

All Three AKT Isoforms Can Upregulate Oxygen Metabolism and Lactate Production in Human Hepatocellular Carcinoma Cell Lines

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
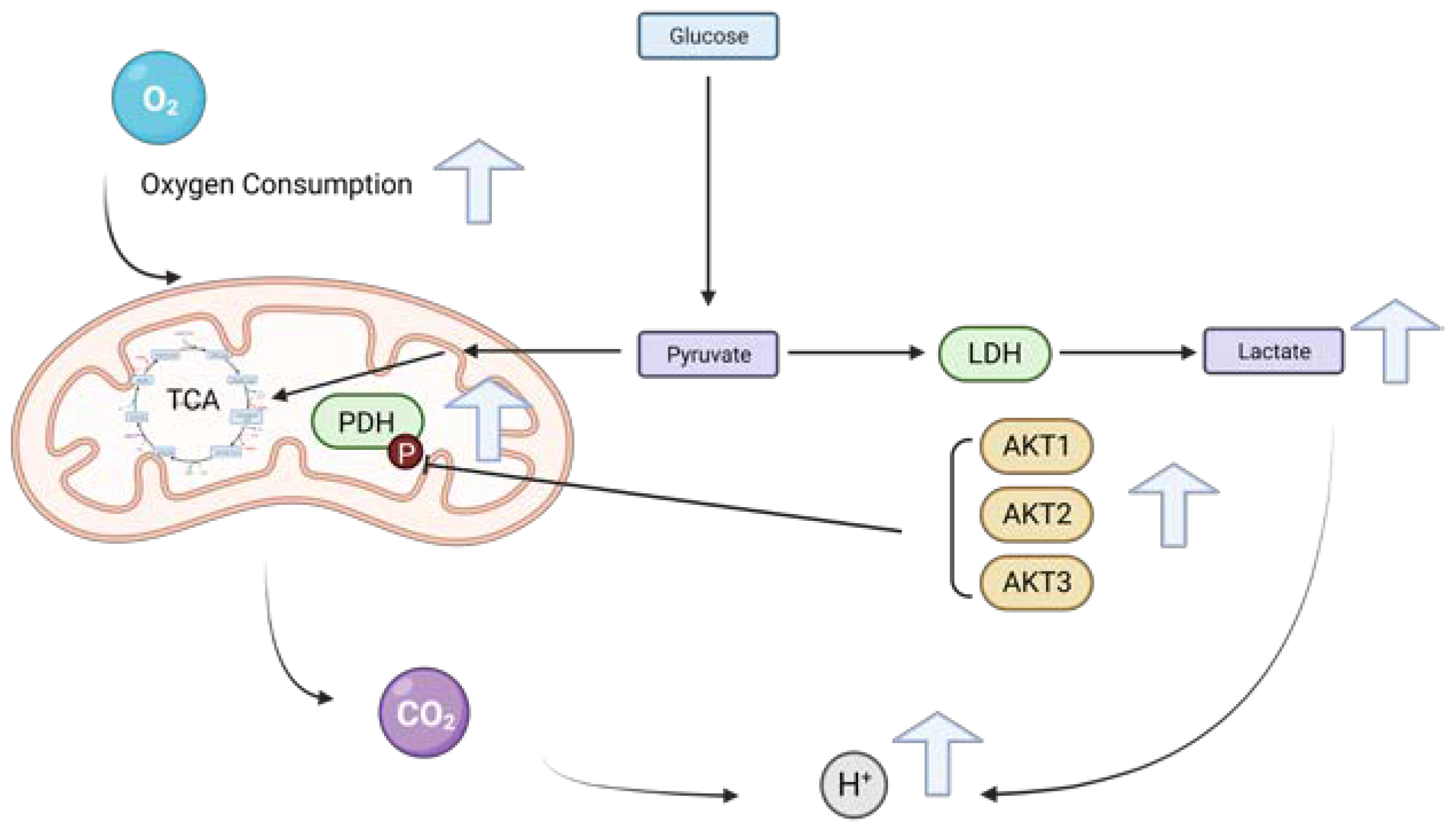
2. Results
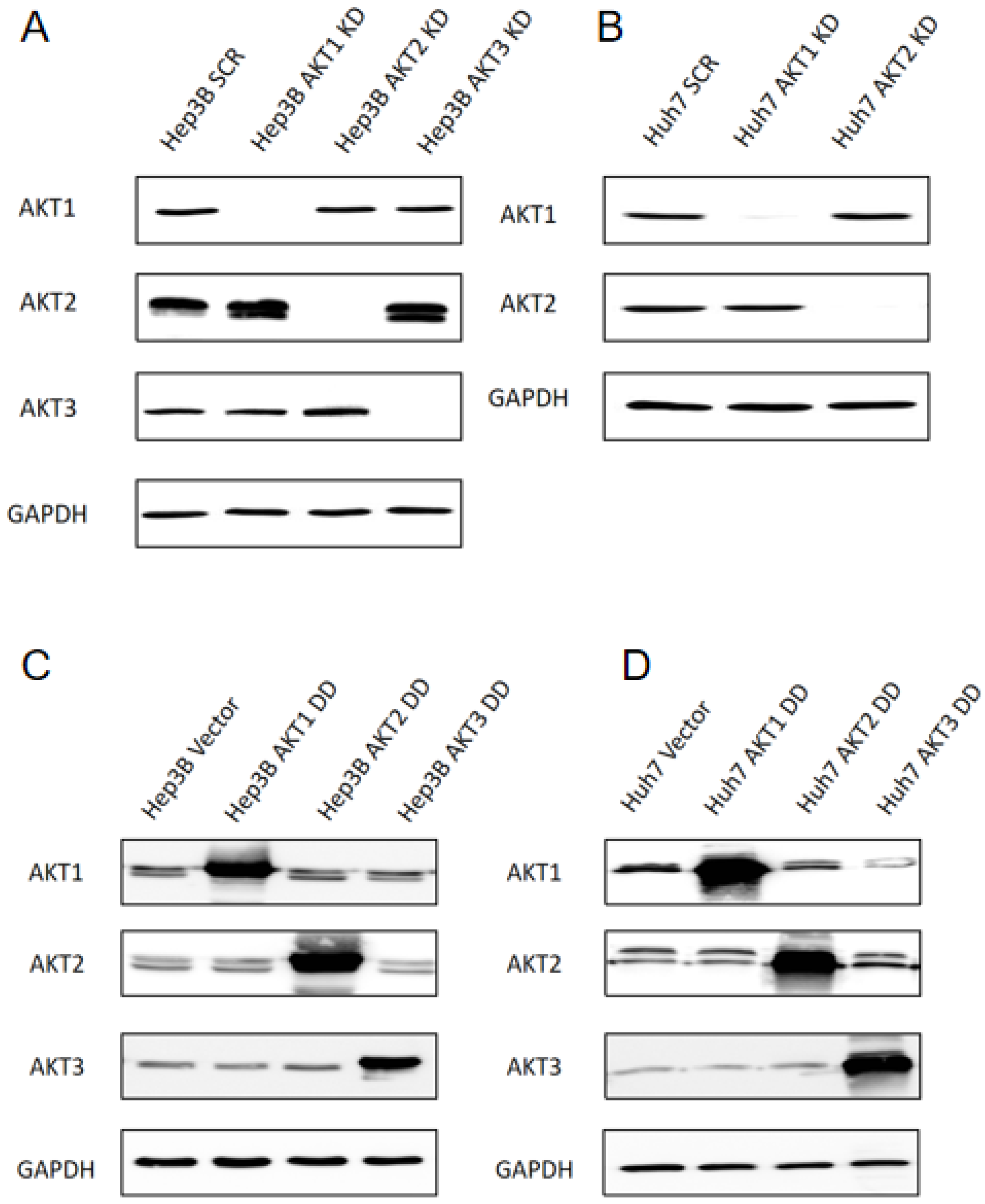
2.1. Generation of Stable Knockdowns and Ectopic Expression of Activated AKT1, AKT2 and AKT3 Mutants in Human HCC Cell Lines
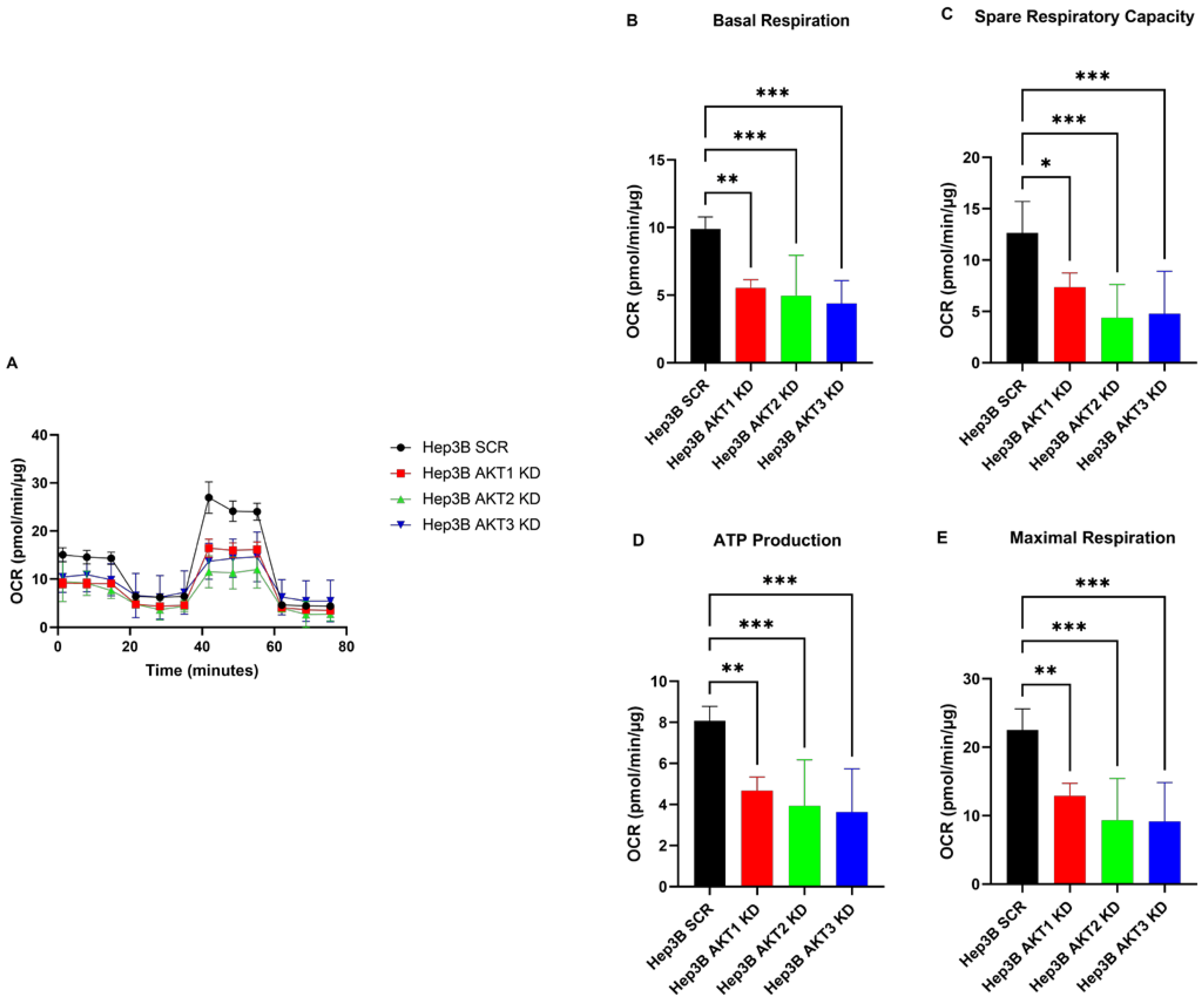
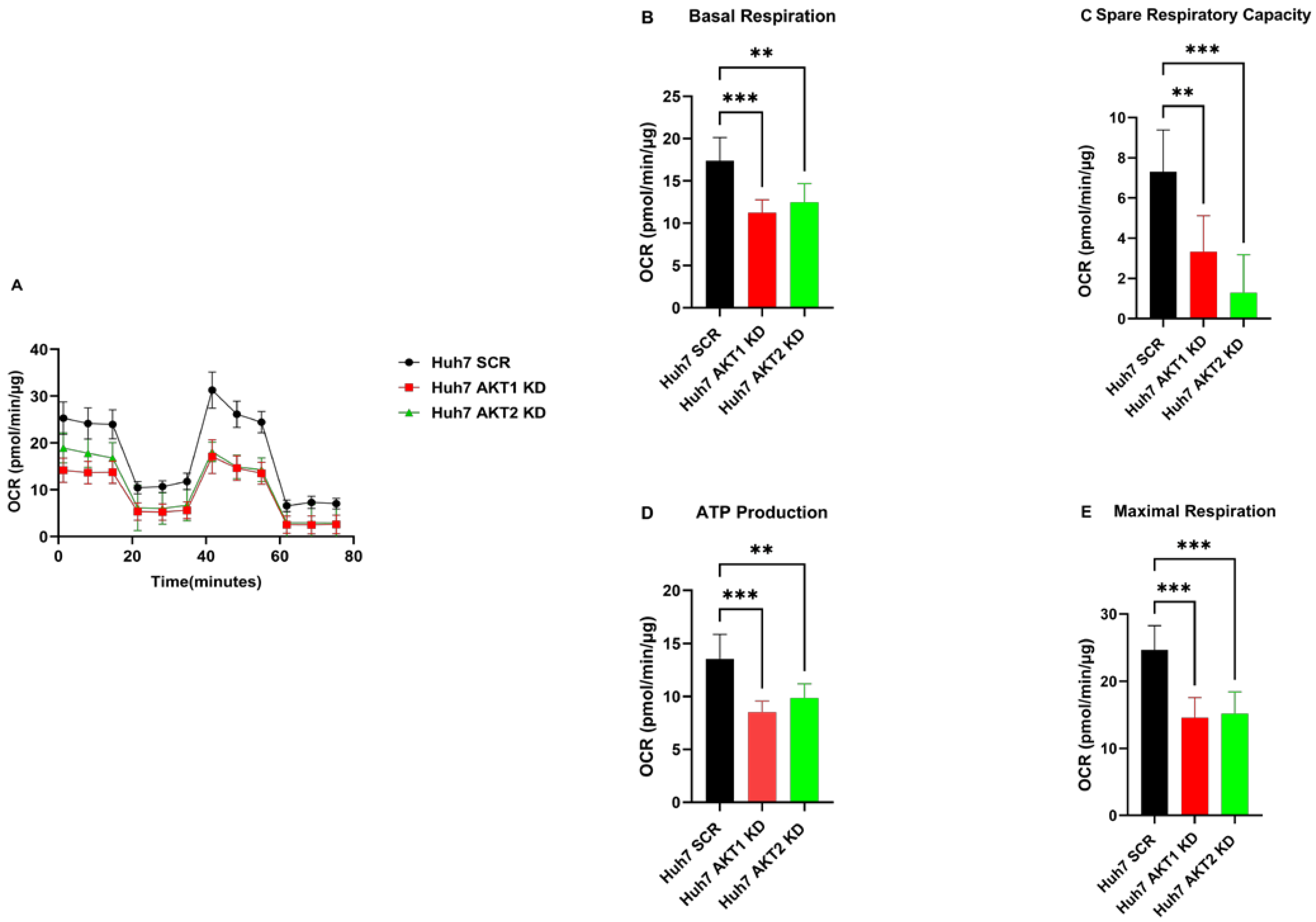
2.2. The Knockdown of AKT1, AKT2 and AKT3 Inhibits the Oxygen Consumption Rate of HCC Cells
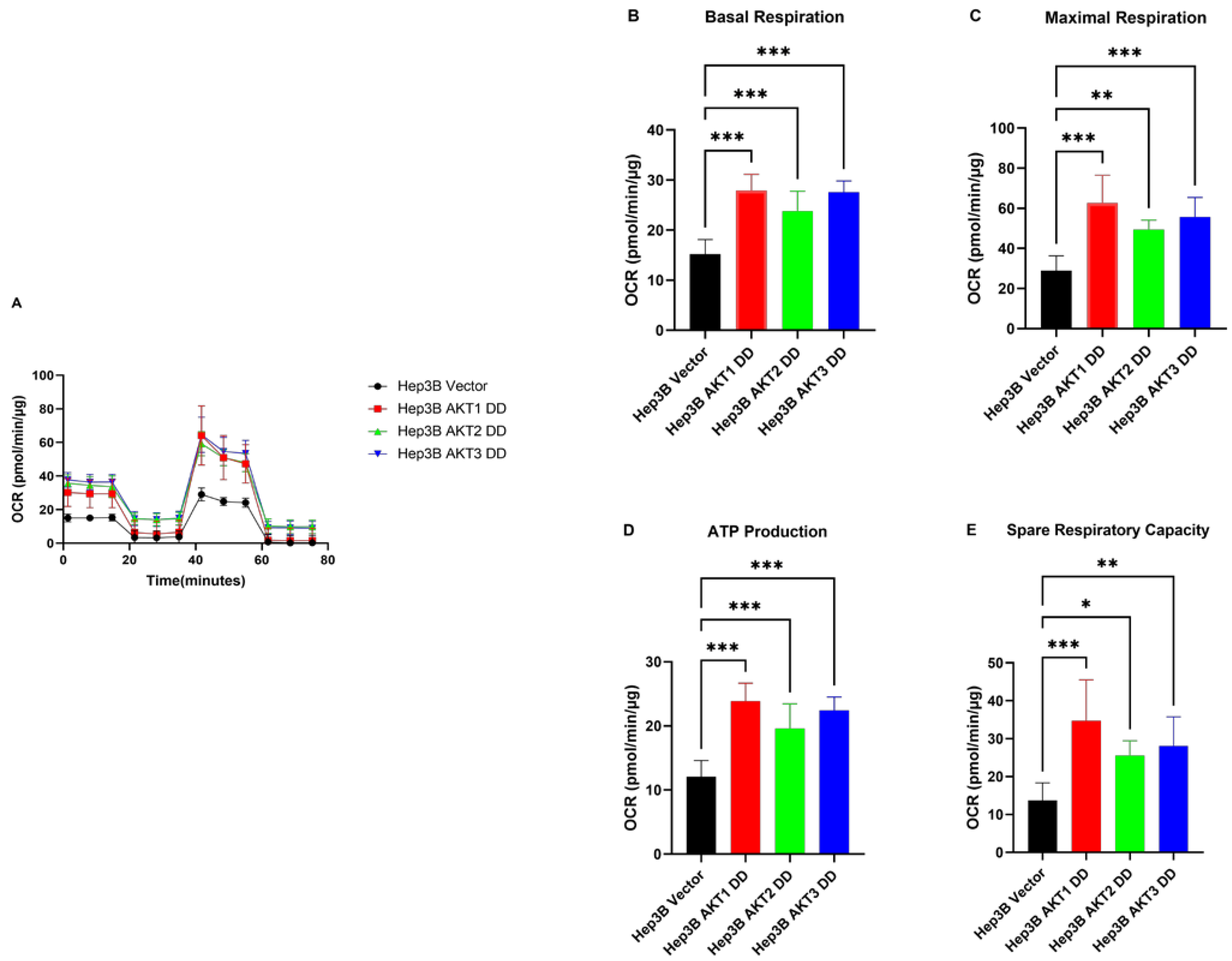
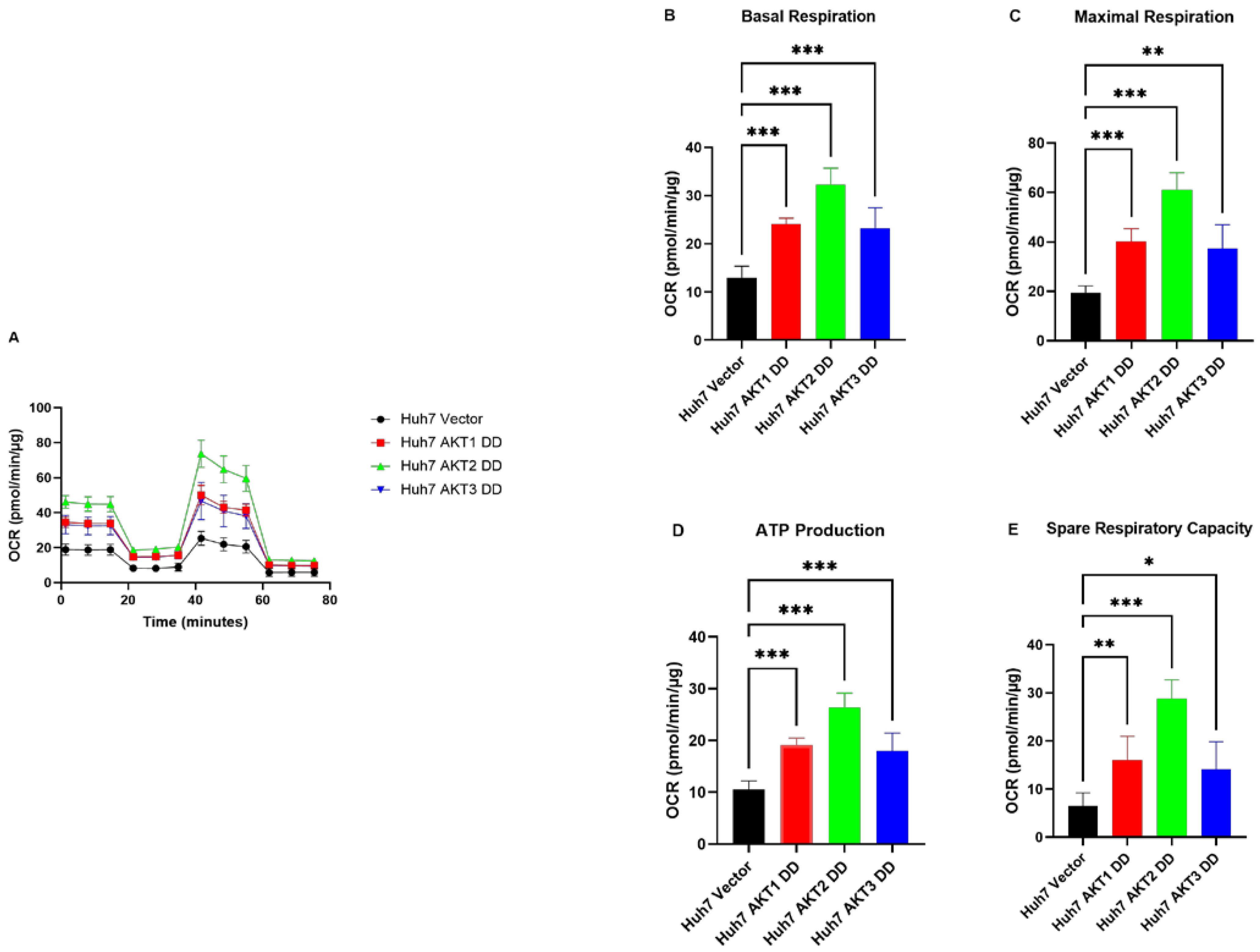
2.3. AKT1, AKT2 and AKT3 Promote OCR in Hep3B and Huh7
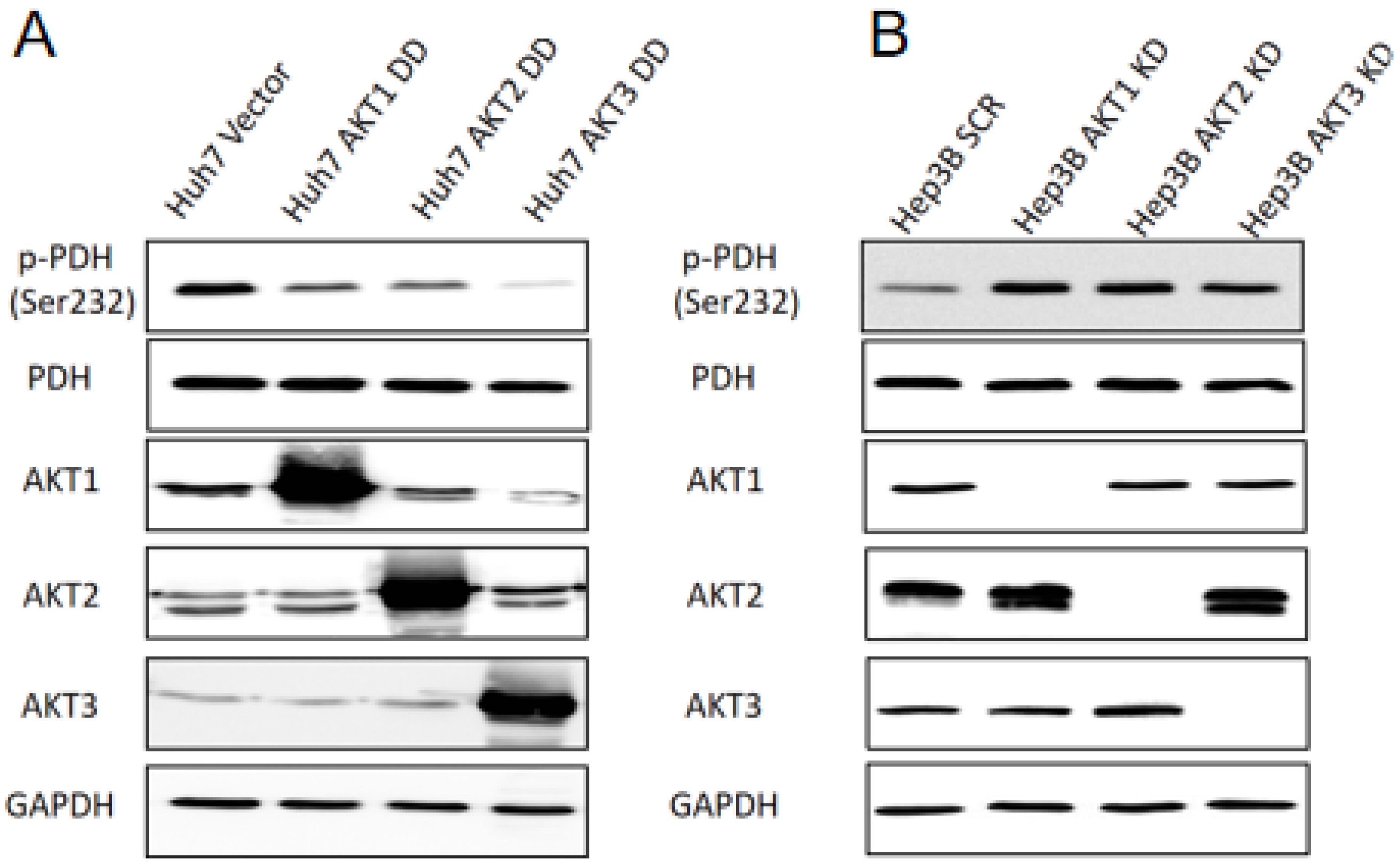
2.4. AKT Isoforms Regulate the Phosphorylation of Pyruvate Dehydrogenase
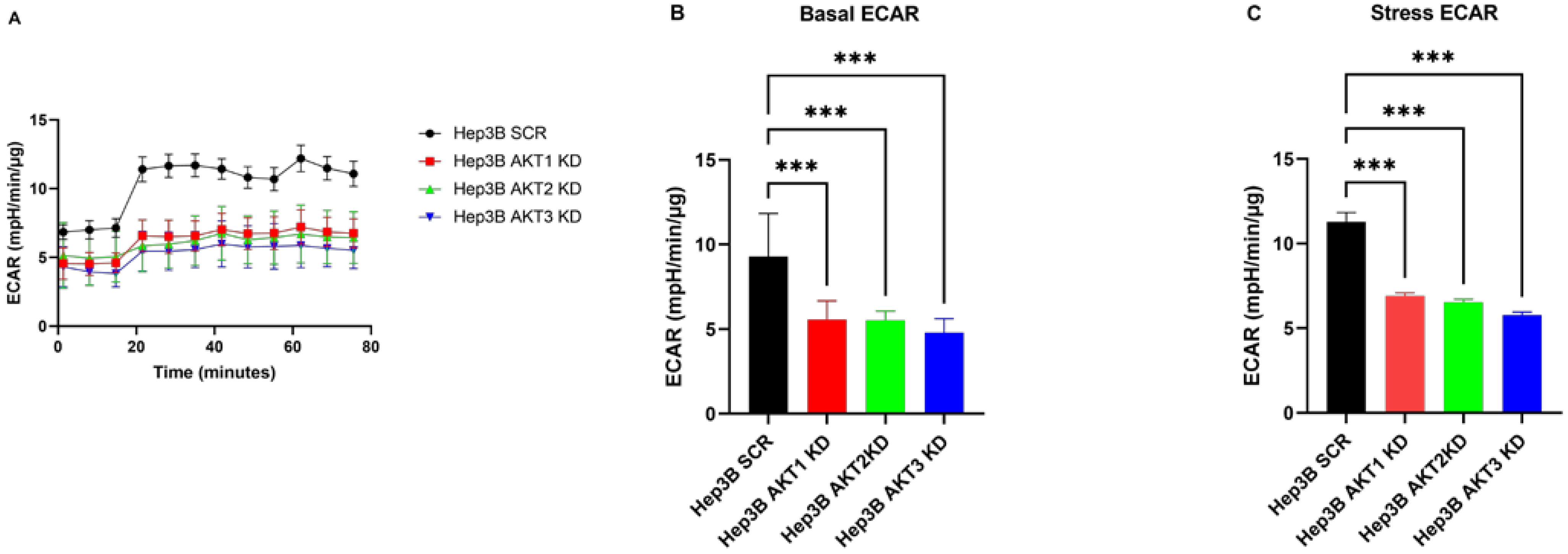
2.5. The Effects of Three AKT Isoforms on ECAR
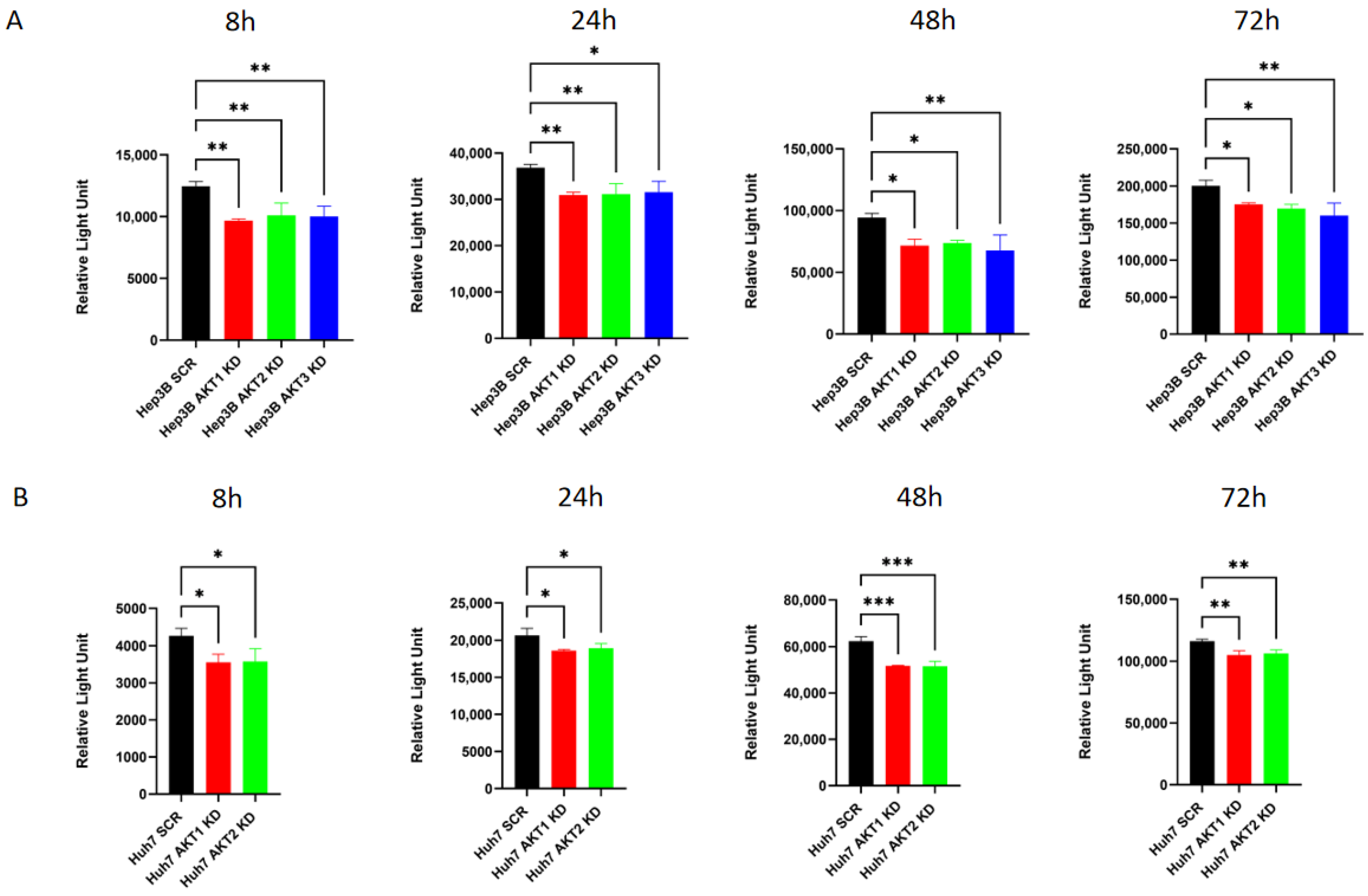
2.6. The Knockdown of AKT Isoforms Is Associated with Decreased Extracellular Lactate Production
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Culture Conditions
4.2. Stable AKT Isoform-Specific Knockdown and Ectopic Expression of Activated AKT Isoforms
4.3. Western Blot Analysis and Densitometric Quantification
4.4. Seahorse Metabolic Flux Measurement
4.5. Bradford Assay for Normalization of Seahorse Assay Data
4.6. Lactate Determination
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choo, S.P.; Tan, W.L.; Goh, B.K.P.; Tai, W.M.; Zhu, A.X. Comparison of hepatocellular carcinoma in Eastern versus Western populations. Cancer 2016, 122, 3430–3446. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski, A. Hepatocellular carcinoma. Lancet 2022, 400, 1345–1362. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Filliol, A.; Schwabe, R.F. Liver cancer metabolism: A hexokinase from the stars. Nat. Metab. 2022, 4, 1225–1226. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Zucman-Rossi, J. Genetics of hepatocellular carcinoma: The next generation. J. Hepatol. 2014, 60, 224–226. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Buontempo, F.; Ersahin, T.; Missiroli, S.; Senturk, S.; Etro, D.; Ozturk, M.; Capitani, S.; Cetin-Atalay, R.; Neri, M.L. Inhibition of Akt signaling in hepatoma cells induces apoptotic cell death independent of Akt activation status. Investig. New Drugs 2011, 29, 1303–1313. [Google Scholar] [CrossRef]
- Hu, T.H.; Huang, C.C.; Lin, P.R.; Chang, H.W.; Ger, L.P.; Lin, Y.W.; Changchien, C.S.; Lee, C.M.; Tai, M.H. Expression and prognostic role of tumor suppressor gene PTEN/MMAC1/TEP1 in hepatocellular carcinoma. Cancer 2003, 97, 1929–1940. [Google Scholar] [CrossRef]
- Nakanishi, K.; Sakamoto, M.; Yamasaki, S.; Todo, S.; Hirohashi, S. Akt phosphorylation is a risk factor for early disease recurrence and poor prognosis in hepatocellular carcinoma. Cancer 2005, 103, 307–312. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- An, J.; Oh, M.; Kim, S.-Y.; Oh, Y.-J.; Oh, B.; Oh, J.-H.; Kim, W.; Jung, J.H.; Kim, H.I.; Kim, J.-S.; et al. PET-Based Radiogenomics Supports mTOR Pathway Targeting for Hepatocellular Carcinoma. Clin. Cancer Res. 2022, 28, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, N.; Gavin, M.; Quinn, W.; Luongo, T.; Gelfer, R.; Baur, J.; Titchenell, P. The role of skeletal muscle Akt in the regulation of muscle mass and glucose homeostasis. Mol. Metab. 2019, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Grabinski, N.; Ewald, F.; Hofmann, B.T.; Staufer, K.; Schumacher, U.; Nashan, B.; Jücker, M. Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol. Cancer 2012, 11, 85. [Google Scholar] [CrossRef] [PubMed]
- Grottke, A.; Ewald, F.; Lange, T.; Nörz, D.; Herzberger, C.; Bach, J.; Grabinski, N.; Gräser, L.; Höppner, F.; Nashan, B.; et al. Downregulation of AKT3 Increases Migration and Metastasis in Triple Negative Breast Cancer Cells by Upregulating S100A4. PLoS ONE 2016, 11, e0146370. [Google Scholar] [CrossRef] [PubMed]
- Hinz, N.; Baranowsky, A.; Horn, M.; Kriegs, M.; Sibbertsen, F.; Smit, D.J.; Clezardin, P.; Lange, T.; Schinke, T.; Jücker, M. Knockdown of AKT3 Activates HER2 and DDR Kinases in Bone-Seeking Breast Cancer Cells, Promotes Metastasis In Vivo and Attenuates the TGFbeta/CTGF Axis. Cells 2021, 10, 430. [Google Scholar] [CrossRef] [PubMed]
- Priolo, C.; Pyne, S.; Rose, J.; Regan, E.R.; Zadra, G.; Photopoulos, C.; Cacciatore, S.; Schultz, D.; Scaglia, N.; McDunn, J.; et al. AKT1 and MYC induce distinctive metabolic fingerprints in human prostate cancer. Cancer Res. 2014, 74, 7198–7204. [Google Scholar] [CrossRef]
- Li, H.; Lu, S.; Chen, Y.; Zheng, L.; Chen, L.; Ding, H.; Ding, J.; Lou, D.; Liu, F.; Zheng, B. AKT2 phosphorylation of hexokinase 2 at T473 promotes tumorigenesis and metastasis in colon cancer cells via NF-kappaB, HIF1alpha, MMP2, and MMP9 upregulation. Cell. Signal. 2019, 58, 99–110. [Google Scholar] [CrossRef]
- Kim, M.; Kim, Y.Y.; Jee, H.J.; Bae, S.S.; Jeong, N.Y.; Um, J.-H.; Yun, J. Akt3 knockdown induces mitochondrial dysfunction in human cancer cells. Acta Biochim. Biophys. Sin. 2016, 48, 447–453. [Google Scholar] [CrossRef]
- Tong, M.; Wong, T.-L.; Zhao, H.; Zheng, Y.; Xie, Y.-N.; Li, C.-H.; Zhou, L.; Che, N.; Yun, J.-P.; Man, K.; et al. Loss of tyrosine catabolic enzyme HPD promotes glutamine anaplerosis through mTOR signaling in liver cancer. Cell. Rep. 2021, 37, 109976. [Google Scholar] [CrossRef]
- Chen, L.; Cheng, X.; Tu, W.; Qi, Z.; Li, H.; Liu, F.; Yang, Y.; Zhang, Z.; Wang, Z. Apatinib inhibits glycolysis by suppressing the VEGFR2/AKT1/SOX5/GLUT4 signaling pathway in ovarian cancer cells. Cell. Oncol. 2019, 42, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, M.J.; Bu, J.; Deng, J.L.; Jiang, B.Y.; Jiang, L.D.; He, X.J. miR-485-3p regulated by MALAT1 inhibits osteosarcoma glycolysis and metastasis by directly suppressing c-MET and AKT3/mTOR signalling. Life Sci. 2021, 268, 118925. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Vaira, V.; Caino, M.C.; Tang, H.-Y.; Seo, J.H.; Kossenkov, A.V.; Ottobrini, L.; Martelli, C.; Lucignani, G.; Bertolini, I.; et al. Mitochondrial Akt Regulation of Hypoxic Tumor Reprogramming. Cancer Cell. 2016, 30, 257–272. [Google Scholar] [CrossRef]
- Park, G.B.; Jeong, J.Y.; Kim, D. GLUT5 regulation by AKT1/3-miR-125b-5p downregulation induces migratory activity and drug resistance in TLR-modified colorectal cancer cells. Carcinogenesis 2020, 41, 1329–1340. [Google Scholar] [CrossRef]
- Wan, H.; Xu, L.; Zhang, H.; Wu, F.; Zeng, W.; Li, T. High expression of NEK2 promotes gastric cancer progression via activating AKT signaling. J. Physiol. Biochem. 2021, 77, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Zhao, J.; Qu, L.; Duan, Z.; Fu, R.; Zhu, C.; Fan, D. Ginsenoside Rh4 suppresses aerobic glycolysis and the expression of PD-L1 via targeting AKT in esophageal cancer. Biochem. Pharmacol. 2020, 178, 114038. [Google Scholar] [CrossRef]
- Nogueira, V.; Park, Y.; Chen, C.-C.; Xu, P.-Z.; Chen, M.-L.; Tonic, I.; Unterman, T.; Hay, N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008, 14, 458–470. [Google Scholar] [CrossRef]
- Nogueira, V.; Patra, K.C.; Hay, N. Selective eradication of cancer displaying hyperactive Akt by exploiting the metabolic consequences of Akt activation. Elife 2018, 7, e32213. [Google Scholar] [CrossRef]
- Chen, J.; Guccini, I.; Di Mitri, D.; Brina, D.; Revandkar, A.; Sarti, M.; Pasquini, E.; Alajati, A.; Pinton, S.; Losa, M.; et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat. Genet. 2018, 50, 219–228. [Google Scholar] [CrossRef]
- Patel, M.S.; Korotchkina, L.G. Regulation of the pyruvate dehydrogenase complex. Biochem. Soc. Trans. 2006, 34, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, A.; Sarcar, B.; Miller, C.R.; Kim, S.-H.; Nakano, I.; Forsyth, P.; Chinnaiyan, P. Ras-mediated modulation of pyruvate dehydrogenase activity regulates mitochondrial reserve capacity and contributes to glioblastoma tumorigenesis. Neuro. Oncol. 2015, 17, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Cerniglia, G.J.; Dey, S.; Gallagher-Colombo, S.M.; Daurio, N.A.; Tuttle, S.; Busch, T.M.; Lin, A.; Sun, R.; Esipova, T.V.; Vinogradov, S.A.; et al. The PI3K/Akt Pathway Regulates Oxygen Metabolism via Pyruvate Dehydrogenase (PDH)-E1alpha Phosphorylation. Mol. Cancer Ther. 2015, 14, 1928–1938. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, E.; Tomlinson, I.P. Mitochondrial tumour suppressors: A genetic and biochemical update. Nat. Rev. Cancer 2005, 5, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sánchez, R.; Rodriguez-Enriquez, S.; Marín-Hernández, Á.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 2009, 19, 25–31. [Google Scholar] [CrossRef]
- Hosios, A.M.; Manning, B.D. Cancer Signaling Drives Cancer Metabolism: AKT and the Warburg Effect. Cancer Res. 2021, 81, 4896–4898. [Google Scholar] [CrossRef]
- Killock, D. Novel ICI-TKI combination improves HCC outcomes. Nat. Rev. Clin. Oncol. 2023, 20, 733. [Google Scholar] [CrossRef]
- Méndez-Blanco, C.; Fondevila, F.; García-Palomo, A.; González-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, R.; Rozeman, E.A.; Kreutz, M.; Renner, K.; Blank, C.U. Targeting tumor-associated acidity in cancer immunotherapy. Cancer Immunol. Immunother. 2018, 67, 1331–1348. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Choi, S.Y.; Niu, X.; Kang, N.; Xue, H.; Killam, J.; Wang, Y. Lactic Acid and an Acidic Tumor Microenvironment suppress Anticancer Immunity. Int. J. Mol. Sci. 2020, 21, 8363. [Google Scholar] [CrossRef] [PubMed]
- Kuchuk, O.; Tuccitto, A.; Citterio, D.; Huber, V.; Camisaschi, C.; Milione, M.; Vergani, B.; Villa, A.; Alison, M.R.; Carradori, S.; et al. pH regulators to target the tumor immune microenvironment in human hepatocellular carcinoma. Oncoimmunology 2018, 7, e1445452. [Google Scholar] [CrossRef]
- Turner, N.C.; Oliveira, M.; Howell, S.J.; Dalenc, F.; Cortes, J.; Moreno, H.L.G.; Hu, X.; Jhaveri, K.; Krivorotko, P.; Loibl, S.; et al. Capivasertib in Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2023, 388, 2058–2070. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, L.-Y.; Smit, D.J.; Popova, N.V.; Horn, S.; Velasquez, L.N.; Huber, S.; Jücker, M. All Three AKT Isoforms Can Upregulate Oxygen Metabolism and Lactate Production in Human Hepatocellular Carcinoma Cell Lines. Int. J. Mol. Sci. 2024, 25, 2168. https://doi.org/10.3390/ijms25042168
Tian L-Y, Smit DJ, Popova NV, Horn S, Velasquez LN, Huber S, Jücker M. All Three AKT Isoforms Can Upregulate Oxygen Metabolism and Lactate Production in Human Hepatocellular Carcinoma Cell Lines. International Journal of Molecular Sciences. 2024; 25(4):2168. https://doi.org/10.3390/ijms25042168
Chicago/Turabian StyleTian, Ling-Yu, Daniel J. Smit, Nadezhda V. Popova, Stefan Horn, Lis Noelia Velasquez, Samuel Huber, and Manfred Jücker. 2024. "All Three AKT Isoforms Can Upregulate Oxygen Metabolism and Lactate Production in Human Hepatocellular Carcinoma Cell Lines" International Journal of Molecular Sciences 25, no. 4: 2168. https://doi.org/10.3390/ijms25042168
APA StyleTian, L.-Y., Smit, D. J., Popova, N. V., Horn, S., Velasquez, L. N., Huber, S., & Jücker, M. (2024). All Three AKT Isoforms Can Upregulate Oxygen Metabolism and Lactate Production in Human Hepatocellular Carcinoma Cell Lines. International Journal of Molecular Sciences, 25(4), 2168. https://doi.org/10.3390/ijms25042168