orhan, A.; Vogelsang, R.P.; Andersen, M.B.; Madsen, M.T.; Hölmich, E.R.; Raskov, H.; Gögenur, I. The Prognostic Value of Tumour-Infiltrating Lymphocytes in Pancreatic Cancer: A Systematic Review and Meta-Analysis. Eur. J. Cancer 2020, 132, 71–84. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune Cell Infiltration as an Indicator of the Immune Microenvironment of Pancreatic Cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of Unique Neoantigen Qualities in Long-Term Survivors of Pancreatic Cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Fryer, R.A.; Barlett, B.; Galustian, C.; Dalgleish, A.G. Mechanisms Underlying Gemcitabine Resistance in Pancreatic Cancer and Sensitisation by the iMiDTM Lenalidomide. Anticancer Res. 2011, 31, 3747–3756. [Google Scholar] [PubMed]
- Koh, E.-K.; Lee, H.-R.; Son, W.-C.; Park, G.-Y.; Kim, J.; Bae, J.-H.; Park, Y.-S. Combinatorial Immunotherapy with Gemcitabine and Ex Vivo-Expanded NK Cells Induces Anti-Tumor Effects in Pancreatic Cancer. Sci. Rep. 2023, 13, 7656. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Galarza, F.F.; McCabe, A.; Melo dos Santos, E.J.; Jones, A.R.; Middleton, D. A Snapshot of Human Leukocyte Antigen (HLA) Diversity Using Data from the Allele Frequency Net Database. Hum. Immunol. 2021, 82, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Myers, N.B.; Wormstall, E.; Hansen, T.H. Differences among Various Class I Molecules in Competition for Beta2m in Vivo. Immunogenetics 1996, 43, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Neisig, A.; Melief, C.J.M.; Neefjes, J. Reduced Cell Surface Expression of HLA-C Molecules Correlates with Restricted Peptide Binding and Stable TAP Interaction1. J. Immunol. 1998, 160, 171–179. [Google Scholar] [CrossRef]
- Yarzabek, B.; Zaitouna, A.J.; Olson, E.; Silva, G.N.; Geng, J.; Geretz, A.; Thomas, R.; Krishnakumar, S.; Ramon, D.S.; Raghavan, M. Variations in HLA-B Cell Surface Expression, Half-Life and Extracellular Antigen Receptivity. eLife 2018, 7, e34961. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Oda, K.; Yokota, S.; Takatsuki, A.; Ikehara, Y. Brefeldin A Causes Disassembly of the Golgi Complex and Accumulation of Secretory Proteins in the Endoplasmic Reticulum. J. Biol. Chem. 1988, 263, 18545–18552. [Google Scholar] [CrossRef]
- Ladasky, J.J.; Shum, B.P.; Canavez, F.; Seuánez, H.N.; Parham, P. Residue 3 of Beta2-Microglobulin Affects Binding of Class I MHC Molecules by the W6/32 Antibody. Immunogenetics 1999, 49, 312–320. [Google Scholar] [CrossRef]
- Parham, P.; Barnstable, C.J.; Bodmer, W.F. Use of a Monoclonal Antibody (W6/32) in Structural Studies of HLA-A,B,C, Antigens. J. Immunol. 1979, 123, 342–349. [Google Scholar] [CrossRef]
- Ljunggren, H.G.; Stam, N.J.; Ohlén, C.; Neefjes, J.J.; Höglund, P.; Heemels, M.T.; Bastin, J.; Schumacher, T.N.; Townsend, A.; Kärre, K. Empty MHC Class I Molecules Come out in the Cold. Nature 1990, 346, 476–480. [Google Scholar] [CrossRef]
- Bouvier, M.; Wiley, D.C. Importance of Peptide Amino and Carboxyl Termini to the Stability of MHC Class I Molecules. Science 1994, 265, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Micheletti, F.; Bazzaro, M.; Canella, A.; Marastoni, M.; Traniello, S.; Gavioli, R. The Lifespan of Major Histocompatibility Complex Class I/Peptide Complexes Determines the Efficiency of Cytotoxic T-Lymphocyte Responses. Immunology 1999, 96, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Kaseke, C.; Park, R.J.; Singh, N.K.; Koundakjian, D.; Bashirova, A.; Garcia Beltran, W.F.; Takou Mbah, O.C.; Ma, J.; Senjobe, F.; Urbach, J.M.; et al. HLA Class-I-Peptide Stability Mediates CD8+ T Cell Immunodominance Hierarchies and Facilitates HLA-Associated Immune Control of HIV. Cell Rep. 2021, 36, 109378. [Google Scholar] [CrossRef] [PubMed]
- Garstka, M.A.; Fish, A.; Celie, P.H.N.; Joosten, R.P.; Janssen, G.M.C.; Berlin, I.; Hoppes, R.; Stadnik, M.; Janssen, L.; Ovaa, H.; et al. The First Step of Peptide Selection in Antigen Presentation by MHC Class I Molecules. Proc. Natl. Acad. Sci. USA 2015, 112, 1505–1510. [Google Scholar] [CrossRef] [PubMed]
- Montealegre, S.; Venugopalan, V.; Fritzsche, S.; Kulicke, C.; Hein, Z.; Springer, S. Dissociation of Β2-Microglobulin Determines the Surface Quality Control of Major Histocompatibility Complex Class I Molecules. FASEB J. 2015, 29, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Trolle, T.; McMurtrey, C.P.; Sidney, J.; Bardet, W.; Osborn, S.C.; Kaever, T.; Sette, A.; Hildebrand, W.H.; Nielsen, M.; Peters, B. The Length Distribution of Class I Restricted T Cell Epitopes Is Determined by Both Peptide Supply and MHC Allele Specific Binding Preference. J. Immunol. 2016, 196, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Abualrous, E.T.; Saini, S.K.; Ramnarayan, V.R.; Ilca, F.T.; Zacharias, M.; Springer, S. The Carboxy Terminus of the Ligand Peptide Determines the Stability of the MHC Class I Molecule H-2Kb: A Combined Molecular Dynamics and Experimental Study. PLoS ONE 2015, 10, e0135421. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 Update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Zhao, W.; Sher, X. Systematically Benchmarking Peptide-MHC Binding Predictors: From Synthetic to Naturally Processed Epitopes. PLoS Comput. Biol. 2018, 14, e1006457. [Google Scholar] [CrossRef]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying Hub Objects and Sub-Networks from Complex Interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C. Role of eIF2α Kinases in Translational Control and Adaptation to Cellular Stress. Cold Spring Harb. Perspect. Biol. 2018, 10, a032870. [Google Scholar] [CrossRef] [PubMed]
- Kesmir, C.; van Noort, V.; de Boer, R.J.; Hogeweg, P. Bioinformatic Analysis of Functional Differences between the Immunoproteasome and the Constitutive Proteasome. Immunogenetics 2003, 55, 437–449. [Google Scholar] [CrossRef][Green Version]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]
- Padrón, L.J.; Maurer, D.M.; O’Hara, M.H.; O’Reilly, E.M.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; et al. Sotigalimab and/or Nivolumab with Chemotherapy in First-Line Metastatic Pancreatic Cancer: Clinical and Immunologic Analyses from the Randomized Phase 2 PRINCE Trial. Nat. Med. 2022, 28, 1167–1177. [Google Scholar] [CrossRef]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef]
- Gravett, A.M.; Trautwein, N.; Stevanović, S.; Dalgleish, A.G.; Copier, J. Gemcitabine Alters the Proteasome Composition and Immunopeptidome of Tumour Cells. Oncoimmunology 2018, 7, e1438107. [Google Scholar] [CrossRef] [PubMed]
- Sawasdee, N.; Thepmalee, C.; Sujjitjoon, J.; Yongpitakwattana, P.; Junking, M.; Poungvarin, N.; Yenchitsomanus, P.-T.; Panya, A. Gemcitabine Enhances Cytotoxic Activity of Effector T-Lymphocytes against Chemo-Resistant Cholangiocarcinoma Cells. Int. Immunopharmacol. 2020, 78, 106006. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef]
- Jongsma, M.L.M.; Guarda, G.; Spaapen, R.M. The Regulatory Network behind MHC Class I Expression. Mol. Immunol. 2019, 113, 16–21. [Google Scholar] [CrossRef]
- Aydemir, N.; Bilaloğlu, R. Genotoxicity of Two Anticancer Drugs, Gemcitabine and Topotecan, in Mouse Bone Marrow in Vivo. Mutat. Res. 2003, 537, 43–51. [Google Scholar] [CrossRef]
- Lee, K.; Kim, D.-E.; Jang, K.-S.; Kim, S.-J.; Cho, S.; Kim, C. Gemcitabine, a Broad-Spectrum Antiviral Drug, Suppresses Enterovirus Infections through Innate Immunity Induced by the Inhibition of Pyrimidine Biosynthesis and Nucleotide Depletion. Oncotarget 2017, 8, 115315–115325. [Google Scholar] [CrossRef]
- Mullen, N.J.; Shukla, S.K.; Thakur, R.; Kollala, S.S.; Wang, D.; Chaika, N.; LaBreck, D.A.; Mallareddy, J.R.; Price, D.H.; Natarajan, A.; et al. DHODH Inhibition Enhances the Efficacy of Immune Checkpoint Blockade by Increasing Cancer Cell Antigen Presentation. bioRxiv 2023. [Google Scholar] [CrossRef]
- Chung, D.-H.; Golden, J.E.; Adcock, R.S.; Schroeder, C.E.; Chu, Y.-K.; Sotsky, J.B.; Cramer, D.E.; Chilton, P.M.; Song, C.; Anantpadma, M.; et al. Discovery of a Broad-Spectrum Antiviral Compound That Inhibits Pyrimidine Biosynthesis and Establishes a Type 1 Interferon-Independent Antiviral State. Antimicrob. Agents Chemother. 2016, 60, 4552–4562. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; de Ruiter, P.E.; Metselaar, H.J.; Kwekkeboom, J.; de Jonge, J.; Tilanus, H.W.; Janssen, H.L.A.; van der Laan, L.J.W. Mycophenolic Acid Augments Interferon-Stimulated Gene Expression and Inhibits Hepatitis C Virus Infection in Vitro and in Vivo. Hepatology 2012, 55, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Alexandraki, A.; Strati, K. Decitabine Treatment Induces a Viral Mimicry Response in Cervical Cancer Cells and Further Sensitizes Cells to Chemotherapy. Int. J. Mol. Sci. 2022, 23, 14042. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Yau, H.L.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Pugh, T.J.; Jones, P.A.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Khan, D.H.; Hurren, R.; Xu, M.; Na, Y.; Kang, H.; Mirali, S.; Wang, X.; Gronda, M.; Jitkova, Y.; et al. Venetoclax Enhances T Cell-Mediated Antileukemic Activity by Increasing ROS Production. Blood 2021, 138, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Kubiniok, P.; Marcu, A.; Bichmann, L.; Kuchenbecker, L.; Schuster, H.; Hamelin, D.J.; Duquette, J.D.; Kovalchik, K.A.; Wessling, L.; Kohlbacher, O.; et al. Understanding the Constitutive Presentation of MHC Class I Immunopeptidomes in Primary Tissues. iScience 2022, 25, 103768. [Google Scholar] [CrossRef] [PubMed]
- Isa, A.; Nehlin, J.O.; Sabir, H.J.; Andersen, T.E.; Gaster, M.; Kassem, M.; Barington, T. Impaired Cell Surface Expression of HLA-B Antigens on Mesenchymal Stem Cells and Muscle Cell Progenitors. PLoS ONE 2010, 5, e10900. [Google Scholar] [CrossRef]
- Kurokohchi, K.; Carrington, M.; Mann, D.L.; Simonis, T.B.; Alexander-Miller, M.A.; Feinstone, S.M.; Akatsuka, T.; Berzofsky, J.A. Expression of HLA Class I Molecules and the Transporter Associated with Antigen Processing in Hepatocellular Carcinoma. Hepatology 1996, 23, 1181–1188. [Google Scholar] [CrossRef]
- Apps, R.; Meng, Z.; Del Prete, G.Q.; Lifson, J.D.; Zhou, M.; Carrington, M. Relative Expression Levels of the HLA Class-I Proteins in Normal and HIV-Infected Cells. J. Immunol. 2015, 194, 3594–3600. [Google Scholar] [CrossRef]
- Dellgren, C.; Nehlin, J.O.; Barington, T. Cell Surface Expression Level Variation between Two Common Human Leukocyte Antigen Alleles, HLA-A2 and HLA-B8, Is Dependent on the Structure of the C Terminal Part of the Alpha 2 and the Alpha 3 Domains. PLoS ONE 2015, 10, e0135385. [Google Scholar] [CrossRef]
- Olson, E.; Geng, J.; Raghavan, M. Polymorphisms of HLA-B: Influences on Assembly and Immunity. Curr. Opin. Immunol. 2020, 64, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.; Ceccarelli, T.; Raghavan, M. Endo-Lysosomal Assembly Variations among Human Leukocyte Antigen Class I (HLA Class I) Allotypes. eLife 2023, 12, e79144. [Google Scholar] [CrossRef]
- Del Val, M.; Lázaro, S.; Ramos, M.; Antón, L.C. Are Membrane Proteins Favored over Cytosolic Proteins in TAP-Independent Processing Pathways? Mol. Immunol. 2013, 55, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Ilca, F.T.; Drexhage, L.Z.; Brewin, G.; Peacock, S.; Boyle, L.H. Distinct Polymorphisms in HLA Class I Molecules Govern Their Susceptibility to Peptide Editing by TAPBPR. Cell Rep. 2019, 29, 1621–1632.e3. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Szeto, C.; Gras, S. The Pockets Guide to HLA Class I Molecules. Biochem. Soc. Trans. 2021, 49, 2319–2331. [Google Scholar] [CrossRef]
- Abi Habib, J.; Lesenfants, J.; Vigneron, N.; Van den Eynde, B.J. Functional Differences between Proteasome Subtypes. Cells 2022, 11, 421. [Google Scholar] [CrossRef]
- Guillaume, B.; Chapiro, J.; Stroobant, V.; Colau, D.; Van Holle, B.; Parvizi, G.; Bousquet-Dubouch, M.-P.; Théate, I.; Parmentier, N.; Van den Eynde, B.J. Two Abundant Proteasome Subtypes That Uniquely Process Some Antigens Presented by HLA Class I Molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 18599–18604. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Pletscher-Frankild, S.; Jensen, L.J.; Mann, M. Mass Spectrometry of Human Leukocyte Antigen Class I Peptidomes Reveals Strong Effects of Protein Abundance and Turnover on Antigen Presentation. Mol. Cell. Proteom. 2015, 14, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.C.-C.; Laurent-Rolle, M.; Cresswell, P. Translational Regulation of Viral RNA in the Type I Interferon Response. Curr. Res. Virol. Sci. 2021, 2, 100012. [Google Scholar] [CrossRef]
- Ohtsuka, M.; Inoko, H.; Kulski, J.K.; Yoshimura, S. Major Histocompatibility Complex (Mhc) Class Ib Gene Duplications, Organization and Expression Patterns in Mouse Strain C57BL/6. BMC Genom. 2008, 9, 178. [Google Scholar] [CrossRef]
- Fortier, M.-H.; Caron, É.; Hardy, M.-P.; Voisin, G.; Lemieux, S.; Perreault, C.; Thibault, P. The MHC Class I Peptide Repertoire Is Molded by the Transcriptome. J. Exp. Med. 2008, 205, 595–610. [Google Scholar] [CrossRef]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved Predictions of MHC Antigen Presentation by Concurrent Motif Deconvolution and Integration of MS MHC Eluted Ligand Data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Zhang, Q.-B.; Ye, R.-F.; Ye, L.-Y.; Zhang, Q.-Y.; Dai, N.-G. Isocorydine Decrease Gemcitabine-Resistance by Inhibiting Epithelial-Mesenchymal Transition via STAT3 in Pancreatic Cancer Cells. Am. J. Transl. Res. 2020, 12, 3702–3714. [Google Scholar]
- Zhang, Z.; Han, H.; Rong, Y.; Zhu, K.; Zhu, Z.; Tang, Z.; Xiong, C.; Tao, J. Hypoxia Potentiates Gemcitabine-Induced Stemness in Pancreatic Cancer Cells through AKT/Notch1 Signaling. J. Exp. Clin. Cancer Res. 2018, 37, 291. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Z.; Gao, C.; Wu, S.; Duan, Q.; Wu, H.; Wang, C.; Shen, Q.; Yin, T. Combination Chemotherapy of Valproic Acid (VPA) and Gemcitabine Regulates STAT3/Bmi1 Pathway to Differentially Potentiate the Motility of Pancreatic Cancer Cells. Cell Biosci. 2019, 9, 50, Erratum in Cell Biosci. 2023, 13, 206. [Google Scholar] [CrossRef]
- Sliker, B.H.; Goetz, B.T.; Barnes, R.; King, H.; Maurer, H.C.; Olive, K.P.; Solheim, J.C. HLA-B Influences Integrin Beta-1 Expression and Pancreatic Cancer Cell Migration. Exp. Cell Res. 2020, 390, 111960. [Google Scholar] [CrossRef]
- King, R.S. Gemcitabine. New First-Line Therapy for Pancreatic Cancer. Cancer Pract. 1996, 4, 353–354. [Google Scholar]
- Toschi, L.; Finocchiaro, G.; Bartolini, S.; Gioia, V.; Cappuzzo, F. Role of Gemcitabine in Cancer Therapy. Future Oncol. 2005, 1, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Wen, X.; Wang, Y.; Lin, M.; Lai, J. Gemcitabine and Checkpoint Blockade Exhibit Synergistic Anti-Tumor Effects in a Model of Murine Lung Carcinoma. Int. Immunopharmacol. 2020, 86, 106694. [Google Scholar] [CrossRef]
- Glorieux, C.; Xia, X.; You, X.; Wang, Z.; Han, Y.; Yang, J.; Noppe, G.; de Meester, C.; Ling, J.; Robert, A.; et al. Cisplatin and Gemcitabine Exert Opposite Effects on Immunotherapy with PD-1 Antibody in K-Ras-Driven Cancer. J. Adv. Res. 2021, 40, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Kuai, J.; Shen, G. Low-Dose Metronomic Gemcitabine Pretreatments Overcome the Resistance of Breast Cancer to Immune Checkpoint Therapy. Immunotherapy 2023, 15, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Dammeijer, F.; De Gooijer, C.J.; van Gulijk, M.; Lukkes, M.; Klaase, L.; Lievense, L.A.; Waasdorp, C.; Jebbink, M.; Bootsma, G.P.; Stigt, J.A.; et al. Immune Monitoring in Mesothelioma Patients Identifies Novel Immune-Modulatory Functions of Gemcitabine Associating with Clinical Response. eBioMedicine 2021, 64, 103160. [Google Scholar] [CrossRef] [PubMed]
- Le, H.K.; Graham, L.; Cha, E.; Morales, J.K.; Manjili, M.H.; Bear, H.D. Gemcitabine Directly Inhibits Myeloid Derived Suppressor Cells in BALB/c Mice Bearing 4T1 Mammary Carcinoma and Augments Expansion of T Cells from Tumor-Bearing Mice. Int. Immunopharmacol. 2009, 9, 900–909. [Google Scholar] [CrossRef]
- Homma, Y.; Taniguchi, K.; Nakazawa, M.; Matsuyama, R.; Mori, R.; Takeda, K.; Ichikawa, Y.; Tanaka, K.; Endo, I. Changes in the Immune Cell Population and Cell Proliferation in Peripheral Blood after Gemcitabine-Based Chemotherapy for Pancreatic Cancer. Clin. Transl. Oncol. 2014, 16, 330–335. [Google Scholar] [CrossRef]
- Minz, A.P.; Mohapatra, D.; Dutta, M.; Sethi, M.; Parida, D.; Mohapatra, A.P.; Mishra, S.; Kar, S.; Sasmal, P.K.; Senapati, S. Statins Abrogate Gemcitabine-Induced PD-L1 Expression in Pancreatic Cancer-Associated Fibroblasts and Cancer Cells with Improved Therapeutic Outcome. Cancer Immunol. Immunother. 2023, 72, 4261–4278. [Google Scholar] [CrossRef]
- Doi, T.; Ishikawa, T.; Okayama, T.; Oka, K.; Mizushima, K.; Yasuda, T.; Sakamoto, N.; Katada, K.; Kamada, K.; Uchiyama, K.; et al. The JAK/STAT Pathway Is Involved in the Upregulation of PD-L1 Expression in Pancreatic Cancer Cell Lines. Oncol. Rep. 2017, 37, 1545–1554. [Google Scholar] [CrossRef]
- Wang, H.; He, X.; Fang, D.; Wang, X.; Guan, J.; Shi, Z.-W.; Chen, X. Gemcitabine-Facilitated Modulation of the Tumor Microenvironment and PD-1/PD-L1 Blockade Generate a Synergistic Antitumor Effect in a Murine Hepatocellular Carcinoma Model. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101853. [Google Scholar] [CrossRef]
- Zhang, H.; Dai, Z.; Wu, W.; Wang, Z.; Zhang, N.; Zhang, L.; Zeng, W.-J.; Liu, Z.; Cheng, Q. Regulatory Mechanisms of Immune Checkpoints PD-L1 and CTLA-4 in Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 184. [Google Scholar] [CrossRef]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer Vaccines as Promising Immuno-Therapeutics: Platforms and Current Progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA Neoantigen Vaccines Stimulate T Cells in Pancreatic Cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Sturm, T.; Sautter, B.; Wörner, T.P.; Stevanović, S.; Rammensee, H.-G.; Planz, O.; Heck, A.J.R.; Aebersold, R. Mild Acid Elution and MHC Immunoaffinity Chromatography Reveal Similar Albeit Not Identical Profiles of the HLA Class I Immunopeptidome. J. Proteome Res. 2021, 20, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Sette, A.; Vitiello, A.; Reherman, B.; Fowler, P.; Nayersina, R.; Kast, W.M.; Melief, C.J.; Oseroff, C.; Yuan, L.; Ruppert, J.; et al. The Relationship between Class I Binding Affinity and Immunogenicity of Potential Cytotoxic T Cell Epitopes. J. Immunol. 1994, 153, 5586–5592. [Google Scholar] [CrossRef]
- O’Donnell, T.J.; Rubinsteyn, A.; Laserson, U. MHCflurry 2.0: Improved Pan-Allele Prediction of MHC Class I-Presented Peptides by Incorporating Antigen Processing. Cell Syst. 2020, 11, 42–48.e7, Erratum in Cell Syst. 2020, 11, 418–419. [Google Scholar] [CrossRef]
- Di Marco, M.; Schuster, H.; Backert, L.; Ghosh, M.; Rammensee, H.-G.; Stevanović, S. Unveiling the Peptide Motifs of HLA-C and HLA-G from Naturally Presented Peptides and Generation of Binding Prediction Matrices. J. Immunol. 2017, 199, 2639–2651. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larson, A.C.; Knoche, S.M.; Brumfield, G.L.; Doty, K.R.; Gephart, B.D.; Moore-Saufley, P.R.; Solheim, J.C. Gemcitabine Modulates HLA-I Regulation to Improve Tumor Antigen Presentation by Pancreatic Cancer Cells. Int. J. Mol. Sci. 2024, 25, 3211. https://doi.org/10.3390/ijms25063211
Larson AC, Knoche SM, Brumfield GL, Doty KR, Gephart BD, Moore-Saufley PR, Solheim JC. Gemcitabine Modulates HLA-I Regulation to Improve Tumor Antigen Presentation by Pancreatic Cancer Cells. International Journal of Molecular Sciences. 2024; 25(6):3211. https://doi.org/10.3390/ijms25063211
Chicago/Turabian StyleLarson, Alaina C., Shelby M. Knoche, Gabrielle L. Brumfield, Kenadie R. Doty, Benjamin D. Gephart, Promise R. Moore-Saufley, and Joyce C. Solheim. 2024. "Gemcitabine Modulates HLA-I Regulation to Improve Tumor Antigen Presentation by Pancreatic Cancer Cells" International Journal of Molecular Sciences 25, no. 6: 3211. https://doi.org/10.3390/ijms25063211
APA StyleLarson, A. C., Knoche, S. M., Brumfield, G. L., Doty, K. R., Gephart, B. D., Moore-Saufley, P. R., & Solheim, J. C. (2024). Gemcitabine Modulates HLA-I Regulation to Improve Tumor Antigen Presentation by Pancreatic Cancer Cells. International Journal of Molecular Sciences, 25(6), 3211. https://doi.org/10.3390/ijms25063211