
Structural Insights into the Penicillin-Binding Protein 4 (DacB) from Mycobacterium tuberculosis


Abstract
1. Introduction
2. Results and Discussion
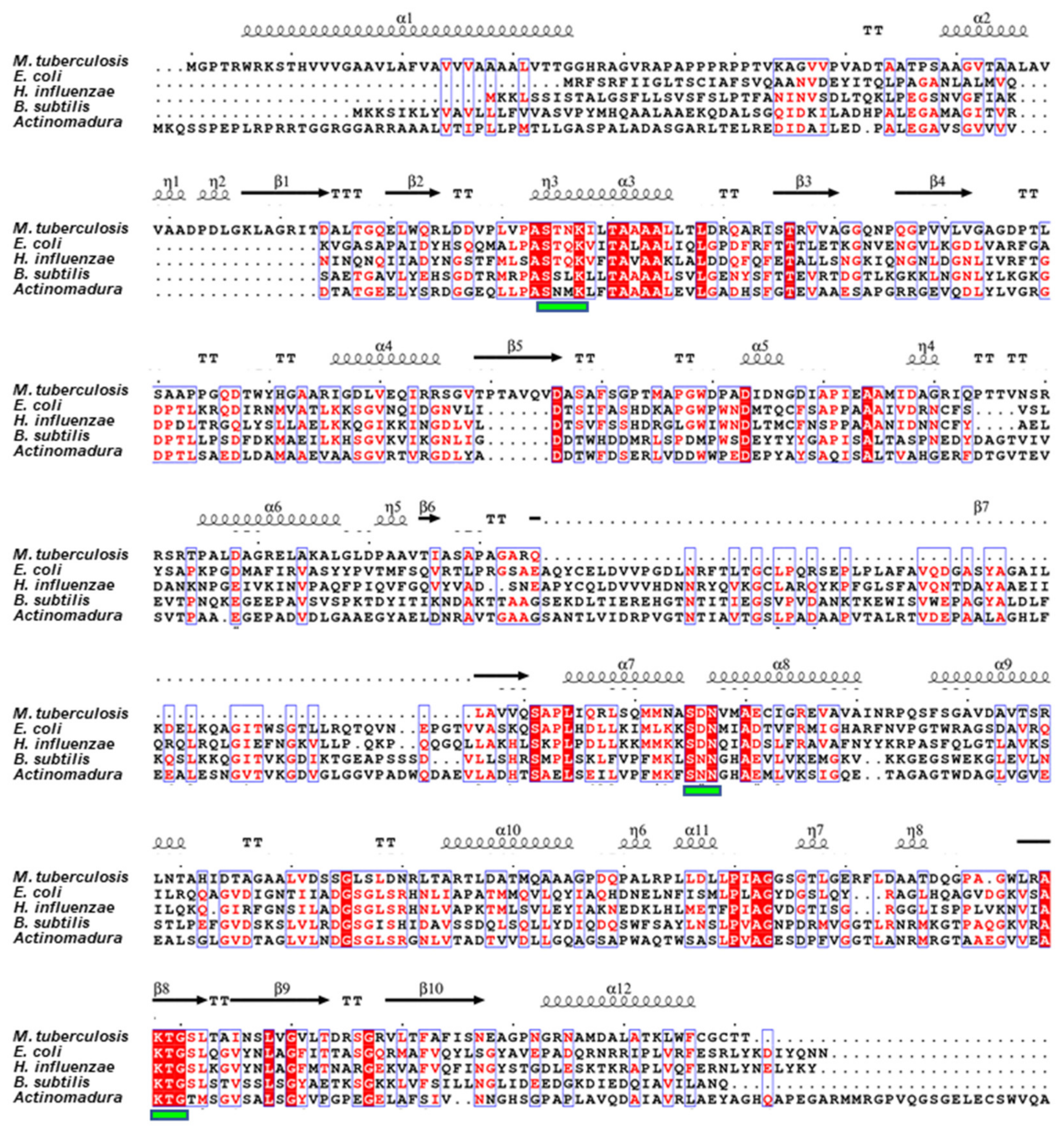
2.1. Sequence Analysis
2.2. Oligomeric State of MtbPBP4 in Solution
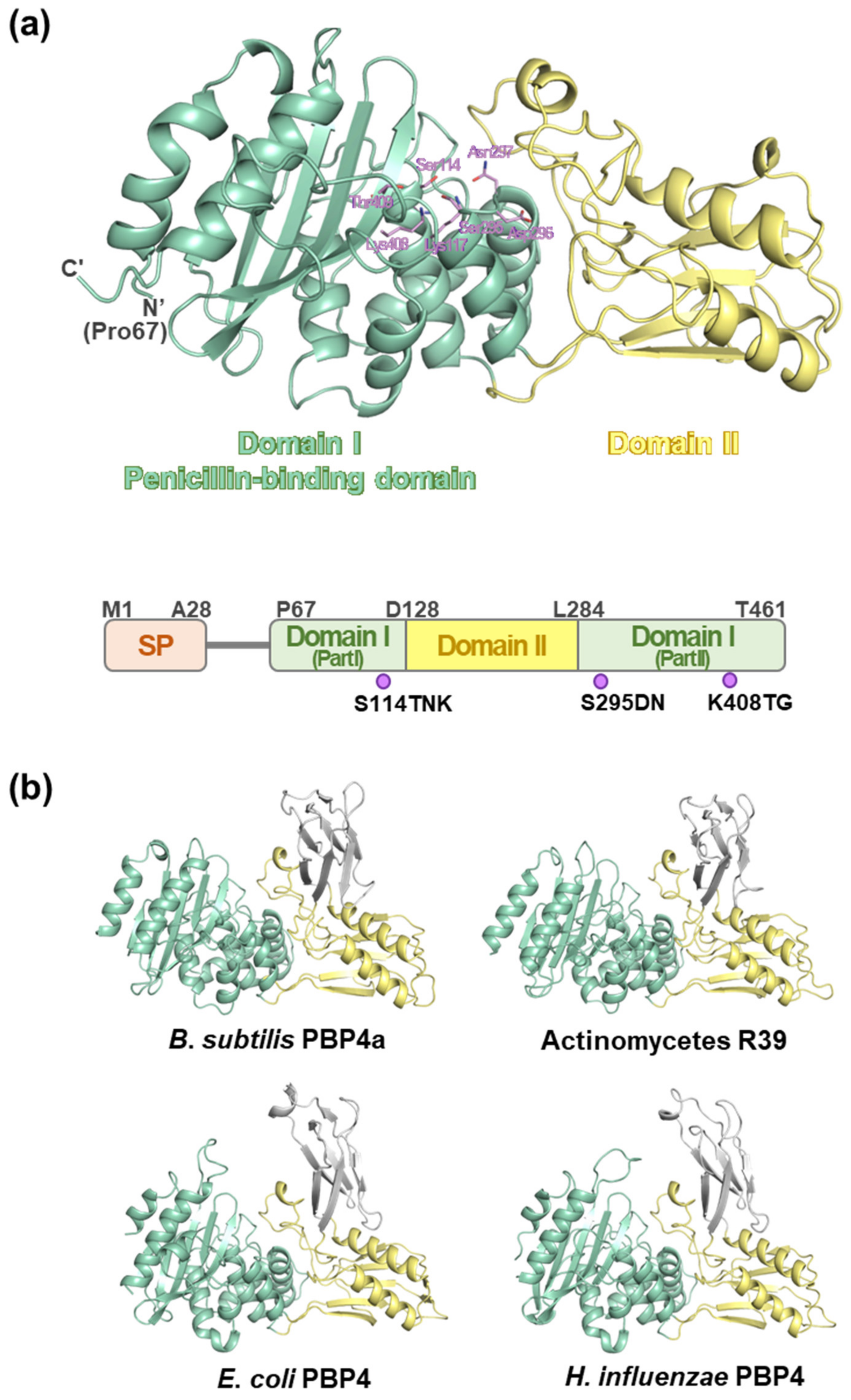
2.3. Overall Structure of MtbPBP4
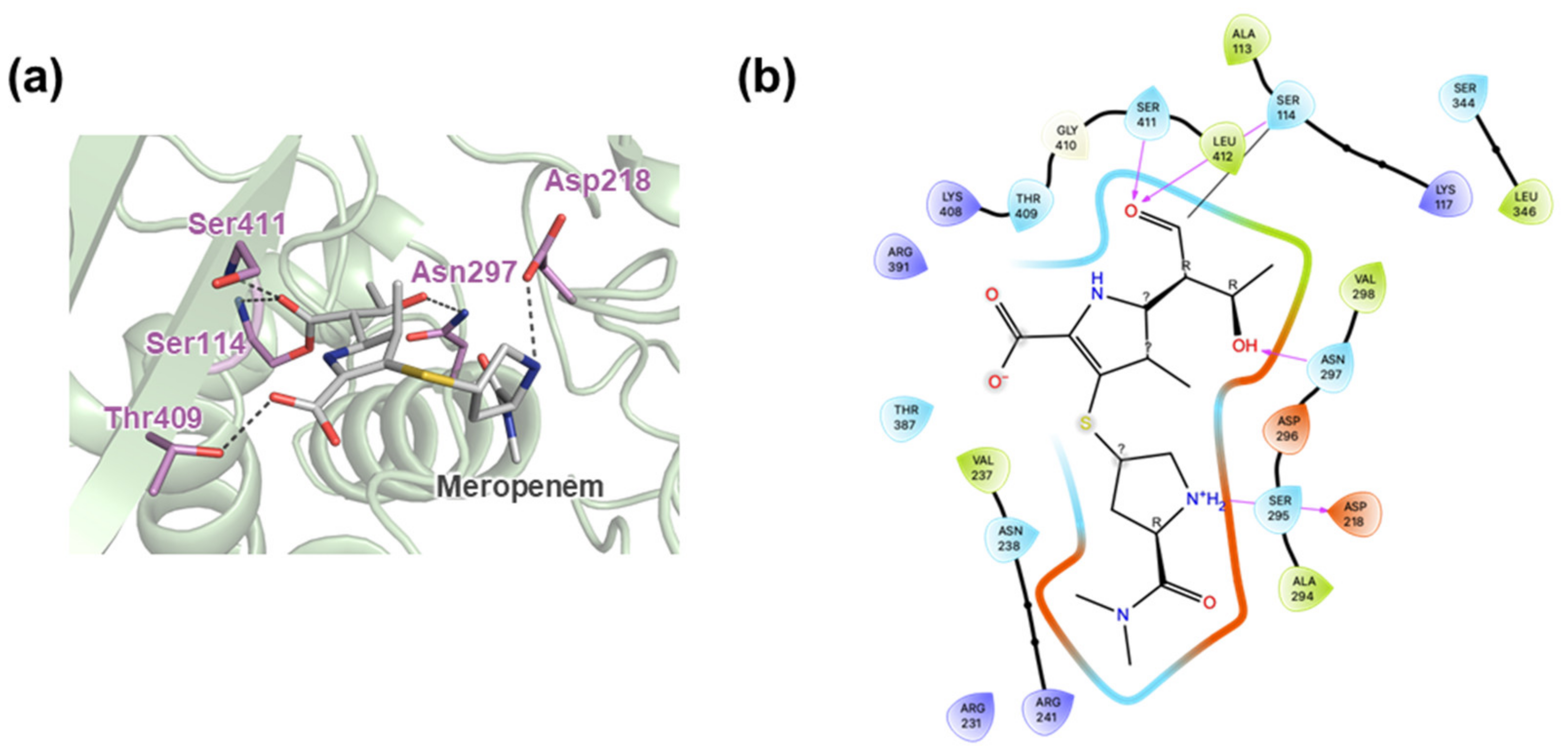
2.4. Active Site Cleft of MtbPBP4
3. Materials and Methods
3.1. Cloning, Expression, and Purification of MtbPBP4
3.2. Oligomeric State Determination of MtbPBP4
3.3. Structure Prediction of MtbPBP4
3.4. MD Simulation
3.5. Covalent Docking
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McBryde, E.S.; Meehan, M.T.; Doan, T.N.; Ragonnet, R.; Marais, B.J.; Guernier, V.; Trauer, J.M. The risk of global epidemic replacement with drug-resistant Mycobacterium tuberculosis strains. Int. J. Infect. Dis. 2017, 56, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Briffotaux, J.; Huang, W.; Wang, X.; Gicquel, B. MmpS5/MmpL5 as an efflux pump in Mycobacterium species. Tuberculosis 2017, 107, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Bothra, A.; Arumugam, P.; Panchal, V.; Menon, D.; Srivastava, S.; Shankaran, D.; Nandy, A.; Jaisinghani, N.; Singh, A.; Gokhale, R.S.; et al. Phospholipid homeostasis, membrane tenacity and survival of Mtb in lipid rich conditions is determined by MmpL11 function. Sci. Rep. 2018, 8, 8317. [Google Scholar] [CrossRef] [PubMed]
- Rullas, J.; Dhar, N.; McKinney, J.D.; Garcia-Perez, A.; Lelievre, J.; Diacon, A.H.; Hugonnet, J.E.; Arthur, M.; Angulo-Barturen, I.; Barros-Aguirre, D.; et al. Combinations of beta-Lactam Antibiotics Currently in Clinical Trials Are Efficacious in a DHP-I-Deficient Mouse Model of Tuberculosis Infection. Antimicrob. Agents Chemother. 2015, 59, 4997–4999. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.A.; El-Hay, T.; Wyres, K.L.; Weissbrod, O.; Munsamy, V.; Yanover, C.; Aharonov, R.; Shaham, O.; Conway, T.C.; Goldschmidt, Y.; et al. Paradoxical Hypersusceptibility of Drug-resistant Mycobacteriumtuberculosis to beta-lactam Antibiotics. eBioMedicine 2016, 9, 170–179. [Google Scholar] [CrossRef]
- Honeyborne, I.; Lipman, M.; Zumla, A.; McHugh, T.D. The changing treatment landscape for MDR/XDR-TB—Can current clinical trials revolutionise and inform a brave new world? Int. J. Infect. Dis. 2019, 80, S23–S28. [Google Scholar] [CrossRef]
- Migliori, G.B.; Tiberi, S.; Zumla, A.; Petersen, E.; Chakaya, J.M.; Wejse, C.; Munoz Torrico, M.; Duarte, R.; Alffenaar, J.W.; Schaaf, H.S.; et al. MDR/XDR-TB management of patients and contacts: Challenges facing the new decade. The 2020 clinical update by the Global Tuberculosis Network. Int. J. Infect. Dis. 2020, 92, S15–S25. [Google Scholar] [CrossRef]
- Dheda, K.; Gumbo, T.; Maartens, G.; Dooley, K.E.; Murray, M.; Furin, J.; Nardell, E.A.; Warren, R.M.; the Lancet Respiratory Medicine drug-resistant tuberculosis Commission group. The Lancet Respiratory Medicine Commission: 2019 update: Epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant and incurable tuberculosis. Lancet Respir. Med. 2019, 7, 820–826. [Google Scholar] [CrossRef]
- Faridgohar, M. Finding New Ways to Combat Multidrug-Resistant Tuberculosis. Microb. Drug Resist. 2020, 26, 71–80. [Google Scholar] [CrossRef]
- Lu, Z.; Wang, H.; Zhang, A.; Liu, X.; Zhou, W.; Yang, C.; Guddat, L.; Yang, H.; Schofield, C.J.; Rao, Z. Structures of Mycobacterium tuberculosis Penicillin-Binding Protein 3 in Complex with Five beta-Lactam Antibiotics Reveal Mechanism of Inactivation. Mol. Pharmacol. 2020, 97, 287–294. [Google Scholar] [CrossRef]
- Shalaby, M.W.; Dokla, E.M.E.; Serya, R.A.T.; Abouzid, K.A.M. Penicillin binding protein 2a: An overview and a medicinal chemistry perspective. Eur. J. Med. Chem. 2020, 199, 112312. [Google Scholar] [CrossRef] [PubMed]
- Sharifzadeh, S.; Brown, N.W.; Shirley, J.D.; Bruce, K.E.; Winkler, M.E.; Carlson, E.E. Chemical tools for selective activity profiling of bacterial penicillin-binding proteins. Methods Enzymol. 2020, 638, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Kar, D.; Murugan, R.A.; Mallick, S.; Dutta, M.; Pandey, S.D.; Chowdhury, C.; Ghosh, A.S. A putative low-molecular-mass penicillin-binding protein (PBP) of Mycobacterium smegmatis exhibits prominent physiological characteristics of DD-carboxypeptidase and beta-lactamase. Microbiology 2015, 161, 1081–1091. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Bai, K.; Gu, M.; Xu, X.; Jiang, N.; Chen, Y.; Li, J.; Luo, L. Class A Penicillin-Binding Protein C Is Responsible for Stress Response by Regulation of Peptidoglycan Assembly in Clavibacter michiganensis. Microbiol. Spectr. 2022, 10, e0181622. [Google Scholar] [CrossRef]
- Murphy, S.G.; Murtha, A.N.; Zhao, Z.; Alvarez, L.; Diebold, P.; Shin, J.H.; VanNieuwenhze, M.S.; Cava, F.; Dorr, T. Class A Penicillin-Binding Protein-Mediated Cell Wall Synthesis Promotes Structural Integrity during Peptidoglycan Endopeptidase Insufficiency in Vibrio cholerae. mBio 2021, 12, e03596-20. [Google Scholar] [CrossRef]
- Hamilton, S.M.; Alexander, J.A.N.; Choo, E.J.; Basuino, L.; da Costa, T.M.; Severin, A.; Chung, M.; Aedo, S.; Strynadka, N.C.J.; Tomasz, A.; et al. High-Level Resistance of Staphylococcus aureus to beta-Lactam Antibiotics Mediated by Penicillin-Binding Protein 4 (PBP4). Antimicrob. Agents Chemother. 2017, 61, e02727-16. [Google Scholar] [CrossRef] [PubMed]
- de Leon, S.R.; Daniels, K.; Clarke, A.J. Production and purification of the penicillin-binding protein 3 from Pseudomonas aeruginosa. Protein Expr. Purif. 2010, 73, 177–183. [Google Scholar] [CrossRef]
- Brambilla, L.; Moran-Barrio, J.; Viale, A.M. Low-molecular-mass penicillin binding protein 6b (DacD) is required for efficient GOB-18 metallo-beta-lactamase biogenesis in Salmonella enterica and Escherichia coli. Antimicrob. Agents Chemother. 2014, 58, 205–211. [Google Scholar] [CrossRef]
- Zhang, W.; Li, S.; Ma, L.; Ding, W.; Xu, Y. Identification of a novel carboxypeptidase encoded by Rv3627c that plays a potential role in mycobacteria morphology and cell division. Enzyme Microb. Technol. 2019, 126, 32–40. [Google Scholar] [CrossRef]
- Chatterjee, S.S.; Chen, L.; Gilbert, A.; da Costa, T.M.; Nair, V.; Datta, S.K.; Kreiswirth, B.N.; Chambers, H.F. PBP4 Mediates beta-Lactam Resistance by Altered Function. Antimicrob. Agents Chemother. 2017, 61, e00932-17. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.A.; Hejazi, I.I.; Thakur, S.C.; Athar, F. Domain-wise differentiation of Mycobacterium tuberculosis H(37) Rv hypothetical proteins: A roadmap to discover bacterial survival potentials. Biotechnol. Appl. Biochem. 2022, 69, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Ealand, C.S.; Asmal, R.; Mashigo, L.; Campbell, L.; Kana, B.D. Characterization of putative DD-carboxypeptidase-encoding genes in Mycobacterium smegmatis. Sci. Rep. 2019, 9, 5194. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, Y.; Dairi, T. Discovery of an alternative pathway of peptidoglycan biosynthesis: A new target for pathway specific inhibitors. J. Ind. Microbiol. Biotechnol. 2021, 48, kuab038. [Google Scholar] [CrossRef] [PubMed]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef]
- Kishida, H.; Unzai, S.; Roper, D.I.; Lloyd, A.; Park, S.Y.; Tame, J.R. Crystal structure of penicillin binding protein 4 (dacB) from Escherichia coli, both in the native form and covalently linked to various antibiotics. Biochemistry 2006, 45, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, E.; Duez, C.; Herman, R.; Kerff, F.; Petrella, S.; Anderson, J.W.; Adediran, S.A.; Pratt, R.F.; Frere, J.M.; Charlier, P. Crystal structure of the Bacillus subtilis penicillin-binding protein 4a, and its complex with a peptidoglycan mimetic peptide. J. Mol. Biol. 2007, 371, 528–539. [Google Scholar] [CrossRef]
- Sauvage, E.; Herman, R.; Petrella, S.; Duez, C.; Bouillenne, F.; Frere, J.M.; Charlier, P. Crystal structure of the Actinomadura R39 DD-peptidase reveals new domains in penicillin-binding proteins. J. Biol. Chem. 2005, 280, 31249–31256. [Google Scholar] [CrossRef]
- Kawai, F.; Clarke, T.B.; Roper, D.I.; Han, G.J.; Hwang, K.Y.; Unzai, S.; Obayashi, E.; Park, S.Y.; Tame, J.R. Crystal structures of penicillin-binding proteins 4 and 5 from Haemophilus influenzae. J. Mol. Biol. 2010, 396, 634–645. [Google Scholar] [CrossRef]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Holm, L.; Laiho, A.; Toronen, P.; Salgado, M. DALI shines a light on remote homologs: One hundred discoveries. Protein Sci. 2023, 32, e4519. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Kumar, P.; Arora, K.; Lloyd, J.R.; Lee, I.Y.; Nair, V.; Fischer, E.; Boshoff, H.I.; Barry, C.E., 3rd. Meropenem inhibits D,D-carboxypeptidase activity in Mycobacterium tuberculosis. Mol. Microbiol. 2012, 86, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Levine, S.R.; Beatty, K.E. Investigating beta-Lactam Drug Targets in Mycobacterium tuberculosis Using Chemical Probes. ACS Infect. Dis. 2021, 7, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, L.D.; Dai, W.; Bottomley, S.P. A family of E. coli expression vectors for laboratory scale and high throughput soluble protein production. BMC Biotechnol 2006, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Eschenfeldt, W.H.; Lucy, S.; Millard, C.S.; Joachimiak, A.; Mark, I.D. A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol Biol 2009, 498, 105–115. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking covalent inhibitors: A parameter free approach to pose prediction and scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | PDB Code | Chain | No. of Cα Aligned | RMSD (Å) | Z-Score | % Identity |
|---|---|---|---|---|---|---|
| B. subtilis | 1W5D | A | 345 (361/458:DALI) | 1.370 (1.8: DALI) | 40.8 | 27 |
| Actinomycetes | 1W79 | A | 335 (363/466: DALI) | 1.190 (1.8:DALI) | 40.9 | 30 |
| E. coli | 2EX2 | A | 330 (355/456:DALI) | 2.518 (2.3:DALI) | 37.7 | 28 |
| H. influenzae | 3A3D | A | 326 (354/453:DALI) | 1.250 (2.2: DALI) | 37.8 | 26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, S.-M.; Kim, D.-H. Structural Insights into the Penicillin-Binding Protein 4 (DacB) from Mycobacterium tuberculosis. Int. J. Mol. Sci. 2024, 25, 983. https://doi.org/10.3390/ijms25020983
Kang S-M, Kim D-H. Structural Insights into the Penicillin-Binding Protein 4 (DacB) from Mycobacterium tuberculosis. International Journal of Molecular Sciences. 2024; 25(2):983. https://doi.org/10.3390/ijms25020983
Chicago/Turabian StyleKang, Sung-Min, and Do-Hee Kim. 2024. "Structural Insights into the Penicillin-Binding Protein 4 (DacB) from Mycobacterium tuberculosis" International Journal of Molecular Sciences 25, no. 2: 983. https://doi.org/10.3390/ijms25020983
APA StyleKang, S.-M., & Kim, D.-H. (2024). Structural Insights into the Penicillin-Binding Protein 4 (DacB) from Mycobacterium tuberculosis. International Journal of Molecular Sciences, 25(2), 983. https://doi.org/10.3390/ijms25020983