

The Human Mutation K237_V238del in a Putative Lipid Binding Motif within the V-ATPase a2 Isoform Suggests a Molecular Mechanism Underlying Cutis Laxa



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
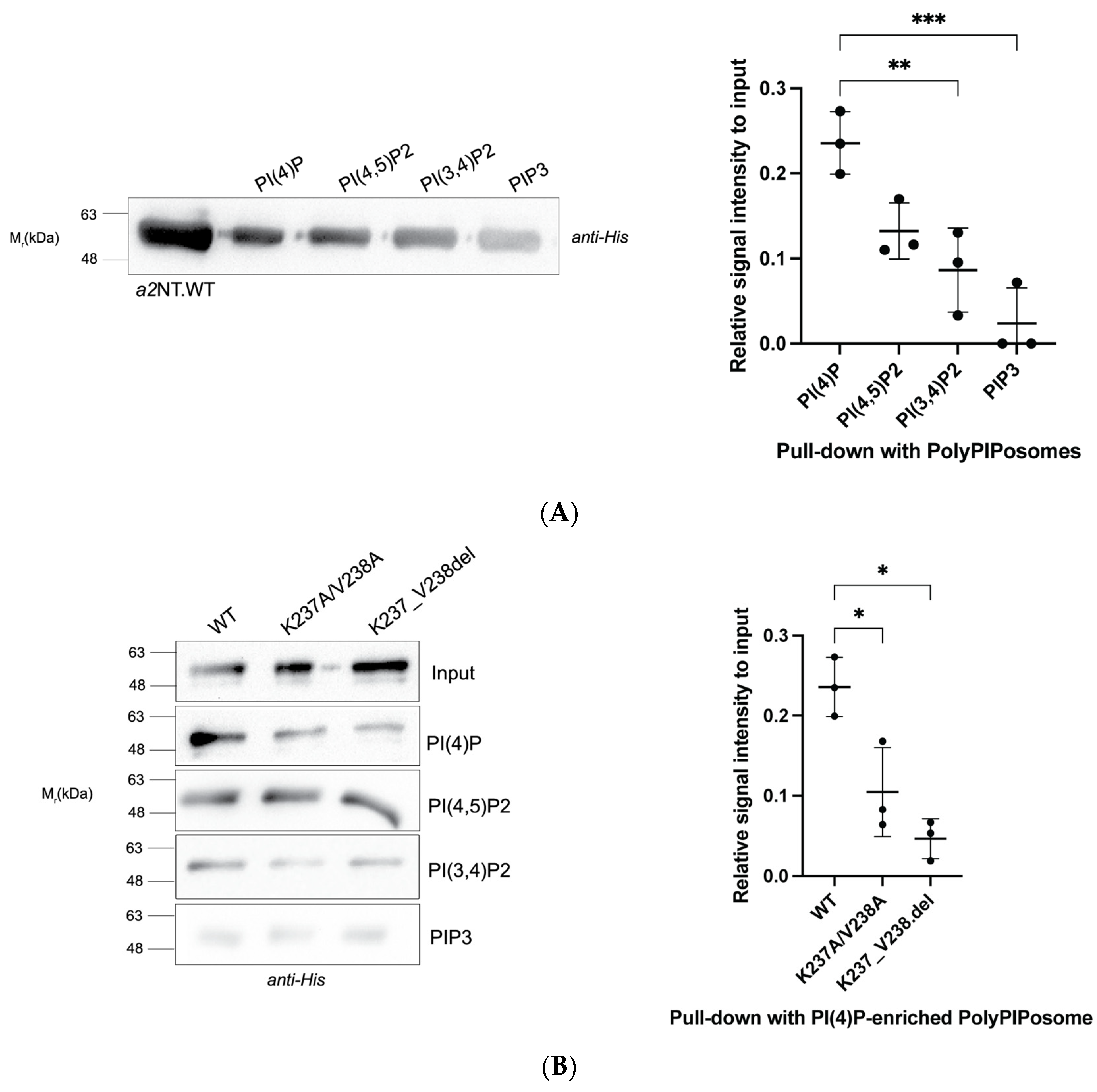
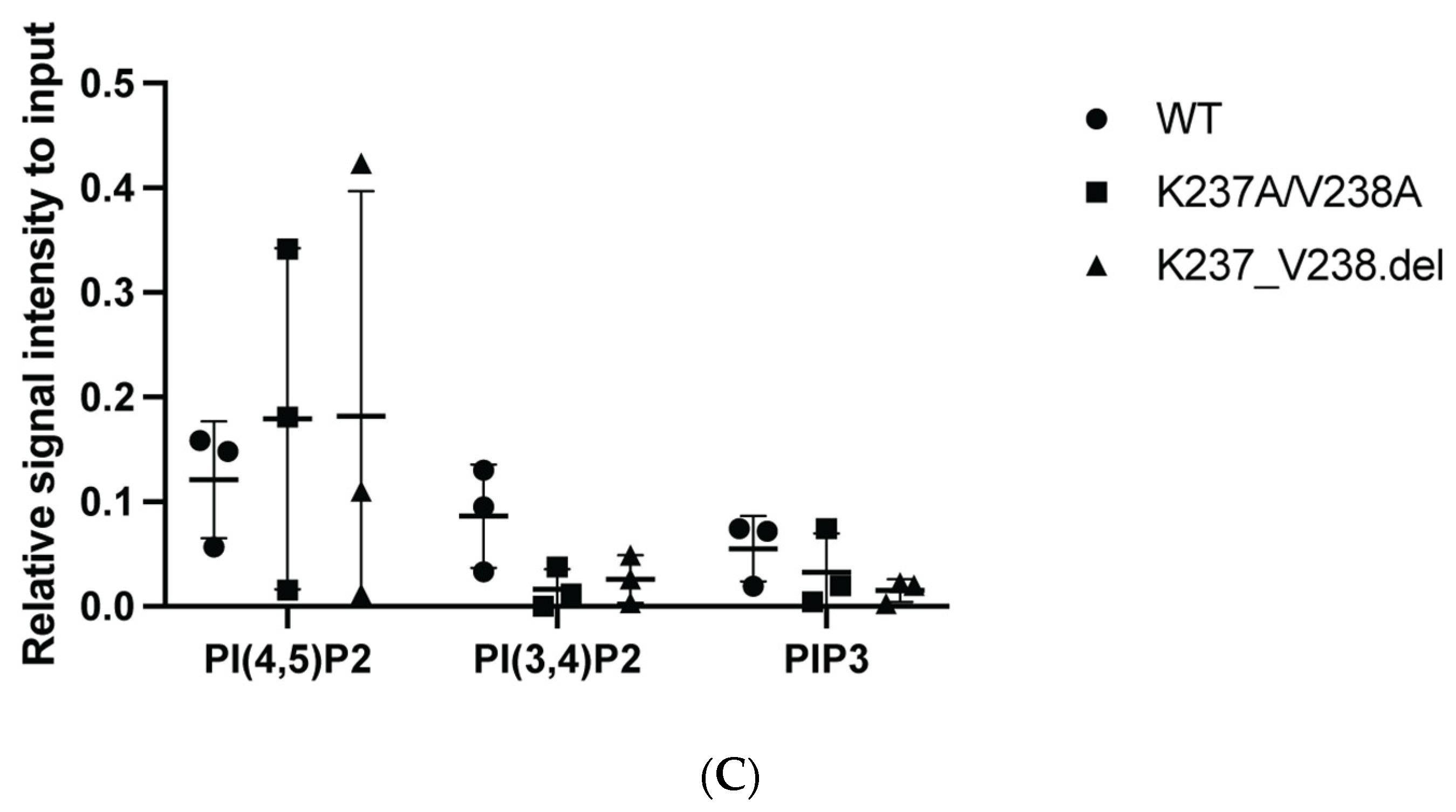
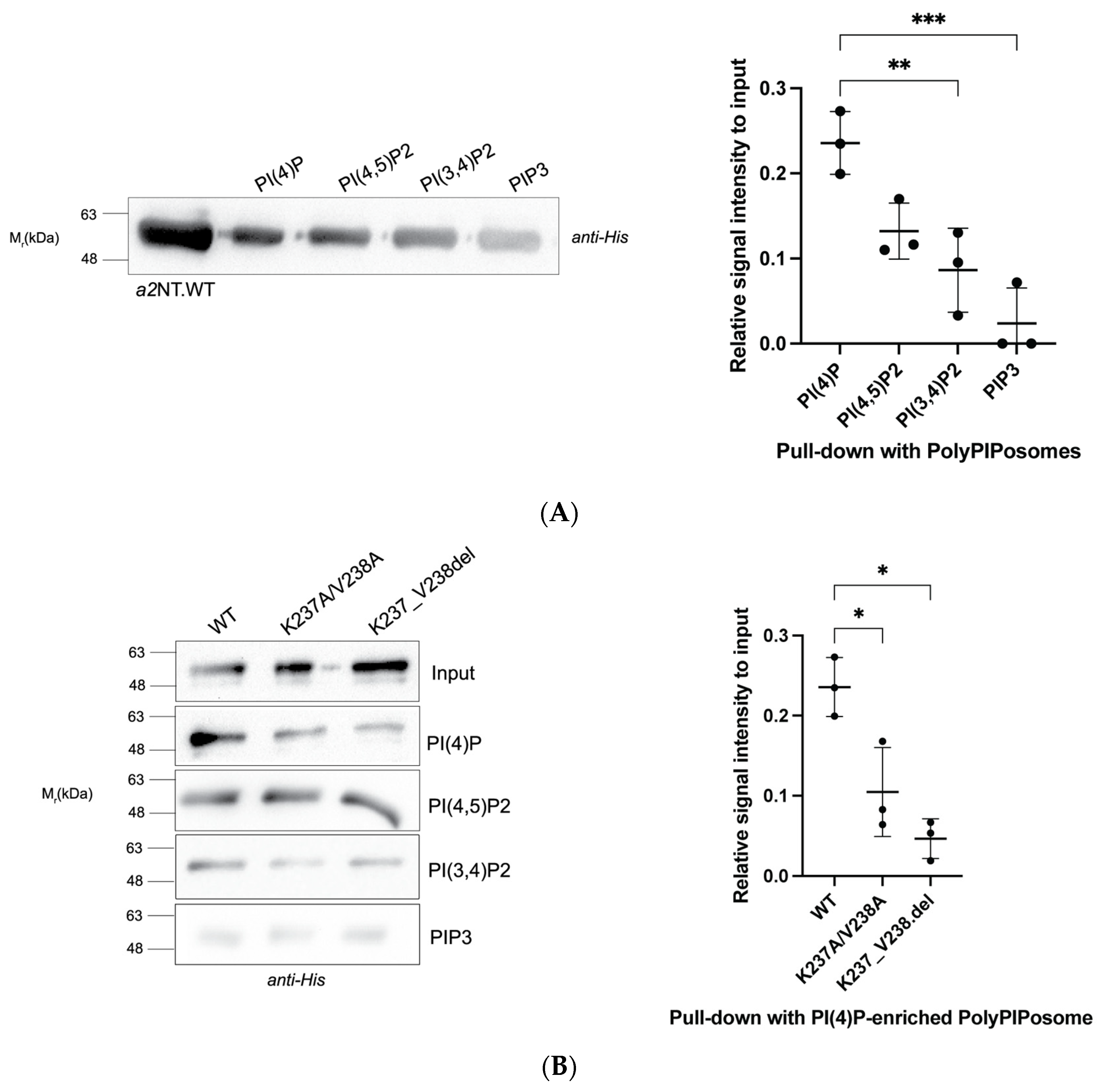
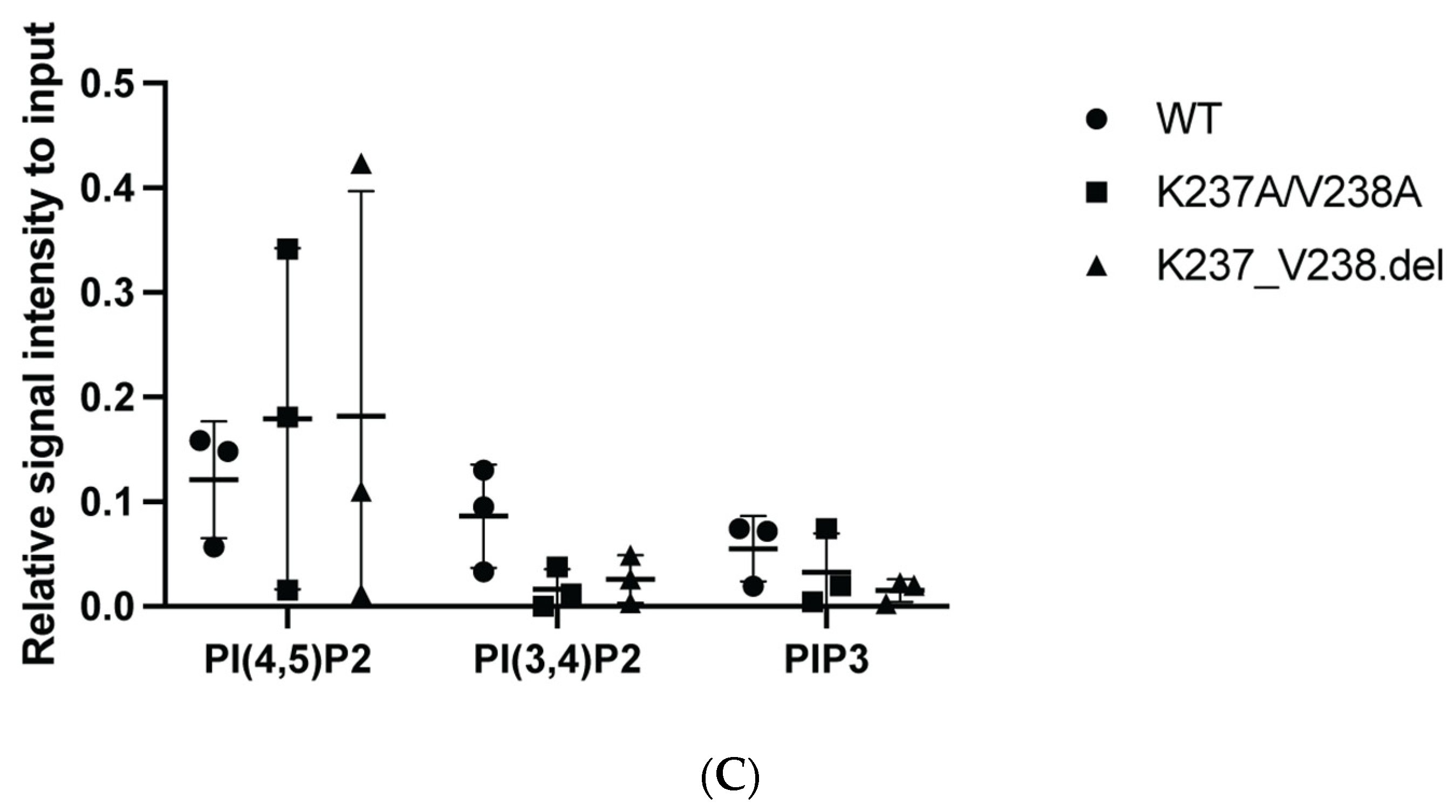
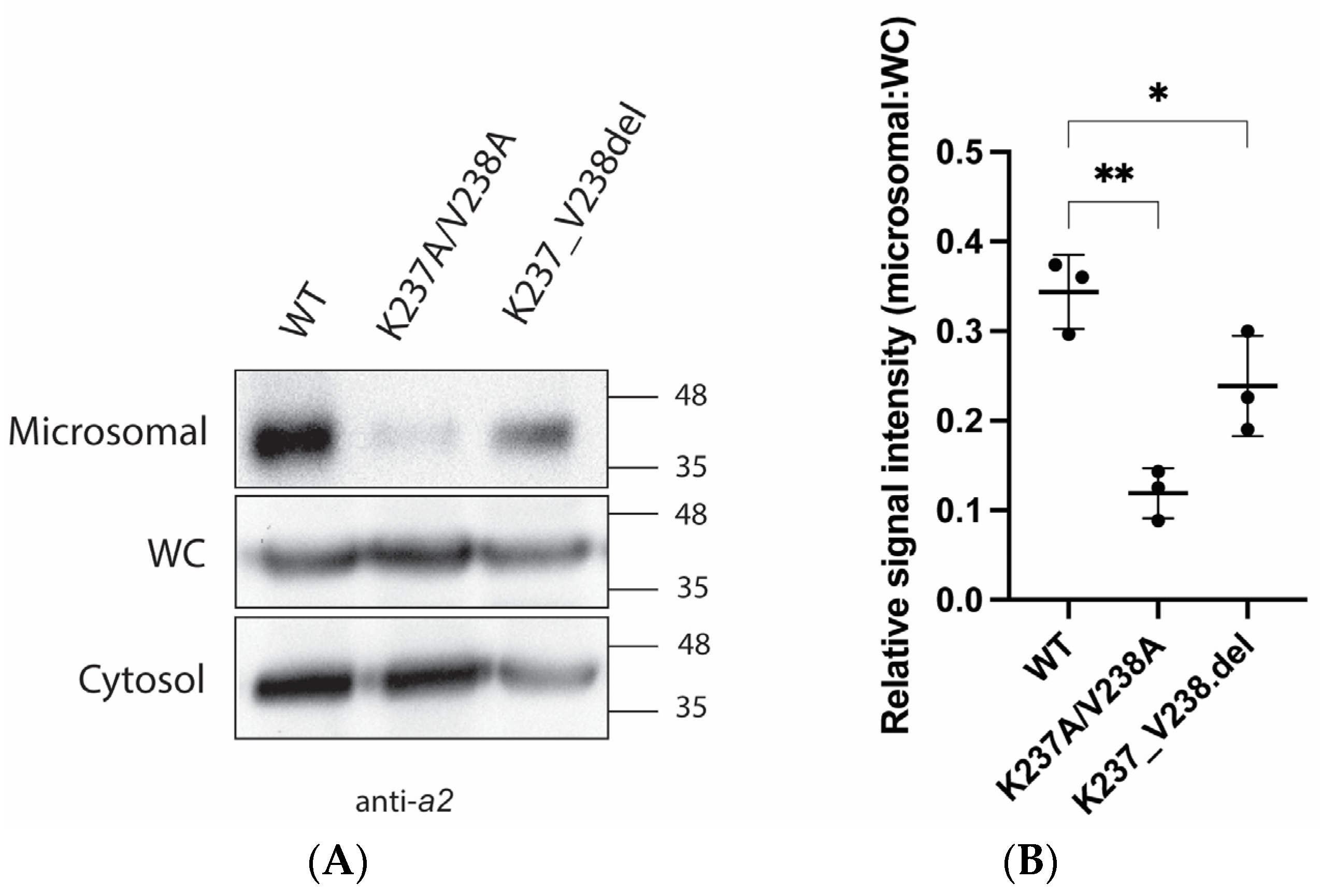
2.1. Mutations within the Putative Binding Motif reduced Interaction of a2 with PI(4)P-Enriched Liposomes In Vitro
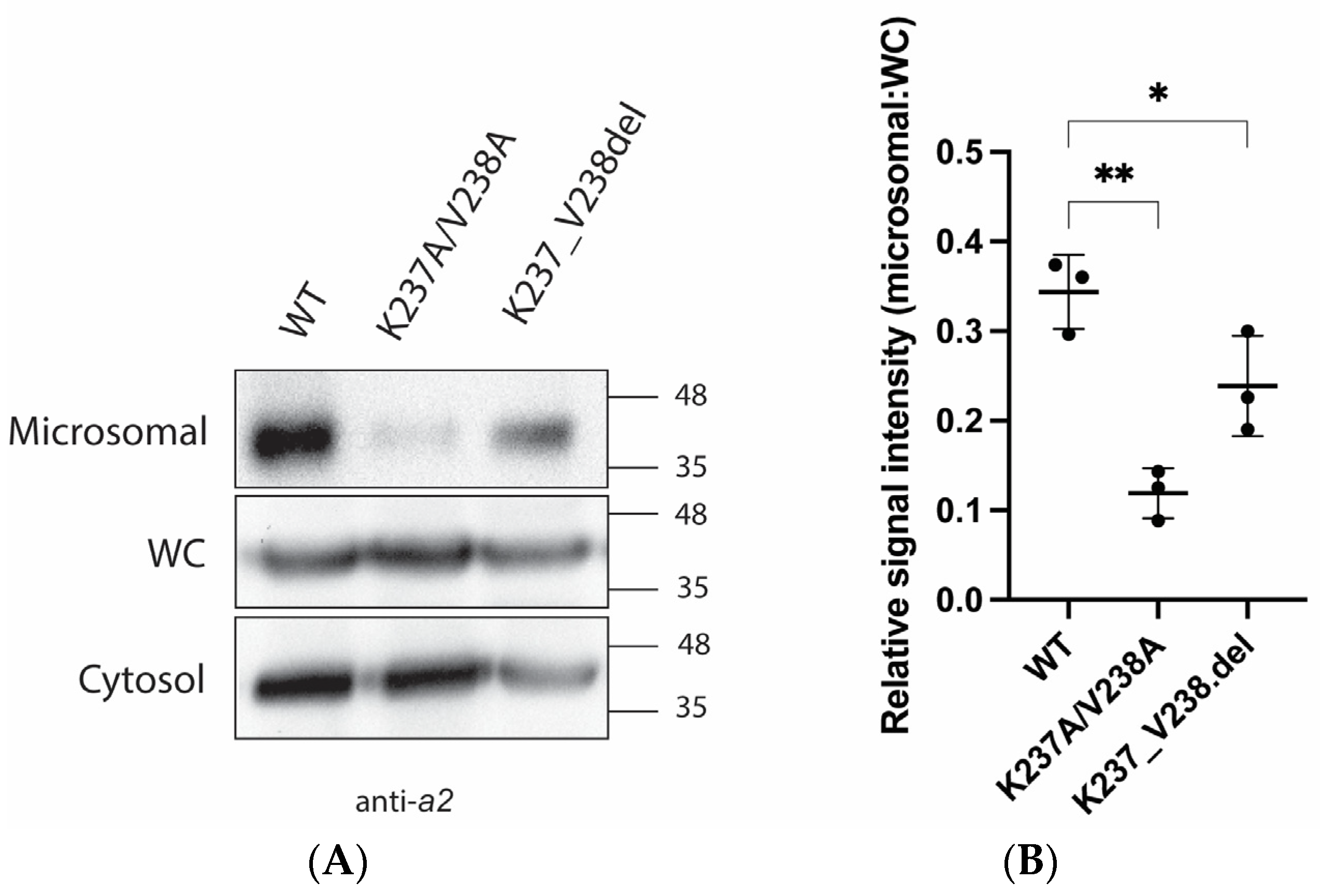
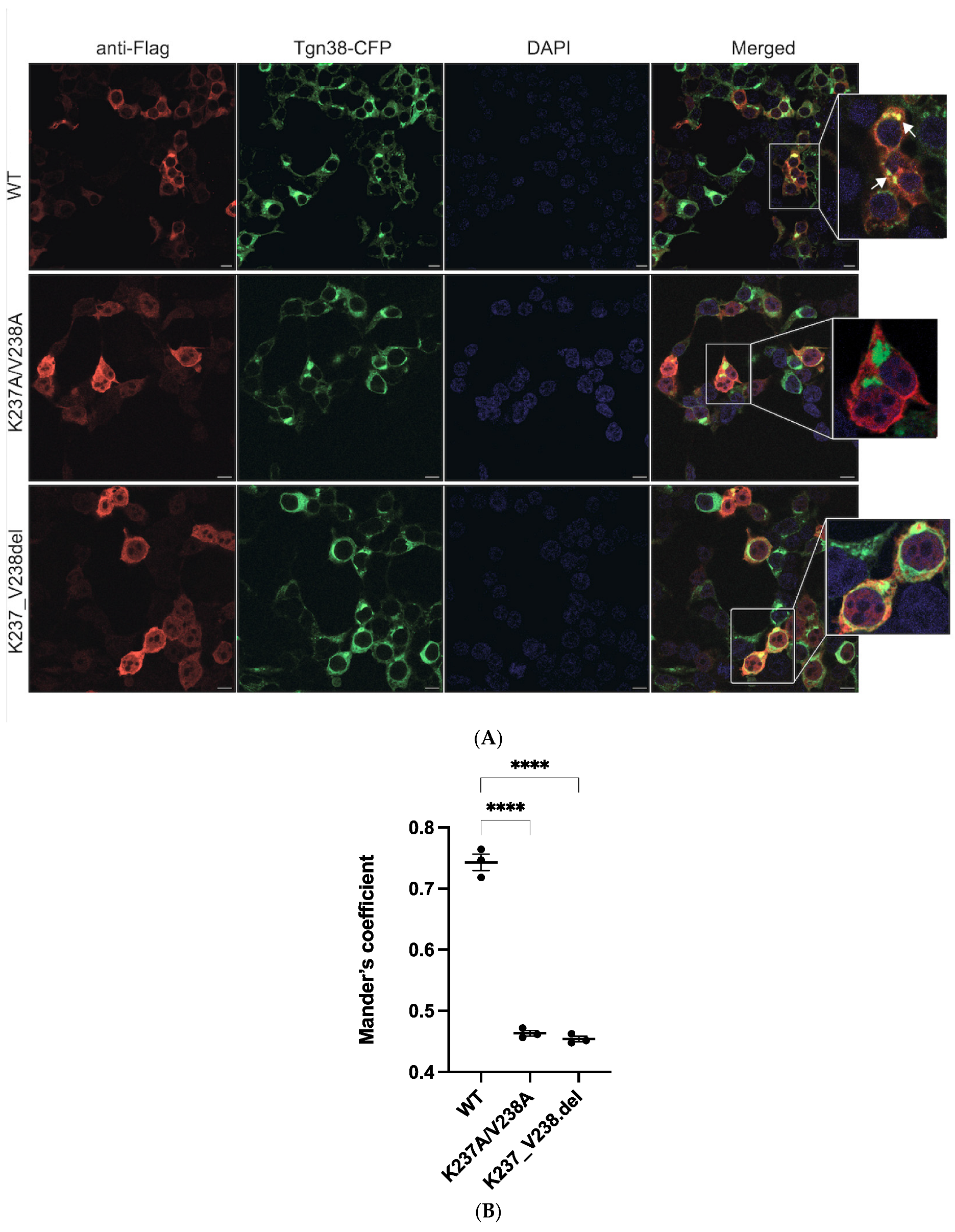
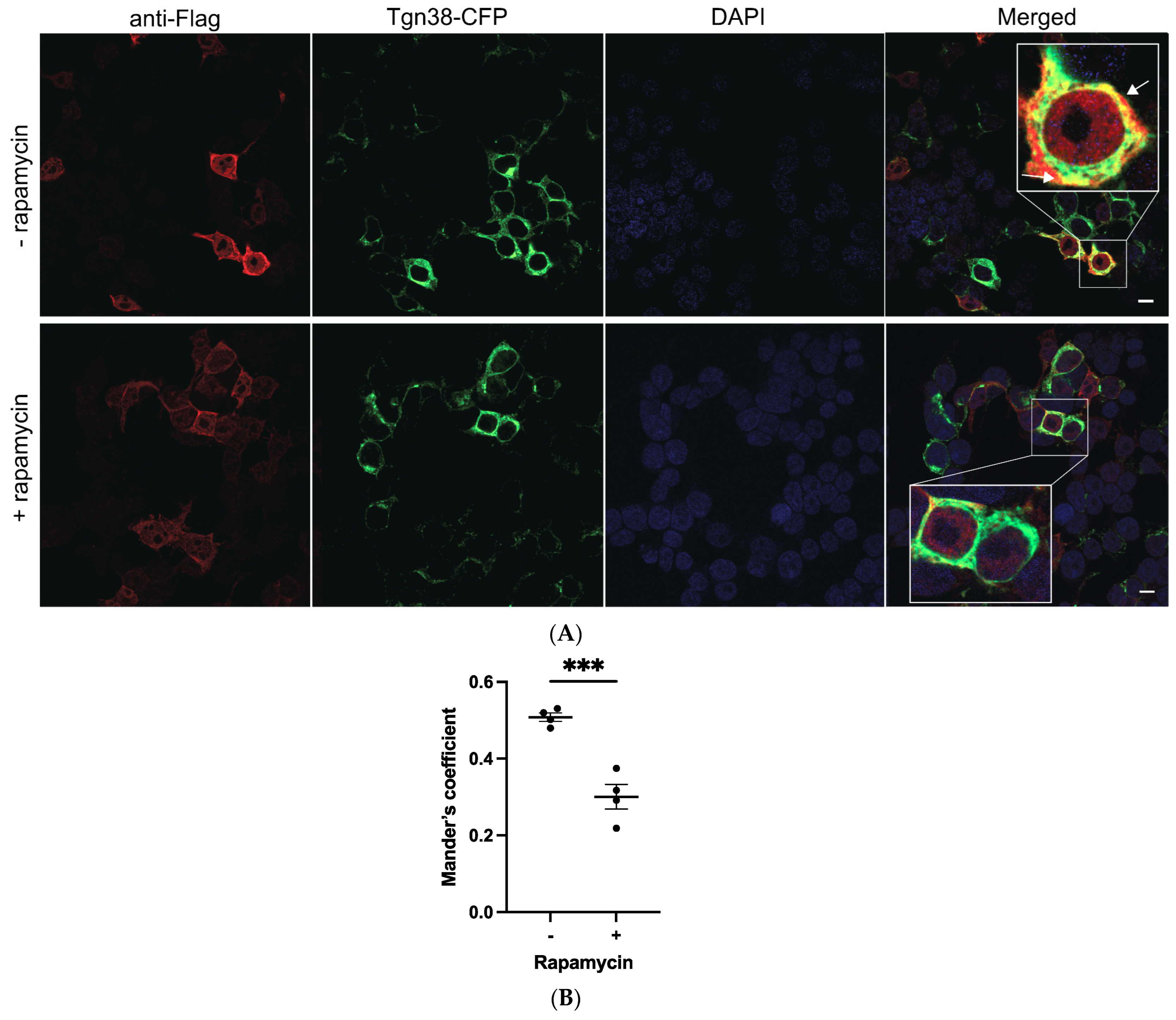
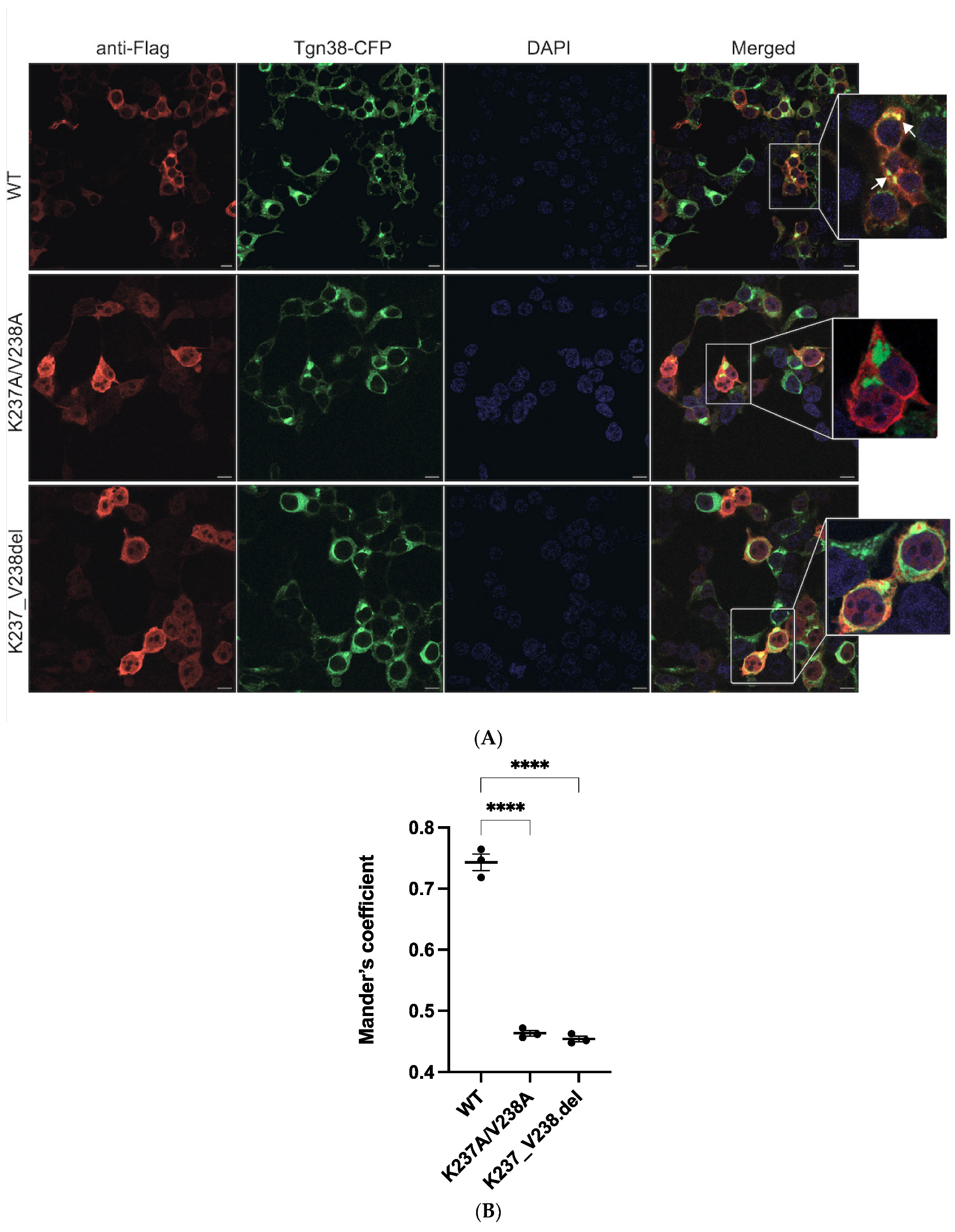
2.2. Mutations K237A/V238A and K237_V238del Reduce a2NT Golgi Localization
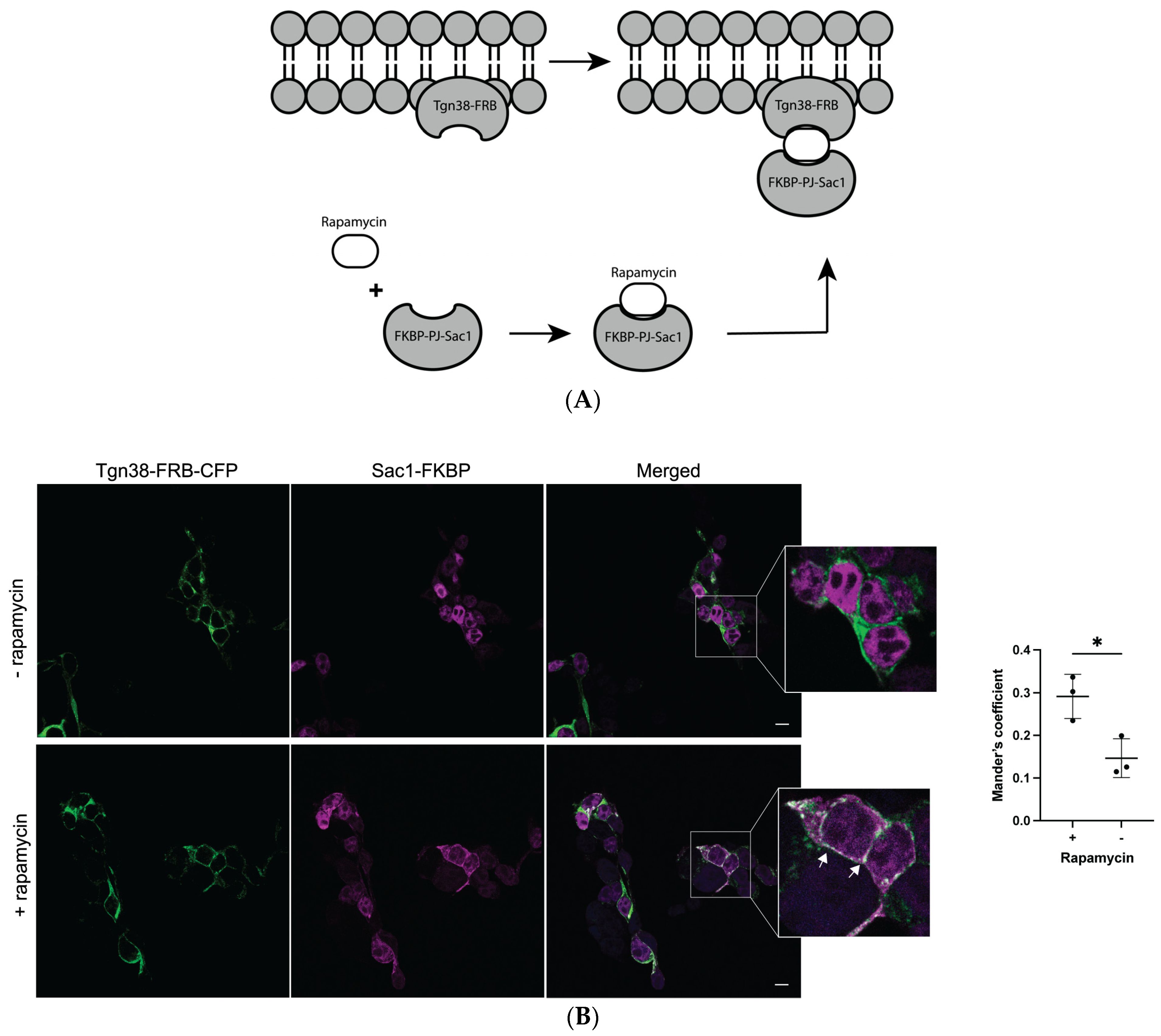
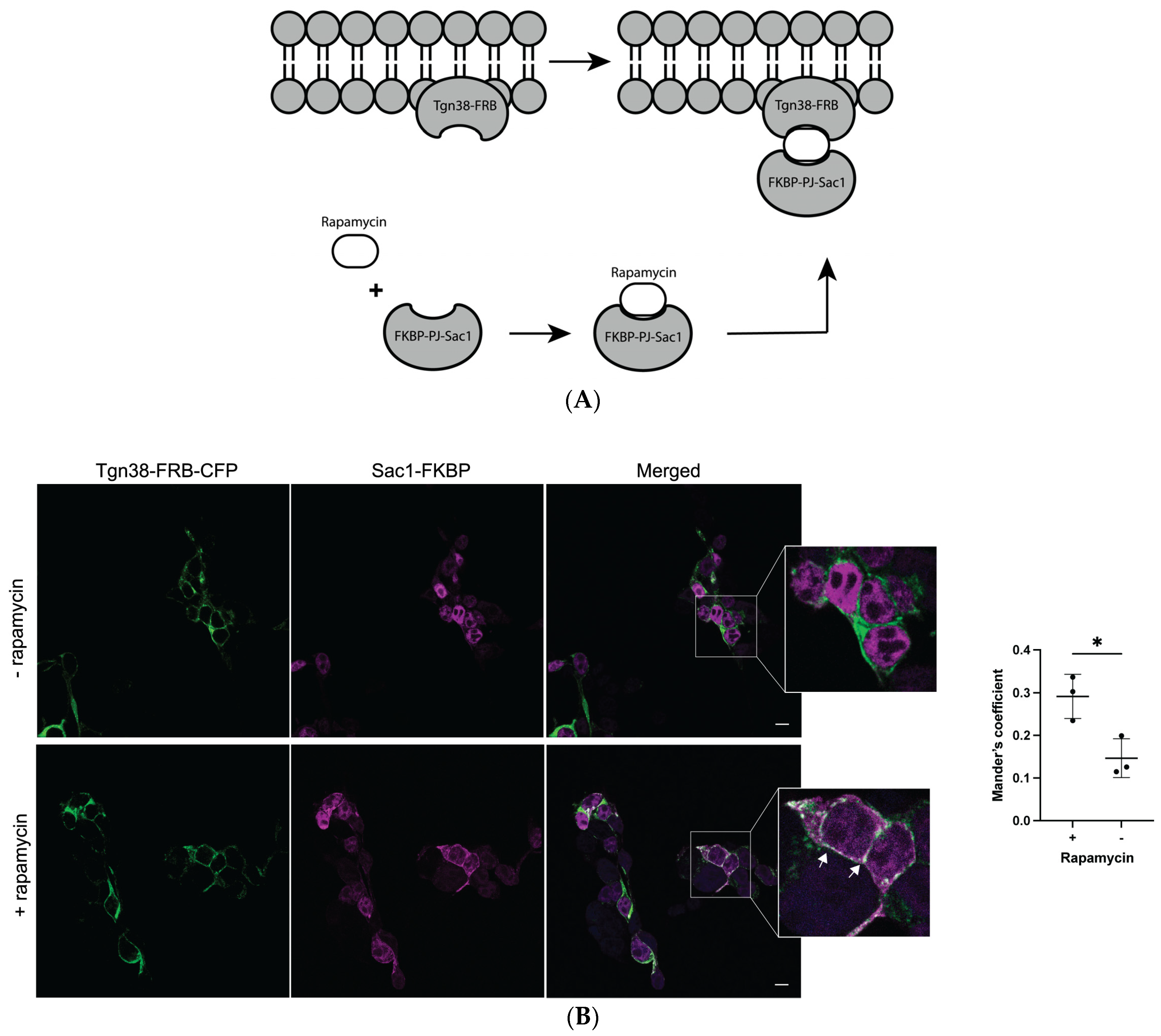
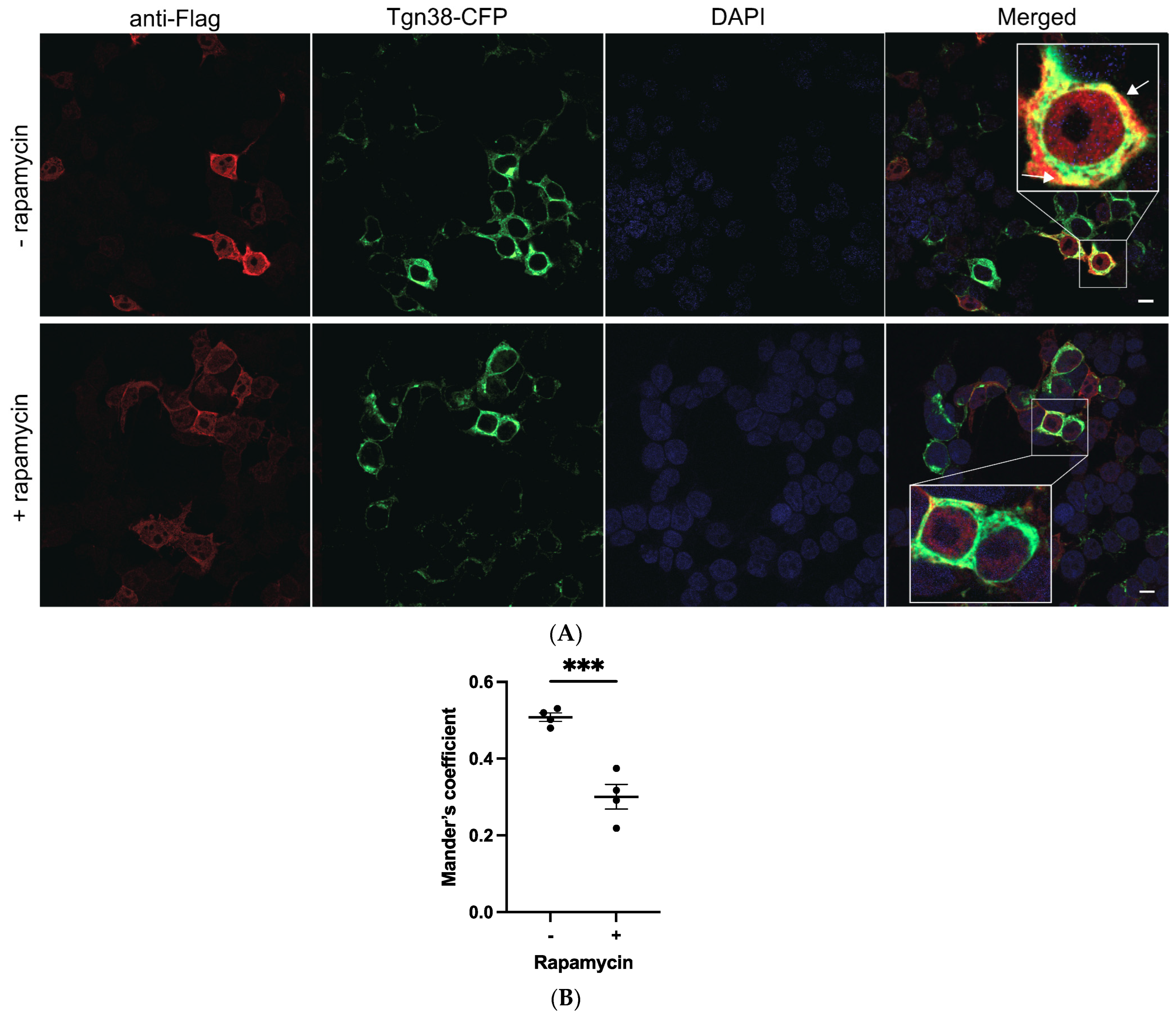
2.3. Depletion of Golgi PI(4)P Impairs a2NT Recruitment to Golgi
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of Human a2NT Wildtype and Mutants K237A/V238A, K237_V238.del from E. coli
4.2. PolyPIPosome Pull-Down Assay
4.3. HEK293 Transfection and Cellular Fractionation
4.4. Immunofluorescence
4.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kibak, H.; Taiz, L.; Starke, T.; Bernasconi, P.; Gogarten, J.P. Evolution of structure and function of V-ATPases. J. Bioenerg. Biomembr. 1992, 24, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Futai, M.; Sun-Wada, G.H.; Wada, Y.; Matsumoto, N.; Nakanishi-Matsui, M. Vacuolar-type ATPase: A proton pump to lysosomal trafficking. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 17. [Google Scholar] [CrossRef]
- Nelson, N. Structure and function of V-ATPases in endocytic and secretory organelles. J. Exp. Biol. 1992, 172, 149–153. [Google Scholar] [CrossRef]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Futai, M.; Oka, T.; Sun-Wada, G.; Moriyama, Y.; Kanazawa, H.; Wada, Y. Luminal acidification of diverse organelles by V-ATPase in animal cells. J. Exp. Biol. 2000, 203 Pt 1, 107–116. [Google Scholar] [CrossRef]
- Zhao, J.; Benlekbir, S.; Rubinstein, J.L. Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature 2015, 521, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Kartner, N.; Manolson, M.F. V-ATPase subunit interactions: The long road to therapeutic targeting. Curr. Protein Pept. Sci. 2012, 13, 164–179. [Google Scholar] [CrossRef]
- Wada, Y.; Sun-Wada, G.H.; Tabata, H.; Kawamura, N. Vacuolar-type proton ATPase as regulator of membrane dynamics in multicellular organisms. J. Bioenerg. Biomembr. 2008, 40, 53–57. [Google Scholar] [CrossRef]
- Perzov, N.; Padler-Karavani, V.; Nelson, H.; Nelson, N. Characterization of yeast V-ATPase mutants lacking Vph1p or Stv1p and the effect on endocytosis. J. Exp. Biol. 2002, 205 Pt 9, 1209–1219. [Google Scholar] [CrossRef]
- Finnigan, G.C.; Cronan, G.E.; Park, H.J.; Srinivasan, S.; Quiocho, F.A.; Stevens, T.H. Sorting of the yeast vacuolar-type, proton-translocating ATPase enzyme complex (V-ATPase): Identification of a necessary and sufficient Golgi/endosomal retention signal in Stv1p. J. Biol. Chem. 2012, 287, 19487–19500. [Google Scholar] [CrossRef]
- Manolson, M.F.; Wu, B.; Proteau, D.; Taillon, B.E.; Roberts, B.T.; Hoyt, M.A.; Jones, E.W. STV1 gene encodes functional homologue of 95-kDa yeast vacuolar H(+)-ATPase subunit Vph1p. J. Biol. Chem. 1994, 269, 14064–14074. [Google Scholar] [CrossRef]
- Hinton, A.; Sennoune, S.R.; Bond, S.; Fang, M.; Reuveni, M.; Sahagian, G.G.; Jay, D.; Martinez-Zaguilan, R.; Forgac, M. Function of a subunit isoforms of the V-ATPase in pH homeostasis and in vitro invasion of MDA-MB231 human breast cancer cells. J. Biol. Chem. 2009, 284, 16400–16408. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki-Nishi, S.; Bowers, K.; Nishi, T.; Forgac, M.; Stevens, T.H. The amino-terminal domain of the vacuolar proton-translocating ATPase a subunit controls targeting and in vivo dissociation, and the carboxyl-terminal domain affects coupling of proton transport and ATP hydrolysis. J. Biol. Chem. 2001, 276, 47411–47420. [Google Scholar] [CrossRef] [PubMed]
- Aoto, K.; Kato, M.; Akita, T.; Nakashima, M.; Mutoh, H.; Akasaka, N.; Tohyama, J.; Nomura, Y.; Hoshino, K.; Ago, Y.; et al. ATP6V0A1 encoding the a1-subunit of the V0 domain of vacuolar H(+)-ATPases is essential for brain development in humans and mice. Nat. Commun. 2021, 12, 2107. [Google Scholar] [CrossRef] [PubMed]
- Wallings, R.; Connor-Robson, N.; Wade-Martins, R. LRRK2 interacts with the vacuolar-type H+-ATPase pump a1 subunit to regulate lysosomal function. Hum. Mol. Genet. 2019, 28, 2696–2710. [Google Scholar] [CrossRef] [PubMed]
- Morel, N.; Dedieu, J.C.; Philippe, J.M. Specific sorting of the a1 isoform of the V-H+ATPase a subunit to nerve terminals where it associates with both synaptic vesicles and the presynaptic plasma membrane. J. Cell Sci. 2003, 116 Pt 23, 4751–4762. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, D.; Volk, E.; Bellen, H.J.; Hiesinger, P.R.; Quiocho, F.A. V-ATPase V0 sector subunit a1 in neurons is a target of calmodulin. J. Biol. Chem. 2008, 283, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Marshansky, V. The V-ATPase a2-subunit as a putative endosomal pH-sensor. Biochem. Soc. Trans. 2007, 35 Pt 5, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Merkulova, M.; McKee, M.; Dip, P.V.; Gruber, G.; Marshansky, V. N-terminal domain of the V-ATPase a2-subunit displays integral membrane protein properties. Protein Sci. 2010, 19, 1850–1862. [Google Scholar] [CrossRef]
- Marshansky, V.; Hosokawa, H.; Merkulova, M.; Bakulina, A.; Dip, P.V.; Thaker, Y.R.; Bjargava, A.; Tonra, J.R.; Ausiello, D.A.; Gruber, G. Structural model of a2-subunit N-terminus and its binding interface for Arf-GEF CTH2: Implication for regulation of V-ATPase, CTH2 function and rational drug design. Curr. Top. Membr. 2019, 83, 77–106. [Google Scholar]
- Chu, A.; Zirngibl, R.A.; Manolson, M.F. The V-ATPase a3 Subunit: Structure, Function and Therapeutic Potential of an Essential Biomolecule in Osteoclastic Bone Resorption. Int. J. Mol. Sci. 2021, 22, 6934. [Google Scholar] [CrossRef]
- Matsumoto, N.; Sekiya, M.; Sun-Wada, G.H.; Wada, Y.; Nakanishi-Matsui, M. The lysosomal V-ATPase a3 subunit is involved in localization of Mon1-Ccz1, the GEF for Rab7, to secretory lysosomes in osteoclasts. Sci. Rep. 2022, 12, 8455. [Google Scholar] [CrossRef]
- Manolson, M.F.; Yu, H.; Chen, W.; Yao, Y.; Li, K.; Lees, R.L.; Heersche, J.N. The a3 isoform of the 100-kDa V-ATPase subunit is highly but differentially expressed in large (>or=10 nuclei) and small (<or= nuclei) osteoclasts. J. Biol. Chem. 2003, 278, 49271–49278. [Google Scholar]
- Brown, D.; Sabolic, I.; Gluck, S. Polarized targeting of V-ATPase in kidney epithelial cells. J. Exp. Biol. 1992, 172, 231–243. [Google Scholar] [CrossRef]
- Oka, T.; Murata, Y.; Namba, M.; Yoshimizu, T.; Toyomura, T.; Yamamoto, A.; Sun-Wada, G.H.; Hamasaki, N.; Wada, Y.; Futai, M. a4, a unique kidney-specific isoform of mouse vacuolar H+-ATPase subunit a. J. Biol. Chem. 2001, 276, 40050–40054. [Google Scholar] [CrossRef]
- Pietrement, C.; Sun-Wada, G.H.; Silva, N.D.; McKee, M.; Marshansky, V.; Brown, D.; Futai, M.; Breton, S. Distinct expression patterns of different subunit isoforms of the V-ATPase in the rat epididymis. Biol. Reprod. 2006, 74, 185–194. [Google Scholar] [CrossRef]
- Golder, Z.J.; Karet Frankl, F.E. Extra-renal locations of the a4 subunit of H(+)ATPase. BMC Cell Biol. 2016, 17, 27. [Google Scholar] [CrossRef] [PubMed]
- Kornak, U.; Reynders, E.; Dimopoulou, A.; van Reeuwijk, J.; Fischer, B.; Rajab, A.; Budde, B.; Nurnberg, P.; Foulquier, F.; ARCL Debré-type Study Group; et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat. Genet. 2008, 40, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Van Maldergem, L.; Dobyns, W.; Kornak, U. ATP6V0A2-Related Cutis Laxa. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Beyens, A.; Moreno-Artero, E.; Bodemer, C.; Cox, H.; Gezdirici, A.; Yilmaz Gulec, E.; Kahloul, N.; Khau Van Kien, P.; Ogur, G.; Harroche, A.; et al. ATP6V0A2-related cutis laxa in 10 novel patients: Focus on clinical variability and expansion of the phenotype. Exp. Dermatol. 2019, 28, 1142–1145. [Google Scholar] [CrossRef]
- Hucthagowder, V.; Morava, E.; Kornak, U.; Lefeber, D.J.; Fischer, B.; Dimopoulou, A.; Aldinger, A.; Choi, J.; Davis, E.C.; Abuelo, D.N.; et al. Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum. Mol. Genet. 2009, 18, 2149–2165. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Matsukawa, R.; Takahashi, S.; Kudo, K.; Sun-Wada, G.H.; Wada, Y.; Nakanishi-Matsui, M. V-ATPase a3 isoform mutations identified in osteopetrosis patients abolish its expression and disrupt osteoclast function. Exp. Cell Res. 2020, 389, 111901. [Google Scholar] [CrossRef]
- Ochotny, N.; Voronov, I.; Owen, C.; Aubin, J.E.; Manolson, M.F. The R740S mutation in the V-ATPase a3 subunit results in osteoclast apoptosis and defective early-stage autophagy. J. Cell. Biochem. 2013, 114, 2823–2833. [Google Scholar] [CrossRef]
- Ochotny, N.; Flenniken, A.M.; Owen, C.; Voronov, I.; Zirngibl, R.A.; Osborne, L.R.; Henderson, J.E.; Adamson, S.L.; Rossant, J.; Manolson, M.F.; et al. The V-ATPase a3 subunit mutation R740S is dominant negative and results in osteopetrosis in mice. J. Bone Miner. Res. 2011, 26, 1484–1493. [Google Scholar] [CrossRef]
- Pangrazio, A.; Caldana, M.E.; Lo Iacono, N.; Mantero, S.; Vezzoni, P.; Villa, A.; Sobacchi, C. Autosomal recessive osteopetrosis: Report of 41 novel mutations in the TCIRG1 gene and diagnostic implications. Osteoporos. Int. 2012, 23, 2713–2718. [Google Scholar] [CrossRef]
- Stehberger, P.A.; Schulz, N.; Finberg, K.E.; Karet, F.E.; Giebisch, G.; Lifton, R.P.; Geibel, J.P.; Wagner, C.A. Localization and regulation of the ATP6V0A4 (a4) vacuolar H+-ATPase subunit defective in an inherited form of distal renal tubular acidosis. J. Am. Soc. Nephrol. 2003, 14, 3027–3038. [Google Scholar] [CrossRef]
- Stover, E.H.; Borthwick, K.J.; Bavalia, C.; Eady, N.; Fritz, D.M.; Rungroj, N.; Giersch, A.B.; Morton, C.C.; Axon, P.R.; Akil, I.; et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J. Med. Genet. 2002, 39, 796–803. [Google Scholar] [CrossRef]
- Srinivasan, S.; Vyas, N.K.; Baker, M.L.; Quiocho, F.A. Crystal structure of the cytoplasmic N-terminal domain of subunit I, a homolog of subunit a, of V-ATPase. J. Mol. Biol. 2011, 412, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Kartner, N.; Yao, Y.; Bhargava, A.; Manolson, M.F. Topology, glycosylation and conformational changes in the membrane domain of the vacuolar H+-ATPase a subunit. J. Cell. Biochem. 2013, 114, 1474–1487. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Kane, P.M. Direct interaction of the Golgi V-ATPase a-subunit isoform with PI(4)P drives localization of Golgi V-ATPases in yeast. Mol. Biol. Cell 2017, 28, 2518–2530. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Diakov, T.T.; Xu, T.; Tarsio, M.; Zhu, W.; Couoh-Cardel, S.; Weisman, L.S.; Kane, P.M. The signaling lipid PI(3,5)P(2) stabilizes V(1)-V(o) sector interactions and activates the V-ATPase. Mol. Biol. Cell 2014, 25, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.G. Phosphoinositides in constitutive membrane traffic. Physiol. Rev. 2004, 84, 699–730. [Google Scholar] [CrossRef] [PubMed]
- Payrastre, B. Phosphoinositides: Lipid kinases and phosphatases. Methods Mol. Biol. 2004, 273, 201–212. [Google Scholar]
- Kanaho, Y.; Suzuki, T. Phosphoinositide kinases as enzymes that produce versatile signaling lipids, phosphoinositides. J. Biochem. 2002, 131, 503–509. [Google Scholar] [CrossRef]
- Posor, Y.; Jang, W.; Haucke, V. Phosphoinositides as membrane organizers. Nat. Rev. Mol. Cell Biol. 2022, 23, 797–816. [Google Scholar] [CrossRef] [PubMed]
- Larijani, B.; Pytowski, L.; Vaux, D.J. The enigma of phosphoinositides and their derivatives: Their role in regulation of subcellular compartment morphology. Biochim. Biophys. Acta Biomembr. 2022, 1864, 183780. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.; Yao, Y.; Saffi, G.T.; Chung, J.H.; Botelho, R.J.; Glibowicka, M.; Deber, C.M.; Manolson, M.F. Characterization of a PIP Binding Site in the N-Terminal Domain of V-ATPase a4 and Its Role in Plasma Membrane Association. Int. J. Mol. Sci. 2023, 24, 4867. [Google Scholar] [CrossRef]
- Fischer, B.; Dimopoulou, A.; Egerer, J.; Gardeitchik, T.; Kidd, A.; Jost, D.; Kayserili, H.; Alanay, Y.; Tantcheva-Poor, I.; Mangold, E.; et al. Further characterization of ATP6V0A2-related autosomal recessive cutis laxa. Hum. Genet. 2012, 131, 1761–1773. [Google Scholar] [CrossRef]
- Zewe, J.P.; Wills, R.C.; Sangappa, S.; Goulden, B.D.; Hammond, G.R. SAC1 degrades its lipid substrate PtdIns4P in the endoplasmic reticulum to maintain a steep chemical gradient with donor membranes. eLife 2018, 7, e35588. [Google Scholar] [CrossRef]
- Del Bel, L.M.; Brill, J.A. Sac1, a lipid phosphatase at the interface of vesicular and nonvesicular transport. Traffic 2018, 19, 301–318. [Google Scholar] [CrossRef]
- Kim, S.; Park, J.; Jeon, B.W.; Hwang, G.; Kang, N.Y.; We, Y.; Park, W.Y.; Oh, E.; Kim, J. Chemical control of receptor kinase signaling by rapamycin-induced dimerization. Mol. Plant 2021, 14, 1379–1390. [Google Scholar] [CrossRef]
- Wang, Y.; Barnett, S.F.H.; Le, S.; Guo, Z.; Zhong, X.; Kanchanawong, P.; Yan, J. Label-free Single-Molecule Quantification of Rapamycin-induced FKBP-FRB Dimerization for Direct Control of Cellular Mechanotransduction. Nano Lett. 2019, 19, 7514–7525. [Google Scholar] [CrossRef] [PubMed]
- Dickson, E.J.; Jensen, J.B.; Hille, B. Golgi and plasma membrane pools of PI(4)P contribute to plasma membrane PI(4,5)P2 and maintenance of KCNQ2/3 ion channel current. Proc. Natl. Acad. Sci. USA 2014, 111, E2281–E2290. [Google Scholar] [CrossRef] [PubMed]
- Couoh-Cardel, S.; Milgrom, E.; Wilkens, S. Affinity Purification and Structural Features of the Yeast Vacuolar ATPase Vo Membrane Sector. J. Biol. Chem. 2015, 290, 27959–27971. [Google Scholar] [CrossRef] [PubMed]
- Mitra, C.; Winkley, S.; Kane, P.M. Human V-ATPase a-subunit isoforms bind specifically to distinct phosphoinositide phospholipids. J. Biol. Chem. 2023, 299, 105473. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.B.; Zurzolo, C. Lipids as targeting signals: Lipid rafts and intracellular trafficking. Traffic 2004, 5, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Golub, T.; Wacha, S.; Caroni, P. Spatial and temporal control of signaling through lipid rafts. Curr. Opin. Neurobiol. 2004, 14, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.L.; Feng, S.; Hilgemann, D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 1998, 391, 803–806. [Google Scholar] [CrossRef]
- Zhang, H.; He, C.; Yan, X.; Mirshahi, T.; Logothetis, D.E. Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P2 interactions. Nat. Cell Biol. 1999, 1, 183–188. [Google Scholar] [CrossRef]
- Lopes, C.M.; Zhang, H.; Rohacs, T.; Jin, T.; Yang, J.; Logothetis, D.E. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 2002, 34, 933–944. [Google Scholar] [CrossRef]
- Milburn, C.C.; Deak, M.; Kelly, S.M.; Price, N.C.; Alessi, D.R.; Van Aalten, D.M. Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem. J. 2003, 375 Pt 3, 531–538. [Google Scholar] [CrossRef]
- Mazhab-Jafari, M.T.; Rohou, A.; Schmidt, C.; Bueler, S.A.; Benlekbir, S.; Robinson, C.V.; Rubinstein, J.L. Atomic model for the membrane-embedded VO motor of a eukaryotic V-ATPase. Nature 2016, 539, 118–122. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Lu, M.; Gaugler, A.; Zhang, L.; Gluck, S.L. Phosphatidylinositol 3-kinase-mediated effects of glucose on vacuolar H+-ATPase assembly, translocation, and acidification of intracellular compartments in renal epithelial cells. Mol. Cell. Biol. 2005, 25, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Coto, J.; Schwartz, C.E.; Wang, L. Protein sector analysis for the clustering of disease-associated mutations. BMC Genom. 2014, 15 (Suppl. 11), S4. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, J.S.; Lindquist, S. Mechanisms of protein-folding diseases at a glance. Dis. Model. Mech. 2014, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.; Simon, A.; et al. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Stransky, L.; Cotter, K.; Forgac, M. The Function of V-ATPases in Cancer. Physiol. Rev. 2016, 96, 1071–1091. [Google Scholar] [CrossRef]
- Alves, M.G.O.; Garcia-Garcia, A.; Perez-Sayans, M. V-ATPases and Their Implication in Oral Cancer. In Regulation of Ca2+-ATPases, V-ATPases and F-ATPases, 1st ed.; Chakraborti, S., Dhalla, N.S., Eds.; Springer: Cham, Switzerland, 2016; Volume 14, pp. 393–405. [Google Scholar]
- Pike, J.A.; Styles, I.B.; Rappoport, J.Z.; Heath, J.K. Quantifying receptor trafficking and colocalization with confocal microscopy. Methods 2017, 115, 42–54. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, A.; Yao, Y.; Glibowicka, M.; Deber, C.M.; Manolson, M.F. The Human Mutation K237_V238del in a Putative Lipid Binding Motif within the V-ATPase a2 Isoform Suggests a Molecular Mechanism Underlying Cutis Laxa. Int. J. Mol. Sci. 2024, 25, 2170. https://doi.org/10.3390/ijms25042170
Chu A, Yao Y, Glibowicka M, Deber CM, Manolson MF. The Human Mutation K237_V238del in a Putative Lipid Binding Motif within the V-ATPase a2 Isoform Suggests a Molecular Mechanism Underlying Cutis Laxa. International Journal of Molecular Sciences. 2024; 25(4):2170. https://doi.org/10.3390/ijms25042170
Chicago/Turabian StyleChu, Anh, Yeqi Yao, Miroslawa Glibowicka, Charles M. Deber, and Morris F. Manolson. 2024. "The Human Mutation K237_V238del in a Putative Lipid Binding Motif within the V-ATPase a2 Isoform Suggests a Molecular Mechanism Underlying Cutis Laxa" International Journal of Molecular Sciences 25, no. 4: 2170. https://doi.org/10.3390/ijms25042170
APA StyleChu, A., Yao, Y., Glibowicka, M., Deber, C. M., & Manolson, M. F. (2024). The Human Mutation K237_V238del in a Putative Lipid Binding Motif within the V-ATPase a2 Isoform Suggests a Molecular Mechanism Underlying Cutis Laxa. International Journal of Molecular Sciences, 25(4), 2170. https://doi.org/10.3390/ijms25042170