
A Comparative Biochemical and Pathological Evaluation of Brain Samples from Knock-In Murine Models of Gaucher Disease

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
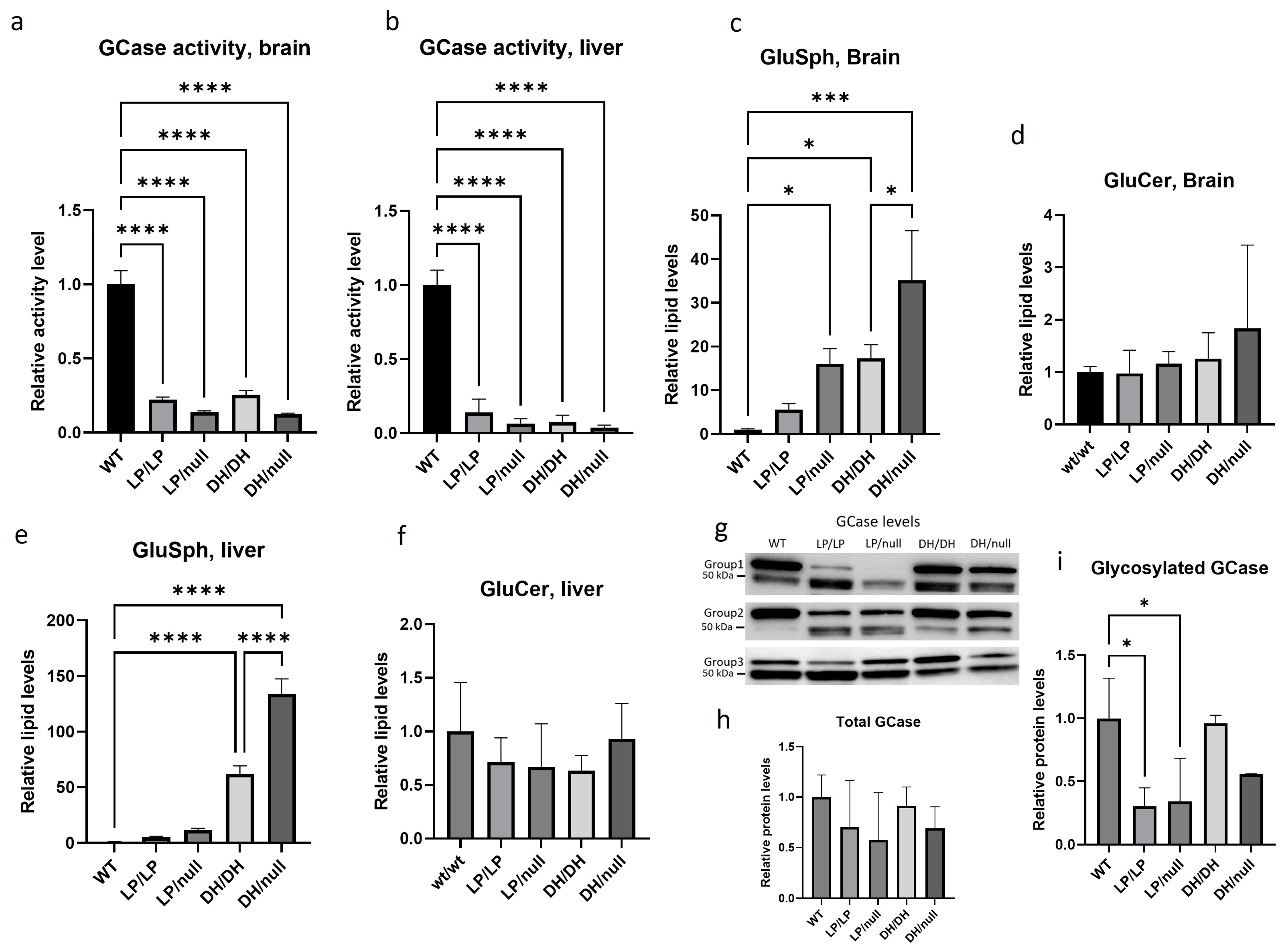
2.1. Evaluation of GCase Pathology in Gba1 Mutant Mice
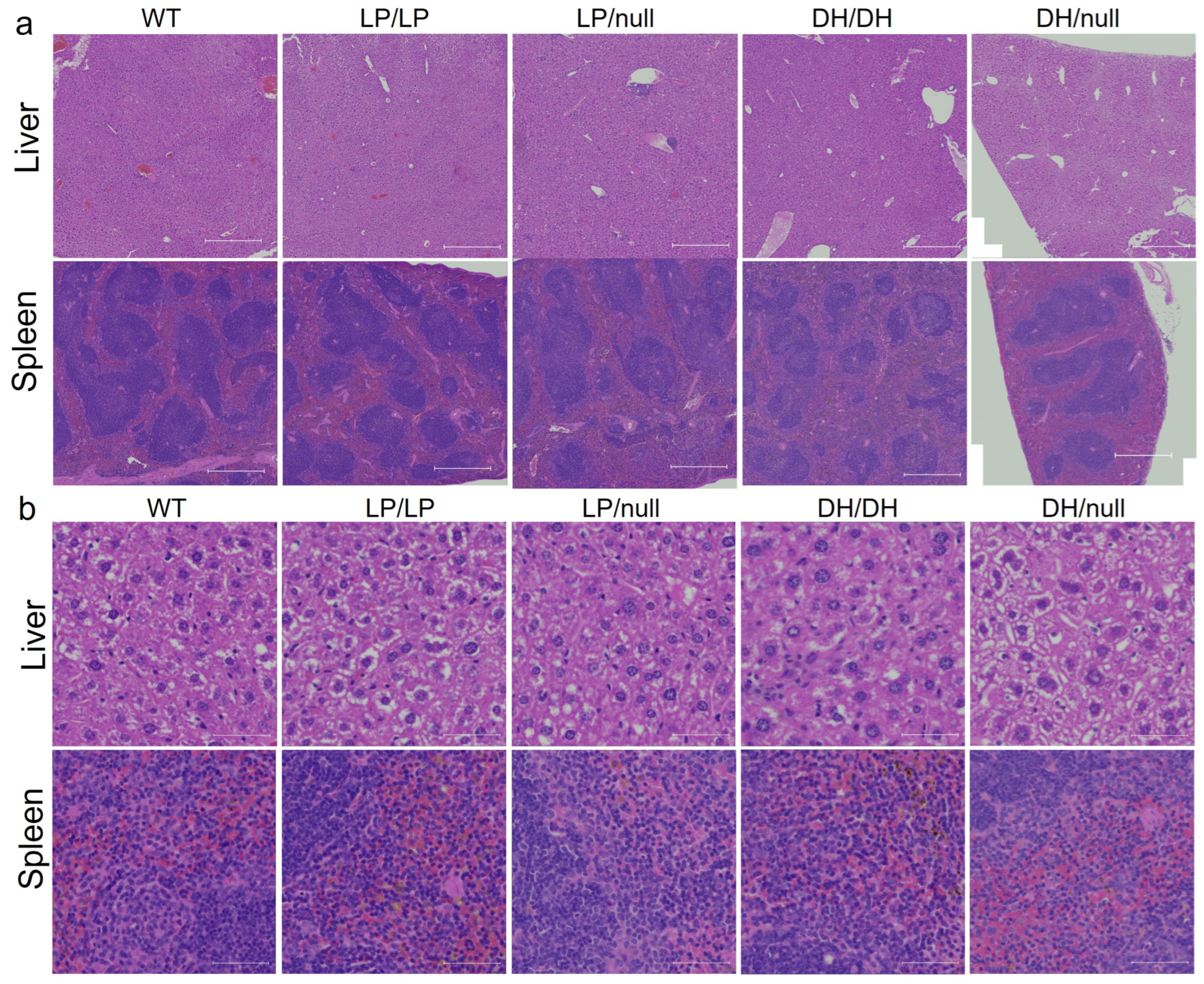
2.2. Evaluation of Visceral Pathology of Gba1 Mutant Mice
2.3. Evaluation of Motor Behavior in the L444P Gba1 Mutant Mice
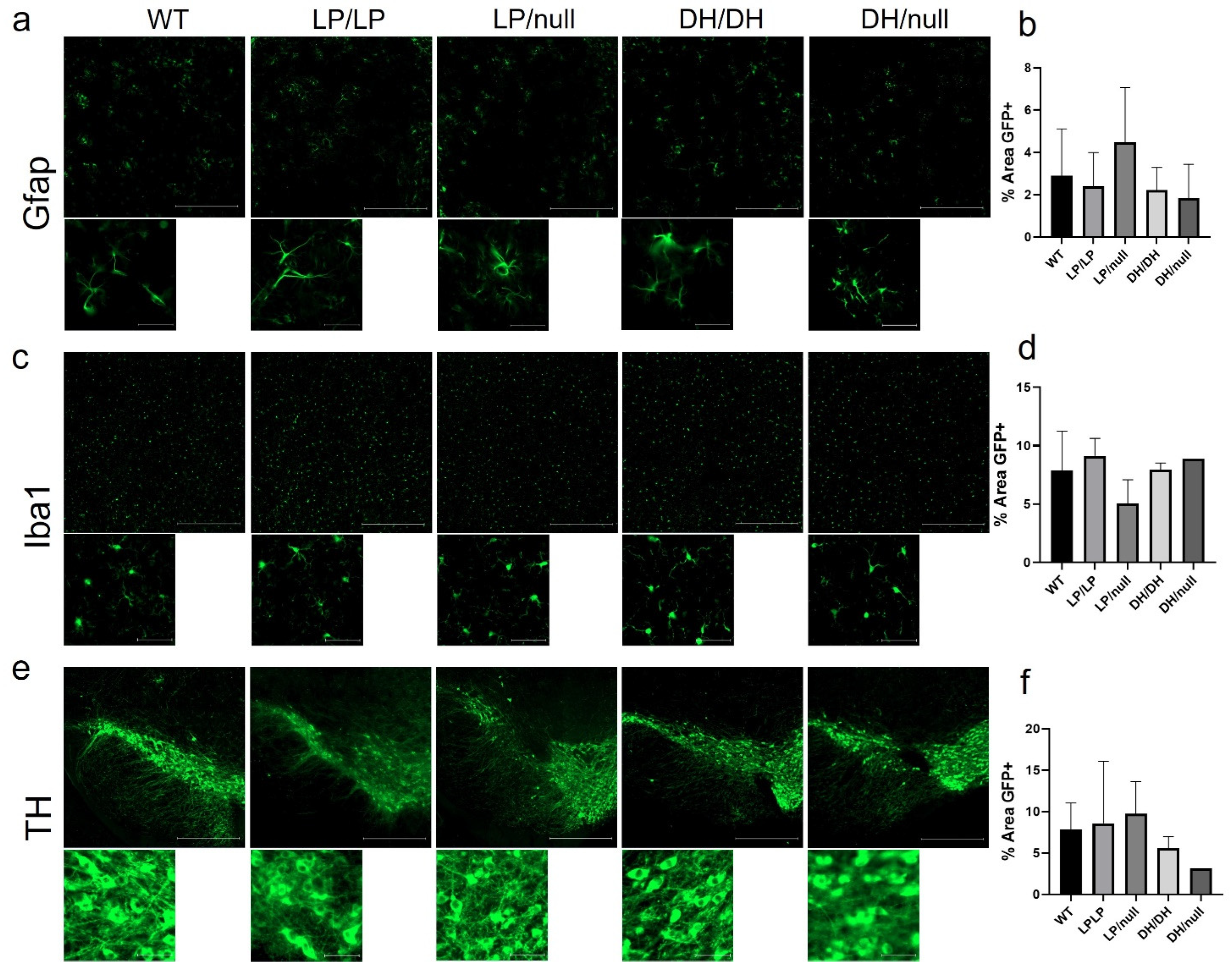
2.4. Evaluation of Neuropathology in the Gba1 Mutant Mice
3. Discussion
4. Materials and Methods
4.1. Mouse Lines
4.2. Motor Behavioral Evaluations
4.3. Tissue Collection
4.4. Immunohistochemistry (IHC)
4.5. IHC Data Processing
4.6. GCase Activity Assays
4.7. GluCer and GluSph Assays
4.8. Immunoblotting
4.9. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neudorfer, O.; Giladi, N.; Elstein, D.; Abrahamov, A.; Turezkite, T.; Aghai, E.; Reches, A.; Bembi, B.; Zimran, A. Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 1996, 89, 691–694. [Google Scholar] [CrossRef]
- Tayebi, N.; Walker, J.; Stubblefield, B.; Orvisky, E.; LaMarca, M.E.; Wong, K.; Rosenbaum, H.; Schiffmann, R.; Bembi, B.; Sidransky, E. Gaucher disease with parkinsonian manifestations: Does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol. Genet. Metab. 2003, 79, 104–109. [Google Scholar] [CrossRef]
- Grabowski, G.A.; Zimran, A.; Ida, H. Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am. J. Hematol. 2015, 90 (Suppl. S1), S12–S18. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Farfel-Becker, T.; Vitner, E.B.; Futerman, A.H. Animal models for Gaucher disease research. Dis. Model. Mech. 2011, 4, 746–752. [Google Scholar] [CrossRef]
- Cho, S.M.; Vardi, A.; Platt, N.; Futerman, A.H. Absence of infiltrating peripheral myeloid cells in the brains of mouse models of lysosomal storage disorders. J. Neurochem. 2019, 148, 625–638. [Google Scholar] [CrossRef]
- Cabasso, O.; Kuppuramalingam, A.; Lelieveld, L.; Van der Lienden, M.; Boot, R.; Aerts, J.M.; Horowitz, M. Animal Models for the Study of Gaucher Disease. Int. J. Mol. Sci. 2023, 24, 16035. [Google Scholar] [CrossRef]
- Tybulewicz, V.L.; Tremblay, M.L.; LaMarca, M.E.; Willemsen, R.; Stubblefield, B.K.; Winfield, S.; Zablocka, B.; Sidransky, E.; Martin, B.M.; Huang, S.P.; et al. Animal model of Gaucher’s disease from targeted disruption of the mouse glucocerebrosidase gene. Nature 1992, 357, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Enquist, I.B.; Lo Bianco, C.; Ooka, A.; Nilsson, E.; Mansson, J.E.; Ehinger, M.; Richter, J.; Brady, R.O.; Kirik, D.; Karlsson, S. Murine models of acute neuronopathic Gaucher disease. Proc. Natl. Acad. Sci. USA 2007, 104, 17483–17488. [Google Scholar] [CrossRef] [PubMed]
- Pewzner-Jung, Y.; Joseph, T.; Blumenreich, S.; Vardi, A.; Ferreira, N.S.; Cho, S.M.; Eilam, R.; Tsoory, M.; Biton, I.E.; Brumfeld, V.; et al. Brain pathology and cerebellar purkinje cell loss in a mouse model of chronic neuronopathic Gaucher disease. Prog. Neurobiol. 2021, 197, 101939. [Google Scholar] [CrossRef] [PubMed]
- Farfel-Becker, T.; Do, J.; Tayebi, N.; Sidransky, E. Can GBA1-Associated Parkinson Disease Be Modeled in the Mouse? Trends Neurosci. 2019, 42, 631–643. [Google Scholar] [CrossRef]
- Farfel-Becker, T.; Vitner, E.B.; Pressey, S.N.; Eilam, R.; Cooper, J.D.; Futerman, A.H. Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease. Hum. Mol. Genet. 2011, 20, 1375–1386. [Google Scholar] [CrossRef]
- Xu, Y.H.; Sun, Y.; Ran, H.; Quinn, B.; Witte, D.; Grabowski, G.A. Accumulation and distribution of alpha-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol. Genet. Metab. 2011, 102, 436–447. [Google Scholar] [CrossRef]
- Liu, Y.; Suzuki, K.; Reed, J.D.; Grinberg, A.; Westphal, H.; Hoffmann, A.; Doring, T.; Sandhoff, K.; Proia, R.L. Mice with type 2 and 3 Gaucher disease point mutations generated by a single insertion mutagenesis procedure. Proc. Natl. Acad. Sci. USA 1998, 95, 2503–2508. [Google Scholar] [CrossRef]
- Mizukami, H.; Mi, Y.; Wada, R.; Kono, M.; Yamashita, T.; Liu, Y.; Werth, N.; Sandhoff, R.; Sandhoff, K.; Proia, R.L. Systemic inflammation in glucocerebrosidase-deficient mice with minimal glucosylceramide storage. J. Clin. Investig. 2002, 109, 1215–1221. [Google Scholar] [CrossRef]
- Fishbein, I.; Kuo, Y.M.; Giasson, B.I.; Nussbaum, R.L. Augmentation of phenotype in a transgenic Parkinson mouse heterozygous for a Gaucher mutation. Brain 2014, 137, 3235–3247. [Google Scholar] [CrossRef]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine Promotes alpha-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef]
- Ginns, E.I.; Mak, S.K.; Ko, N.; Karlgren, J.; Akbarian, S.; Chou, V.P.; Guo, Y.; Lim, A.; Samuelsson, S.; LaMarca, M.L.; et al. Neuroinflammation and alpha-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol. Genet. Metab. 2014, 111, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.H.; Quinn, B.; Witte, D.; Grabowski, G.A. Viable mouse models of acid beta-glucosidase deficiency: The defect in Gaucher disease. Am. J. Pathol. 2003, 163, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Abrahamov, A.; Elstein, D.; Gross-Tsur, V.; Farber, B.; Glaser, Y.; Hadas-Halpern, I.; Ronen, S.; Tafakjdi, M.; Horowitz, M.; Zimran, A. Gaucher’s disease variant characterised by progressive calcification of heart valves and unique genotype. Lancet 1995, 346, 1000–1003. [Google Scholar] [CrossRef] [PubMed]
- Kurolap, A.; Del Toro, M.; Spiegel, R.; Gutstein, A.; Shafir, G.; Cohen, I.J.; Barrabes, J.A.; Feldman, H.B. Gaucher disease type 3c: New patients with unique presentations and review of the literature. Mol. Genet. Metab. 2019, 127, 138–146. [Google Scholar] [CrossRef]
- Sun, Y.; Quinn, B.; Witte, D.P.; Grabowski, G.A. Gaucher disease mouse models: Point mutations at the acid beta-glucosidase locus combined with low-level prosaposin expression lead to disease variants. J. Lipid Res. 2005, 46, 2102–2113. [Google Scholar] [CrossRef]
- Xu, Y.H.; Reboulet, R.; Quinn, B.; Huelsken, J.; Witte, D.; Grabowski, G.A. Dependence of reversibility and progression of mouse neuronopathic Gaucher disease on acid beta-glucosidase residual activity levels. Mol. Genet. Metab. 2008, 94, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Mahoney-Crane, C.L.; Viswanathan, M.; Russell, D.; Curtiss, R.A.C.; Freire, J.; Bobba, S.S.; Coyle, S.D.; Kandebo, M.; Yao, L.; Wan, B.L.; et al. Neuronopathic GBA1L444P Mutation Accelerates Glucosylsphingosine Levels and Formation of Hippocampal Alpha-Synuclein Inclusions. J. Neurosci. 2023, 43, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kim, D.; Kim, S.; Kim, S.; Karuppagounder, S.S.; Kwon, S.H.; Lee, S.; Kam, T.I.; Lee, S.; Ham, S.; et al. alpha-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol. Neurodegener. 2018, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Hwang, H.; Choi, S.; Kwon, S.H.; Lee, S.; Park, J.H.; Kim, S.; Ko, H.S. D409H GBA1 mutation accelerates the progression of pathology in A53T alpha-synuclein transgenic mouse model. Acta Neuropathol. Commun. 2018, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Cecioni, S.; Ashmus, R.A.; Gilormini, P.A.; Zhu, S.; Chen, X.; Shan, X.; Gros, C.; Deen, M.C.; Wang, Y.; Britton, R.; et al. Quantifying lysosomal glycosidase activity within cells using bis-acetal substrates. Nat. Chem. Biol. 2022, 18, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Laqtom, N.N.; Dong, W.; Medoh, U.N.; Cangelosi, A.L.; Dharamdasani, V.; Chan, S.H.; Kunchok, T.; Lewis, C.A.; Heinze, I.; Tang, R.; et al. CLN3 is required for the clearance of glycerophosphodiesters from lysosomes. Nature 2022, 609, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Abu-Remaileh, M.; Wyant, G.A.; Kim, C.; Laqtom, N.N.; Abbasi, M.; Chan, S.H.; Freinkman, E.; Sabatini, D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017, 358, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Enquist, I.B.; Nilsson, E.; Ooka, A.; Mansson, J.E.; Olsson, K.; Ehinger, M.; Brady, R.O.; Richter, J.; Karlsson, S. Effective cell and gene therapy in a murine model of Gaucher disease. Proc. Natl. Acad. Sci. USA 2006, 103, 13819–13824. [Google Scholar] [CrossRef]
- Huh, Y.E.; Usnich, T.; Scherzer, C.R.; Klein, C.; Chung, S.J. GBA1 Variants and Parkinson’s Disease: Paving the Way for Targeted Therapy. J. Mov. Disord. 2023, 16, 261–278. [Google Scholar] [CrossRef]
- Stojkovska, I.; Krainc, D.; Mazzulli, J.R. Molecular mechanisms of alpha-synuclein and GBA1 in Parkinson’s disease. Cell Tissue Res. 2018, 373, 51–60. [Google Scholar] [CrossRef]
- Chen, C.; Hertz, E.; Chen, Y.; Sidransky, E. Targeting protein clearance pathways in GBA1-associated Parkinson disease. Expert Opin. Ther. Targets 2022, 26, 1031–1035. [Google Scholar] [CrossRef]
- Sun, Y.; Grabowski, G.A. Impaired autophagosomes and lysosomes in neuronopathic Gaucher disease. Autophagy 2010, 6, 648–649. [Google Scholar] [CrossRef] [PubMed]
- Corado, C.R.; Pinkstaff, J.; Jiang, X.; Galban, E.M.; Fisher, S.J.; Scholler, O.; Russell, C.; Bagel, J.H.; PA, O.D.; Ory, D.S.; et al. Cerebrospinal fluid and serum glycosphingolipid biomarkers in canine globoid cell leukodystrophy (Krabbe Disease). Mol. Cell. Neurosci. 2020, 102, 103451. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furderer, M.L.; Berhe, B.; Chen, T.C.; Wincovitch, S.; Jiang, X.; Tayebi, N.; Sidransky, E.; Han, T.-U. A Comparative Biochemical and Pathological Evaluation of Brain Samples from Knock-In Murine Models of Gaucher Disease. Int. J. Mol. Sci. 2024, 25, 1827. https://doi.org/10.3390/ijms25031827
Furderer ML, Berhe B, Chen TC, Wincovitch S, Jiang X, Tayebi N, Sidransky E, Han T-U. A Comparative Biochemical and Pathological Evaluation of Brain Samples from Knock-In Murine Models of Gaucher Disease. International Journal of Molecular Sciences. 2024; 25(3):1827. https://doi.org/10.3390/ijms25031827
Chicago/Turabian StyleFurderer, Makaila L., Bahafta Berhe, Tiffany C. Chen, Stephen Wincovitch, Xuntian Jiang, Nahid Tayebi, Ellen Sidransky, and Tae-Un Han. 2024. "A Comparative Biochemical and Pathological Evaluation of Brain Samples from Knock-In Murine Models of Gaucher Disease" International Journal of Molecular Sciences 25, no. 3: 1827. https://doi.org/10.3390/ijms25031827
APA StyleFurderer, M. L., Berhe, B., Chen, T. C., Wincovitch, S., Jiang, X., Tayebi, N., Sidransky, E., & Han, T.-U. (2024). A Comparative Biochemical and Pathological Evaluation of Brain Samples from Knock-In Murine Models of Gaucher Disease. International Journal of Molecular Sciences, 25(3), 1827. https://doi.org/10.3390/ijms25031827