
The Construction of lncRNA/circRNA–miRNA–mRNA Networks Reveals Functional Genes Related to Growth Traits in Schima superba

Abstract
1. Introduction
2. Results
2.1. Quality and Statistics of Whole Transcriptome Sequencing
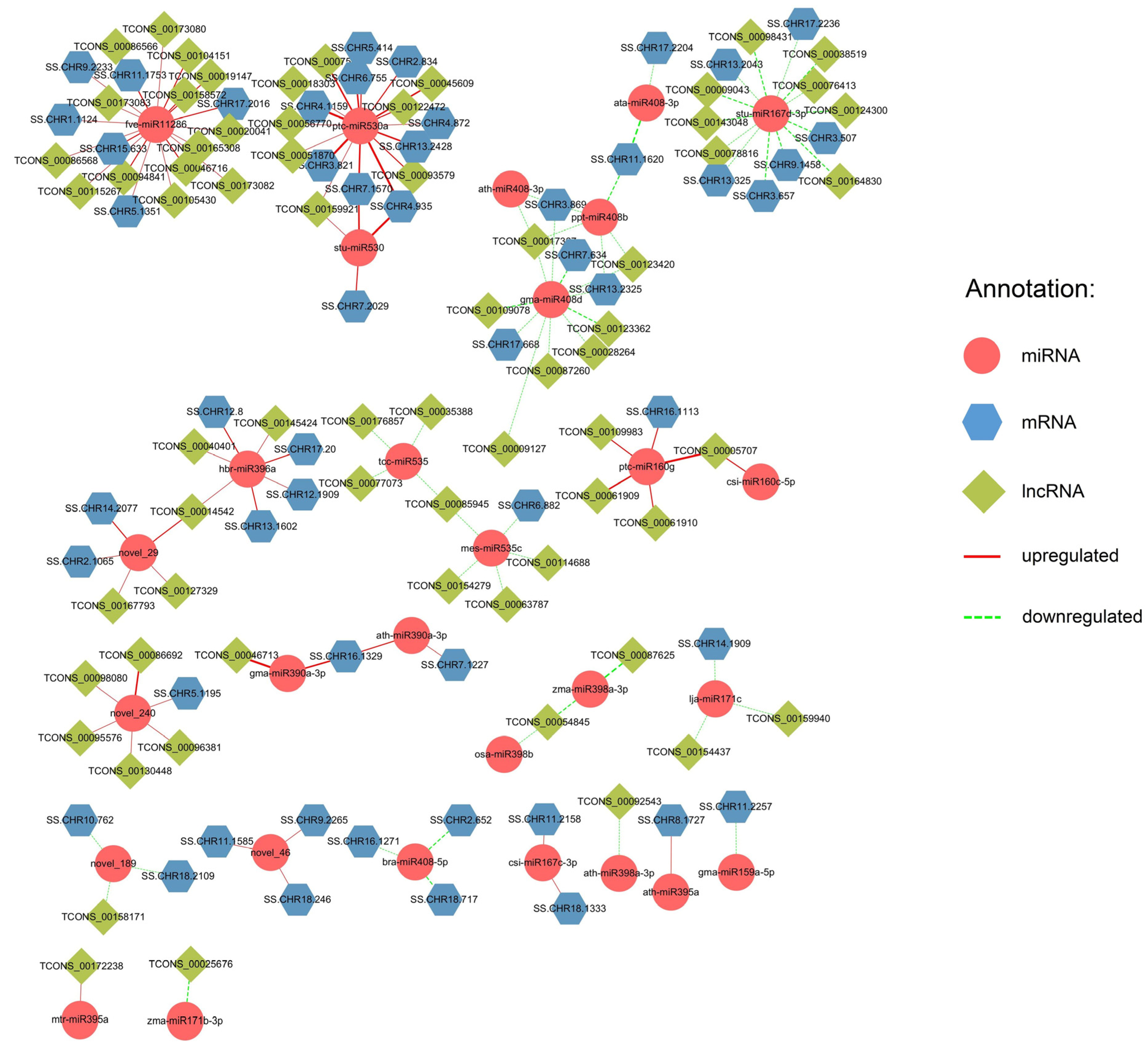
2.2. Regulatory Networks of lncRNA-miRNA-mRNA
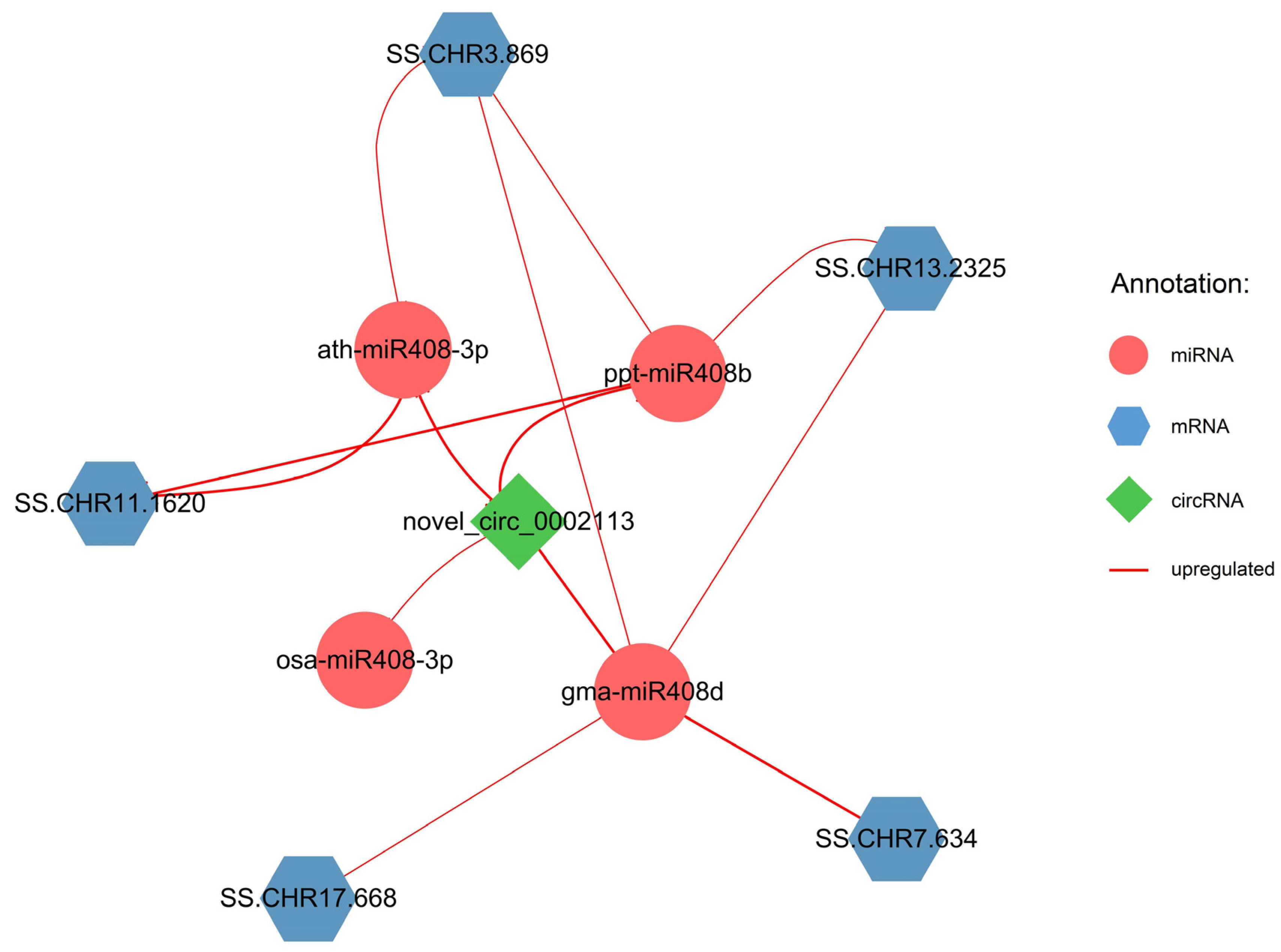
2.3. Regulatory Networks of circRNA–miRNA–mRNA
2.4. Identification and Characteristics of Candidate Genes
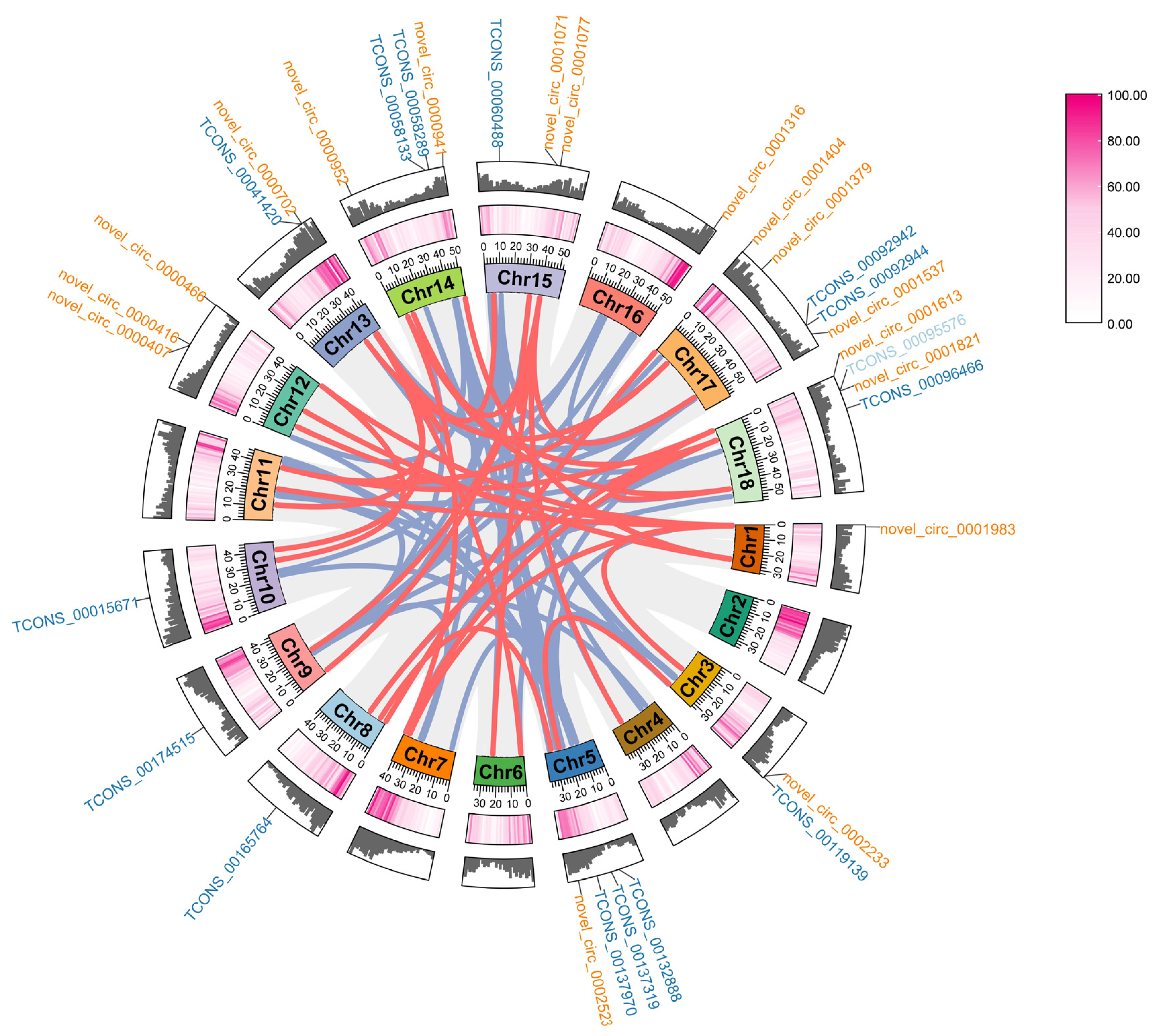
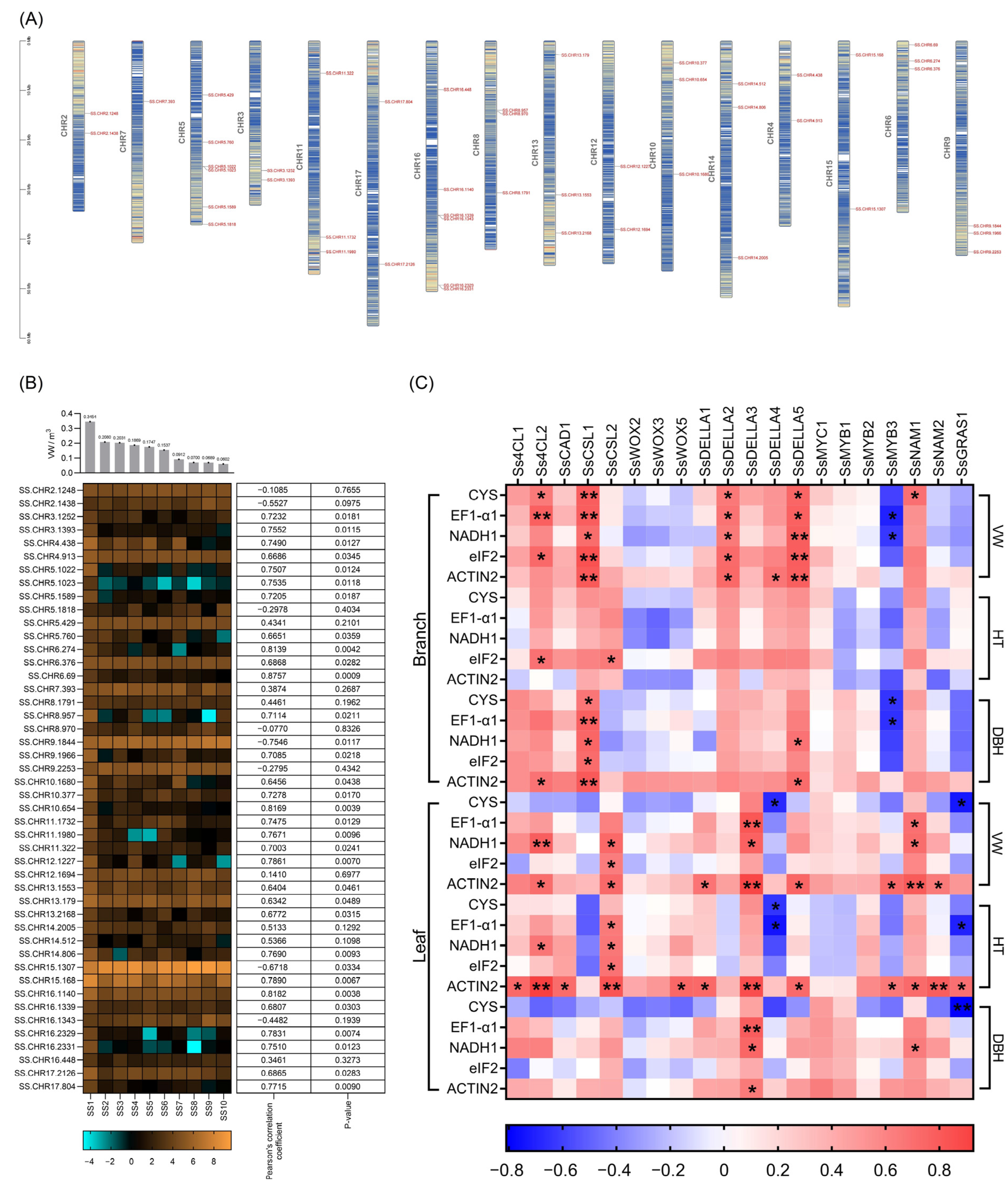
2.5. Identification of Allelic Variations
2.6. Phylogenetic Analysis of Cellulose Synthase and Cellulose Synthase-like Proteins
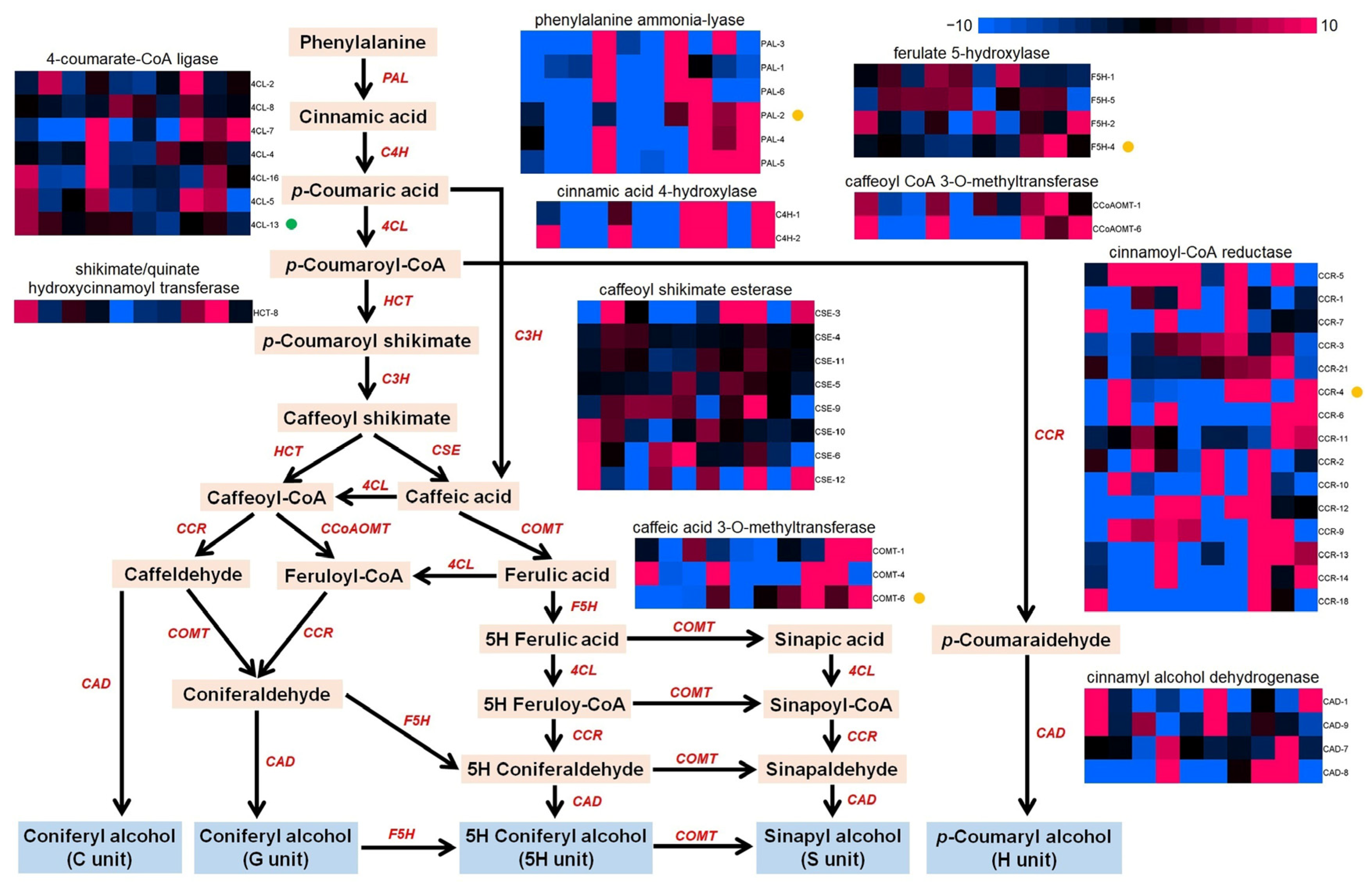
2.7. Differentially Expressed Genes in the Phenylpropane Pathway
3. Discussion
4. Materials and Methods
4.1. Plant Materials and RNA Sampling
4.2. Library Preparation and Sequencing
4.3. Quality Control
4.4. Read Mapping and Assembly
4.5. Identification of mRNAs, lncRNAs, circRNAs, and miRNAs
4.6. Quantification and Differential Expression Analysis
4.7. Construction of the Regulatory Network
4.8. Quantitative Real-Time PCR
4.9. Phylogenetic Analysis
4.10. SNP Analysis
4.11. Heatmap and Gene Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bai, Q.; He, B.; Cai, Y.; Lian, H.; Zhang, Q.; Liang, D.; Wang, Y. Genetic diversity and population structure of Schima superba from southern China. Front. Ecol. Evol. 2022, 10, 879512. [Google Scholar] [CrossRef]
- Zhang, R.; Yang, H.; Zhou, Z.; Shen, B.; Xiao, J.; Wang, B. A high-density genetic map of Schima superba based on its chromosomal characteristics. BMC Plant Biol. 2019, 19, 41. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Huang, P.; Guo, S.; Liu, S.; Hu, X.; Liu, X. Comprehensive analysis of Betula platyphylla Suk. PIF gene family and their potential functions in growth and development. Int. J. Mol. Sci. 2022, 23, 15326. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, Y.; Smarda, P.; Bures, P.; Guo, Q. Transcriptome and proteome associated analysis of flavonoid metabolism in haploid Ginkgo biloba. Int. J. Biol. Macromol. 2023, 224, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Shen, L.; Hou, Q.; Zhou, Z.; Mei, L.; Zhao, H.; Wen, X. Identification of genes and metabolic pathways involved in resin yield in masson pine by integrative analysis of transcriptome, proteome and biochemical characteristics. Int. J. Mol. Sci. 2022, 23, 11420. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, L.; Qi, H.; Xu, M. LncRNA/circRNA-miRNA-mRNA networks regulate the development of root and shoot meristems of Populus. Ind. Crops Prod. 2019, 133, 333–347. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, P.; Zhang, M.; Huang, Z.; Hippolyte, A.R.; Hou, Y.; Lou, X.; Ji, K. Identification, classification and characterization of LBD transcription factor family genes in Pinus massoniana. Int. J. Mol. Sci. 2022, 23, 13215. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Yuan, T.; Yang, K.; Liu, Y.; Li, Y.; Gao, Z. Identification and characterization of CircRNA-associated CeRNA networks in moso bamboo under nitrogen stress. BMC Plant Biol. 2023, 23, 142. [Google Scholar] [CrossRef]
- Chen, Y.; Li, X.; Su, L.; Chen, X.; Zhang, S.; Xu, X.; Zhang, Z.; Chen, Y.; Xu, X.; Lin, Y.; et al. Genome-wide identification and characterization of long non-coding RNAs involved in the early somatic embryogenesis in Dimocarpus longan Lour. BMC Genom. 2018, 19, 805. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, D.; Zhang, S.; Lou, Y.; An, X.; Jiang, Z.; Gao, Z. Transcriptome and miRNAome analysis reveals components regulating tissue differentiation of bamboo shoots. Plant Physiol. 2022, 188, 2182–2198. [Google Scholar] [CrossRef]
- Zaman, F.; Zhang, M.; Wu, R.; Zhang, Q.; Luo, Z.; Yang, S. Recent research advances of small regulatory RNA in fruit crops. Horticulturae 2023, 9, 294. [Google Scholar] [CrossRef]
- Zhang, L.; Fan, D.; Li, H.; Chen, Q.; Zhang, Z.; Liu, M.; Liu, J.; Song, Y.; He, J.; Xu, W.; et al. Characterization and identification of grapevine heat stress-responsive microRNAs revealed the positive regulated function of vvi-miR167 in thermostability. Plant Sci. 2023, 329, 111623. [Google Scholar] [CrossRef]
- Zhu, M.; Dong, Q.; Bing, J.; Songbuerbatu; Zheng, L.; Dorjee, T.; Liu, Q.; Zhou, Y.; Gao, F. Combined lncRNA and mRNA expression profiles identified the lncRNA-miRNA-mRNA modules regulating the cold stress response in Ammopiptanthus nanus. Int. J. Mol. Sci. 2023, 24, 6502. [Google Scholar] [CrossRef]
- Gao, Y.; Cui, Y.; Zhao, R.; Chen, X.; Zhang, J.; Zhao, J.; Kong, L. Cryo-treatment enhances the embryogenicity of mature somatic embryos via the lncRNA-miRNA-mRNA network in white spruce. Int. J. Mol. Sci. 2022, 23, 1111. [Google Scholar] [CrossRef]
- Babaei, S.; Singh, M.B.; Bhalla, P.L. Circular RNAs modulate the floral fate acquisition in soybean shoot apical meristem. BMC Plant Biol. 2023, 23, 322. [Google Scholar] [CrossRef]
- Huang, J.; Wang, Y.; Yu, J.; Li, F.; Yi, L.; Li, Y.; Xie, N.; Wu, Q.; Samarina, L.; Tong, W.; et al. Evolutionary Landscape of Tea Circular RNAs and Its Contribution to Chilling Tolerance of Tea Plant. Int. J. Mol. Sci. 2023, 24, 1478. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, R.; Zhou, Z.; Hua, B.; Huang, S.; Ma, L.; Fan, H. A variation and selection of growth and wood traits for 10-year-old Schima superba. J. Nanjing For. Univ. Nat. Sci. Ed. 2020, 44, 85–92. [Google Scholar]
- Chen, X.; Xu, X.; Zhang, S.; Munir, N.; Zhu, C.; Zhang, Z.; Chen, Y.; Xu, X.; Lin, Y.; Lai, Z. Genome-wide circular RNA profiling and competing endogenous RNA regulatory network analysis provide new insights into the molecular mechanisms underlying early somatic embryogenesis in Dimocarpus longan Lour. Tree Physiol. 2022, 42, 1876–1898. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Lin, Y.; Gao, X.-Q.; Wang, X.-L. Non-Canonical Functions of Splicing Factors in RNA Metabolism. CRC Crit. Rev. Plant Sci. 2020, 39, 493–513. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, P.; Chen, Q.; Wang, J.; Chen, M. Identification and characterization of ncRNA-associated ceRNA networks in Arabidopsis leaf development. BMC Genom. 2018, 19, 607. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liang, W.; Ye, S.; Chen, X.; Zhou, Y.; Lu, J.; Shen, Y.; Wang, X.; Zhou, J.; Yu, C.; et al. Whole-transcriptome analysis reveals autophagy Is involved in early senescence of zj-es mutant rice. Front. Plant Sci. 2022, 13, 899054. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Kong, L.; Zhang, Y.; Wu, M.; Wang, Y.; Li, J.; Jiao, N.; Xu, J.; Chen, J.; Tu, Z. Gene expression analysis revealed Hbr-miR396b as a key piece participating in reaction wood formation of Hevea brasiliensis (rubber tree). Ind. Crops Prod. 2022, 177, 114460. [Google Scholar] [CrossRef]
- Xu, C.; Li, J.; Wang, H.; Liu, H.; Yu, Z.; Zhao, Z. Whole-transcriptome sequencing reveals a ceRNA regulatory network associated with the process of periodic albinism under low temperature in Baiye No. 1 (Camellia sinensis). Int. J. Mol. Sci. 2023, 24, 7162. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhong, Y.; Qi, X. LncRNA TCONS_00021861 is functionally associated with drought tolerance in rice (Oryza sativa L.) via competing endogenous RNA regulation. BMC Plant Biol. 2021, 21, 410. [Google Scholar] [CrossRef] [PubMed]
- Quan, M.; Xiao, L.; Lu, W.; Liu, X.; Song, F.; Si, J.; Du, Q.; Zhang, D. Association genetics in Populus reveal the allelic interactions of Pto-MIR167a and its targets in wood formation. Front. Plant Sci. 2018, 9, 744. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Quan, M.; Du, Q.; Zhang, D. The Interactions between the Long Non-coding RNA NERDL and Its Target Gene Affect Wood Formation in Populus tomentosa. Front. Plant Sci. 2017, 8, 1035. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-L.; Li, Y.-H.; Hsieh, W.-P.; Lin, M.-C.; Ahn, J.H.; Wu, S.-H. HUA ENHANCER1 Is involved in posttranscriptional regulation of positive and negative regulators in Arabidopsis photomorphogenesis. Plant Cell 2014, 26, 2858–2872. [Google Scholar] [CrossRef]
- Mishra, R.; Mohapatra, R.; Mahanty, B.; Joshi, R.K. Analysis of microRNAs and their targets from onion (Allium cepa) using genome survey sequences (GSS) and expressed sequence tags (ESTs). Bioinformation 2019, 15, 907–917. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Z.; Zeng, C.; Dong, X.; Li, M.; Liu, Z.; Yan, M. The elemental defense effect of cadmium on Alternaria brassicicola in Brassica juncea. BMC Plant Biol. 2022, 22, 17. [Google Scholar] [CrossRef]
- Imran, M.; Liu, T.; Wang, Z.; Wang, M.; Liu, S.; Gao, X.; Wang, A.; Liu, S.; Tian, Z.; Zhang, M. Evolutionary conservation of nested MIR159 structural microRNA genes and their promoter characterization in Arabidopsis thaliana. Front. Plant Sci. 2022, 13, 948751. [Google Scholar] [CrossRef]
- Mishra, S.; Chaudhary, R.; Sharma, P. Temporal expression analysis of microRNAs and their target GRAS genes induced by osmotic stress in two contrasting wheat genotypes. Mol. Biol. Rep. 2023, 50, 5621–5633. [Google Scholar] [CrossRef]
- Shi, J.; Zai, W.; Xiong, Z.; Wang, K.; Shui, D.; Jing, Z.g. Small RNA profiling reveals a role of miRNAs in response to Ralstonia solanacearum infection in tomato. J. Plant Growth Regul. 2023, 42, 3342–3355. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.; Qin, T.; Guo, X.; Xu, K.; Xu, C.; Yuan, W. Functional conservation and divergence of miR156 and miR529 during rice development. Crop J. 2023, 11, 692–703. [Google Scholar] [CrossRef]
- Ma, Z.; Li, W.; Wang, H.; Yu, D. WRKY transcription factors WRKY12 and WRKY13 interact with SPL10 to modulate age-mediated flowering. J. Integr. Plant Biol. 2020, 62, 1659–1673. [Google Scholar] [CrossRef]
- Li, Y.; Jin, F.; Wu, X.; da Silva, J.A.T.; Xiong, Y.; Zhang, X.; Ma, G. Identification and function of miRNA-mRNA interaction pairs during lateral root development of hemi-parasitic Santalum album L. seedlings. J. Plant Physiol. 2023, 280, 153866. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Jiang, H.; Lin, Y.; Shang, L.; Wang, M.; Li, D.; Fu, X.; Geisler, M.; Qi, Y.; Gao, Z.; et al. A novel miR167a-OsARF6-OsAUX3 module regulates grain length and weight in rice. Mol. Plant. 2021, 14, 1683–1698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liao, H.; Lucas, W.J. Molecular mechanisms underlying phosphate sensing, signaling, and adaptation in plants. J. Integr. Plant Biol. 2014, 56, 192–220. [Google Scholar] [CrossRef] [PubMed]
- Basu, U.; Parida, S.K. Restructuring plant types for developing tailor-made crops. Plant Biotechnol. J. 2023, 21, 1106–1122. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Wang, S.; Zhang, B.; Shang-Guan, K.; Shi, Y.; Zhang, D.; Liu, X.; Wu, K.; Xu, Z.; Fu, X.; et al. A gibberellin-mediated DELLA-NAC signaling cascade regulates cellulose synthesis in rice. Plant Cell 2015, 27, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tian, Y.; Wu, K.; Ye, Y.; Yu, J.; Zhang, J.; Liu, Q.; Hu, M.; Li, H.; Tong, Y.; et al. Modulating plant growth-metabolism coordination for sustainable agriculture. Nature 2018, 560, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Kong, L.; Wu, X.; Gao, J.; Niu, T.; Li, J.; Li, Z.; Dai, L. GsNAC2 gene enhances saline-alkali stress tolerance by promoting plant growth and regulating glutathione metabolism in Sorghum bicolor. Funct. Plant Biol. 2023, 50, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Negishi, N.; Nanto, K.; Hayashi, K.; Onogi, S.; Kawaoka, A. Transcript abundances of LIM transcription factor, 4CL, CAld5H and CesAs affect wood properties in Eucalyptus globulus. Silvae Genet. 2011, 60, 288–296. [Google Scholar] [CrossRef]
- Cheng, H.; Song, X.; Hu, Y.; Wu, T.; Yang, Q.; An, Z.; Feng, S.; Deng, Z.; Wu, W.; Zeng, X.; et al. Chromosome-level wild Hevea brasiliensis genome provides new tools for genomic-assisted breeding and valuable loci to elevate rubber yield. Plant Biotechnol. J. 2023, 21, 1058–1072. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Dong, X.; Liu, Q.; Wang, R.; Li, Y.; Huang, X.; Zhao, Y. SLAF-Seq technology-based genome-wide association and population structure analyses of ancient Camellia sinensis (L.) Kuntze in Sandu County, China. Forests 2022, 13, 1885. [Google Scholar] [CrossRef]
- Guo, L.; Li, Y.; Lei, Y.; Gao, J.; Song, C.; Guo, D.; Hou, X. Genome-scale investigation and identification of variations associated with early flowering based on whole genome resequencing and transcriptome integrated analysis in tree peony. Sci. Hortic. 2023, 310, 111695. [Google Scholar] [CrossRef]
- Jing, Y.; Bian, L.; Zhang, X.; Zhao, B.; Zheng, R.; Su, S.; Ye, D.; Zheng, X.; El-Kassaby, Y.A.; Shi, J. Genetic diversity and structure of the 4(th) cycle breeding population of Chinese fir (Cunninghamia lanceolata (lamb.) hook). Front. Plant Sci. 2023, 14, 1106615. [Google Scholar] [CrossRef] [PubMed]
- Jose, S.; Lada, R.R. Marker-trait association analysis for postharvest needle retention/abscission in balsam fir (Abies balsamea). Plant Breed. 2019, 138, 647–657. [Google Scholar] [CrossRef]
- Nantongo, J.S.; Potts, B.M.; Klapste, J.; Graham, N.J.; Dungey, H.S.; Fitzgerald, H.; O’Reilly-Wapstra, J.M. Genomic selection for resistance to mammalian bark stripping and associated chemical compounds in radiata pine. G3-Genes Genomes Genet. 2022, 12, jkac245. [Google Scholar] [CrossRef]
- Morton, B.R.; Bi, I.V.; McMullen, M.D.; Gaut, B.S. Variation in mutation dynamics across the maize genome as a function of regional and flanking base composition. Genetics 2006, 172, 569–577. [Google Scholar] [CrossRef]
- Park, S.; Yu, H.-J.; Mun, J.-H.; Lee, S.-C. Genome-wide discovery of DNA polymorphism in Brassica rapa. Mol. Genet. Genom. 2010, 283, 135–145. [Google Scholar] [CrossRef]
- Guo, C.; McDowell, I.C.; Nodzenski, M.; Scholtens, D.M.; Allen, A.S.; Lowe, W.L.; Reddy, T.E. Transversions have larger regulatory effects than transitions. BMC Genom. 2017, 18, 394. [Google Scholar] [CrossRef]
- Parkhomchuk, D.; Borodina, T.; Amstislavskiy, V.; Banaru, M.; Hallen, L.; Krobitsch, S.; Lehrach, H.; Soldatov, A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009, 37, e123. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, J.; Zhao, F. Circular RNA identification based on multiple seed matching. Brief. Bioinform. 2018, 19, 803–810. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Wu, H.-J.; Ma, Y.-K.; Chen, T.; Wang, M.; Wang, X.-J. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef]
- Friedlaender, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: MicroRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- Moxon, S.; Schwach, F.; Dalmay, T.; MacLean, D.; Studholme, D.J.; Moulton, V. A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics 2008, 24, 2252–2253. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L.; et al. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, R.; Zhou, Z. Identification and validation of reference genes for gene expression analysis in Schima superba. Genes 2021, 12, 732. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Patel, V.M.; Coon, M.; Nguyen, T.; Land, S.J.; Ruden, D.M.; Lu, X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012, 3, 35. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP Type | Transition SNPs | Transversion SNPs | ||||
|---|---|---|---|---|---|---|
| C/T | A/G | A/T | A/C | G/T | G/C | |
| Number of allelic sites | 26,704 | 26,579 | 9163 | 8453 | 8444 | 7579 |
| Frequency (%) | 30.72 | 30.58 | 10.54 | 9.73 | 9.71 | 8.72 |
| Total (percent of total) | 53,283 (61.30%) | 33,639 (38.70%) | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Q.; Shi, L.; Li, K.; Xu, F.; Zhang, W. The Construction of lncRNA/circRNA–miRNA–mRNA Networks Reveals Functional Genes Related to Growth Traits in Schima superba. Int. J. Mol. Sci. 2024, 25, 2171. https://doi.org/10.3390/ijms25042171
Bai Q, Shi L, Li K, Xu F, Zhang W. The Construction of lncRNA/circRNA–miRNA–mRNA Networks Reveals Functional Genes Related to Growth Traits in Schima superba. International Journal of Molecular Sciences. 2024; 25(4):2171. https://doi.org/10.3390/ijms25042171
Chicago/Turabian StyleBai, Qingsong, Lingling Shi, Kejian Li, Fang Xu, and Weihua Zhang. 2024. "The Construction of lncRNA/circRNA–miRNA–mRNA Networks Reveals Functional Genes Related to Growth Traits in Schima superba" International Journal of Molecular Sciences 25, no. 4: 2171. https://doi.org/10.3390/ijms25042171
APA StyleBai, Q., Shi, L., Li, K., Xu, F., & Zhang, W. (2024). The Construction of lncRNA/circRNA–miRNA–mRNA Networks Reveals Functional Genes Related to Growth Traits in Schima superba. International Journal of Molecular Sciences, 25(4), 2171. https://doi.org/10.3390/ijms25042171