
Molecular Mechanisms in Pathophysiology of Mucopolysaccharidosis and Prospects for Innovative Therapy

 , , ,
, , , 
Abstract
1. Introduction
2. Accumulation of GAGs and Subsequent Pathophysiological Alterations
2.1. Heparan Sulfate (HS)
2.2. Chondroitin Sulfate (CS) and Keratan Sulfate (KS)
2.3. Dermatan Sulfate (DS)
2.4. Hyaluronic Acid (HA)
2.5. Symptoms Associated with Several Types of GAGs
3. Activated Inflammatory Pathways in MPS: Potential Molecular Targets for Therapeutic Intervention (Figure 1)

4. Development of Innovative Therapies
4.1. Hurdles for Effective ERT in CNS and Bone
4.2. Current Development of Gene Therapy for MPS
4.2.1. Adeno-Associated Virus Vector (AAV)
4.2.2. Retroviral Vectors, including Lentiviral Vector
4.2.3. Sleeping Beauty Transposon System
4.3. Immunomodulatory Drugs Tested in MPS Models (Figure 1)
4.3.1. Monoclonal Antibody against Tumor Necrosis Factor-Alpha (TNF-α)
4.3.2. Interleukin-1 Receptor Antagonist: Anakinra
4.3.3. Anti-Oxidant Drugs
4.3.4. Cathepsin B Inhibitor: CA-074
4.3.5. Pentosan Polysulfate
4.4. Immunomodulatory Drugs Not Yet Tested in MPS Models (Figure 1)
4.4.1. Anti-Oxidant Drug
4.4.2. Caspase-1 Inhibitor
4.4.3. Gasdermin D (GSDMD) Inhibitor: Disulfiram
4.4.4. Inhibitor of TLR4 Signaling: TAK-242
4.4.5. Reagents Capable of Inhibiting NLRP3 Inflammasome
4.4.6. NF-κB (Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells) Inhibitor
4.5. Other Potential Therapies in the Future
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Muenzer, J. Early Initiation of Enzyme Replacement Therapy for the Mucopolysaccharidoses. Mol. Genet. Metab. 2014, 111, 63–72. [Google Scholar] [CrossRef]
- Caruso, R.C.; Kaiser-Kupfer, M.I.; Muenzer, J.; Ludwig, I.H.; Zasloff, M.A.; Mercer, P.A. Electroretinographic Findings in the Mucopolysaccharidoses. Ophthalmology 1986, 93, 1612–1616. [Google Scholar] [CrossRef]
- Poorthuis, B.J.H.M.; Wevers, R.A.; Kleijer, W.J.; Groener, J.E.M.; de Jong, J.G.N.; van Weely, S.; Niezen-Koning, K.E.; Diggelen, O.P. van. The Frequency of Lysosomal Storage Diseases in The Netherlands. Hum. Genet. 1999, 105, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Fujii, T.; Fukushi, M.; Oguma, T.; Shimada, T.; Maeda, M.; Kida, K.; Shibata, Y.; Futatsumori, H.; Montaño, A.M.; et al. Newborn Screening and Diagnosis of Mucopolysaccharidoses. Mol. Genet. Metab. 2013, 110, 42–53. [Google Scholar] [CrossRef]
- Celik, B.; Tomatsu, S.C.; Tomatsu, S.; Khan, S.A. Epidemiology of Mucopolysaccharidoses Update. Diagnostics 2021, 11, 273. [Google Scholar] [CrossRef]
- Applegarth, D.A.; Toone, J.R.; Lowry, R.A.R.B. Incidence of Inborn Errors of Metabolism in British Columbia, 1969–1996. Pediatrics 2000, 105, e10. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, S.; Green, A.; Preece, M.A.; Burton, H. The Incidence of Inherited Metabolic Disorders in the West Midlands, UK. Arch. Dis. Child. 2006, 91, 896. [Google Scholar] [CrossRef]
- Schwartz, N.B.; Domowicz, M.S. Glycobiology of the Nervous System. Adv. Neurobiol. 2014, 9, 89–115. [Google Scholar] [CrossRef]
- Braunlin, E.A.; Harmatz, P.R.; Scarpa, M.; Furlanetto, B.; Kampmann, C.; Loehr, J.P.; Ponder, K.P.; Roberts, W.C.; Rosenfeld, H.M.; Giugliani, R. Cardiac Disease in Patients with Mucopolysaccharidosis: Presentation, Diagnosis and Management. J. Inherit. Metab. Dis. 2011, 34, 1183–1197. [Google Scholar] [CrossRef]
- Sodhi, H.; Panitch, A. Glycosaminoglycans in Tissue Engineering: A Review. Biomolecules 2020, 11, 29. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal Storage Diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Fecarotta, S.; Tarallo, A.; Damiano, C.; Minopoli, N.; Parenti, G. Pathogenesis of Mucopolysaccharidoses, an Update. Int. J. Mol. Sci. 2020, 21, 2515. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Lavery, C.; Giugliani, R.; Harmatz, P.; Scarpa, M.; Węgrzyn, G.; Orii, T. (Eds.) Mucopolysaccharidoses Update (2 Volume Set); Nova Science Publishers Inc.: New York, NY, USA, 2018. [Google Scholar]
- Gurinova, E.; Maksimova, N.; Sukhomyasova, A.L. Clinical Description of a Rare Autosomal Recessive Syndrome in the Yakut Children. Yakut Med. J. 2014, 2, 14–18. [Google Scholar]
- Kondo, H.; Maksimova, N.; Otomo, T.; Kato, H.; Imai, A.; Asano, Y.; Kobayashi, K.; Nojima, S.; Nakaya, A.; Hamada, Y.; et al. Mutation in VPS33A Affects Metabolism of Glycosaminoglycans: A New Type of Mucopolysaccharidosis with Severe Systemic Symptoms. Hum. Mol. Genet. 2016, 26, 173–183. [Google Scholar] [CrossRef]
- Dursun, A.; Yalnizoglu, D.; Gerdan, O.F.; Yucel-Yilmaz, D.; Sagiroglu, M.S.; Yuksel, B.; Gucer, S.; Sivri, S.; Ozgul, R.K. A Probable New Syndrome with the Storage Disease Phenotype Caused by the VPS33A Gene Mutation. Clin. Dysmorphol. 2017, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vasilev, F.; Sukhomyasova, A.; Otomo, T. Mucopolysaccharidosis-Plus Syndrome. Int. J. Mol. Sci. 2020, 21, 421. [Google Scholar] [CrossRef] [PubMed]
- Fratantoni, J.C.; Hall, C.W.; Neufeld, E.F. Hurler and Hunter Syndromes: Mutual Correction of the Defect in Cultured Fibroblasts. Science 1968, 162, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Cantz, M.; Chrambach, A.; Neufeld, E.F. Characterization of the Factor Deficient in the Hunter Syndrome by Polyacrylamide Gel Electrophoresis. Biochem. Biophys. Res. Commun. 1970, 39, 936–942. [Google Scholar] [CrossRef]
- Cantz, M.; Chrambach, A.; Bach, G.; Neufeld, E.F. The Hunter Corrective Factor Purification and Preliminary Characterization. J. Biol. Chem. 1972, 247, 5456–5462. [Google Scholar] [CrossRef]
- Bach, G.; Eisenberg, F.; Cantz, M.; Neufeld, E.F. The Defect in the Hunter Syndrome: Deficiency of Sulfoiduronate Sulfatase. Proc. Natl. Acad. Sci. USA 1973, 70, 2134–2138. [Google Scholar] [CrossRef]
- Hobbs, J.R.; Barrett, A.J.; Chambers, D.; James, D.C.O.; Hugh-Jones, K.; Byrom, N.; Henry, K.; Lucas, C.F.; Rogers, T.R.; Benson, P.F.; et al. Reversal of clinical features of hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 1981, 318, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biol. Blood Marrow Transplant. 2019, 25, e226–e246. [Google Scholar] [CrossRef] [PubMed]
- Sifuentes, M.; Doroshow, R.; Hoft, R.; Mason, G.; Walot, I.; Diament, M.; Okazaki, S.; Huff, K.; Cox, G.F.; Swiedler, S.J.; et al. A Follow-up Study of MPS I Patients Treated with Laronidase Enzyme Replacement Therapy for 6 Years. Mol. Genet. Metab. 2007, 90, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Carvalho, C.G.; Herber, S.; Pinto, L.L.d.C. Recent Advances in Treatment Approaches of Mucopolysaccharidosis VI. Curr. Pharm. Biotechnol. 2011, 12, 956–972. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Wraith, J.E.; Beck, M.; Giugliani, R.; Harmatz, P.; Eng, C.M.; Vellodi, A.; Martin, R.; Ramaswami, U.; Gucsavas-Calikoglu, M.; et al. A Phase II/III Clinical Study of Enzyme Replacement Therapy with Idursulfase in Mucopolysaccharidosis II (Hunter Syndrome). Genet. Med. 2006, 8, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Hendriksz, C.J.; Giugliani, R.; Harmatz, P.; Mengel, E.; Guffon, N.; Valayannopoulos, V.; Parini, R.; Hughes, D.; Pastores, G.M.; Lau, H.A.; et al. Multi-Domain Impact of Elosulfase Alfa in Morquio A Syndrome in the Pivotal Phase III Trial. Mol. Genet. Metab. 2015, 114, 178–185. [Google Scholar] [CrossRef]
- Kaufman, M.B. Pharmaceutical Approval Update. Pharm. Ther. 2018, 43, 83–84. [Google Scholar]
- Solomon, M.; Muro, S. Lysosomal Enzyme Replacement Therapies: Historical Development, Clinical Outcomes, and Future Perspectives. Adv. Drug Deliv. Rev. 2017, 118, 109–134. [Google Scholar] [CrossRef]
- Yilmaz, B.S.; Davison, J.; Jones, S.A.; Baruteau, J. Novel Therapies for Mucopolysaccharidosis Type III. J. Inherit. Metab. Dis. 2021, 44, 129–147. [Google Scholar] [CrossRef]
- Wijburg, F.A.; Heap, F.; Rust, S.; de Ruijter, J.; Tump, E.; Marchal, J.P.; Nestrasil, I.; Shapiro, E.; Jones, S.A.; Alexanderian, D. Long-Term Safety and Clinical Outcomes of Intrathecal Heparan-N-Sulfatase in Patients with Sanfilippo Syndrome Type A. Mol. Genet. Metab. 2021, 134, 317–322. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Boado, R.J.; Giugliani, R.; Schmidt, M. Plasma Pharmacokinetics of Valanafusp Alpha, a Human Insulin Receptor Antibody-Iduronidase Fusion Protein, in Patients with Mucopolysaccharidosis Type I. BioDrugs 2018, 32, 169–176. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug and Gene Targeting to the Brain with Molecular Trojan Horses. Nat. Rev. Drug Discov. 2002, 1, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, H.; Takahashi, K.; Minami, K.; Hirato, T.; Yamamoto, T.; So, S.; Tanizawa, K.; Schmidt, M.; Sato, Y. Treatment of Neuronopathic Mucopolysaccharidoses with Blood–Brain Barrier-Crossing Enzymes: Clinical Application of Receptor-Mediated Transcytosis. Pharmaceutics 2022, 14, 1240. [Google Scholar] [CrossRef] [PubMed]
- Kida, S.; Koshimura, Y.; Yoden, E.; Yoshioka, A.; Morimoto, H.; Imakiire, A.; Tanaka, N.; Tanaka, S.; Mori, A.; Ito, J.; et al. Enzyme Replacement with Transferrin Receptor-Targeted α-L-Iduronidase Rescues Brain Pathology in Mucopolysaccharidosis I Mice. Mol. Ther.-Methods Clin. Dev. 2023, 29, 439–449. [Google Scholar] [CrossRef]
- Boado, R.J.; Lu, J.Z.; Hui, E.K.-W.; Pardridge, W.M. Insulin Receptor Antibody–Sulfamidase Fusion Protein Penetrates the Primate Blood–Brain Barrier and Reduces Glycosoaminoglycans in Sanfilippo Type A Cells. Mol. Pharm. 2014, 11, 2928–2934. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Lu, J.Z.; Hui, E.K.-W.; Lin, H.; Pardridge, W.M. Insulin Receptor Antibody−α-N-Acetylglucosaminidase Fusion Protein Penetrates the Primate Blood–Brain Barrier and Reduces Glycosoaminoglycans in Sanfilippo Type B Fibroblasts. Mol. Pharm. 2016, 13, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Melbouci, M.; Mason, R.W.; Suzuki, Y.; Fukao, T.; Orii, T.; Tomatsu, S. Growth Impairment in Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 124, 1–10. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.L.; Flanigan, K.M. Update in the Mucopolysaccharidoses. Semin. Pediatr. Neurol. 2021, 37, 100874. [Google Scholar] [CrossRef]
- Mandolfo, O.; Parker, H.; Bigger, B. Innate Immunity in Mucopolysaccharide Diseases. Int. J. Mol. Sci. 2022, 23, 1999. [Google Scholar] [CrossRef]
- Wiesinger, A.-M.; Bigger, B.; Giugliani, R.; Scarpa, M.; Moser, T.; Lampe, C.; Kampmann, C.; Lagler, F.B. The Inflammation in the Cytopathology of Patients with Mucopolysaccharidoses-Immunomodulatory Drugs as an Approach to Therapy. Front. Pharmacol. 2022, 13, 863667. [Google Scholar] [CrossRef]
- Costi, S.; Caporali, R.F.; Marino, A. Mucopolysaccharidosis: What Pediatric Rheumatologists and Orthopedics Need to Know. Diagnostics 2022, 13, 75. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, F.; Linhardt, R.J. Analysis of the Glycosaminoglycan Chains of Proteoglycans. J. Histochem. Cytochem. 2020, 69, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, M.; Arunkumar, N.; Kubaski, F.; Mason, R.W.; Tadao, O.; Tomatsu, S. Clinical Presentation and Diagnosis of Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 125, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Sawamoto, K.; Mason, R.W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; Tomatsu, S. Enzyme Replacement Therapy for Mucopolysaccharidoses; Past, Present, and Future. J. Hum. Genet. 2019, 64, 1153–1171. [Google Scholar] [CrossRef] [PubMed]
- Anikiej-Wiczenbach, P.; Mański, A.; Milska-Musa, K.; Limanówka, M.; Wierzba, J.; Jamsheer, A.; Cyske, Z.; Gaffke, L.; Pierzynowska, K.; Węgrzyn, G. Highly Diverse Phenotypes of Mucopolysaccharidosis Type IIIB Sibling Patients: Effects of an Additional Mutation in the AUTS2 Gene. J. Appl. Genet. 2022, 63, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J. Overview of the Mucopolysaccharidoses. Rheumatology 2011, 50 (Suppl. S5), v4–v12. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.; Brown, J.R.; Lorey, F.; Dickson, P.I.; Crawford, B.E.; Esko, J.D. Glycan-Based Biomarkers for Mucopolysaccharidoses. Mol. Genet. Metab. 2014, 111, 73–83. [Google Scholar] [CrossRef]
- Verheyen, S.; Blatterer, J.; Speicher, M.R.; Bhavani, G.S.; Boons, G.-J.; Ilse, M.-B.; Andrae, D.; Sproß, J.; Vaz, F.M.; Kircher, S.G.; et al. Novel Subtype of Mucopolysaccharidosis Caused by Arylsulfatase K (ARSK) Deficiency. J. Med. Genet. 2021, 59, 957–964. [Google Scholar] [CrossRef]
- Natowicz, M.R.; Short, M.P.; Wang, Y.; Dickersin, G.R.; Gebhardt, M.C.; Rosenthal, D.I.; Sims, K.B.; Rosenberg, A.E. Clinical and Biochemical Manifestations of Hyaluronidase Deficiency. N. Engl. J. Med. 1996, 335, 1029–1033. [Google Scholar] [CrossRef]
- Sofronova, V.; Gotovtseva, L.; Danilova, A.; Sukhomyasova, A.; Moriwaki, T.; Terawaki, S.; Otomo, T.; Maksimova, N. Prenatal Diagnosis of Mucopolysaccharidosis-Plus Syndrome (MPSPS). Genes 2023, 14, 1581. [Google Scholar] [CrossRef]
- Medeiros, G.F.; Mendes, A.; Castro, R.A.B.; Baú, E.C.; Nader, H.B.; Dietrich, C.P. Distribution of Sulfated Glycosaminoglycans in the Animal Kingdom: Widespread Occurrence of Heparin-like Compounds in Invertebrates. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2000, 1475, 287–294. [Google Scholar] [CrossRef]
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavão, M.S.; Tzanakakis, G.N.; Karamanos, N.K. Glycosaminoglycans: Key Players in Cancer Cell Biology and Treatment. FEBS J. 2012, 279, 1177–1197. [Google Scholar] [CrossRef]
- Prydz, K. Determinants of Glycosaminoglycan (GAG) Structure. Biomolecules 2015, 5, 2003–2022. [Google Scholar] [CrossRef]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan Sulfate Proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef]
- Dyer, D.P.; Salanga, C.L.; Volkman, B.F.; Kawamura, T.; Handel, T.M. The Dependence of Chemokine–Glycosaminoglycan Interactions on Chemokine Oligomerization. Glycobiology 2016, 26, 312–326. [Google Scholar] [CrossRef]
- Rajasekaran, D.; Keeler, C.; Syed, M.A.; Jones, M.C.; Harrison, J.K.; Wu, D.; Bhandari, V.; Hodsdon, M.E.; Lolis, E.J. A Model of GAG/MIP-2/CXCR2 Interfaces and Its Functional Effects. Biochemistry 2012, 51, 5642–5654. [Google Scholar] [CrossRef]
- Sepuru, K.M.; Rajarathnam, K. Structural Basis of a Chemokine Heterodimer Binding to Glycosaminoglycans. Biochem. J. 2021, 478, 1009–1021. [Google Scholar] [CrossRef]
- Pontejo, S.M.; Murphy, P.M. Two Glycosaminoglycan-Binding Domains of the Mouse Cytomegalovirus-Encoded Chemokine MCK-2 Are Critical for Oligomerization of the Full-Length Protein. J. Biol. Chem. 2017, 292, 9613–9626. [Google Scholar] [CrossRef]
- Xie, M.; Li, J. Heparan Sulfate Proteoglycan—A Common Receptor for Diverse Cytokines. Cell. Signal. 2019, 54, 115–121. [Google Scholar] [CrossRef]
- Pasquale, V.D.; Pavone, L.M. Heparan Sulfate Proteoglycans: The Sweet Side of Development Turns Sour in Mucopolysaccharidoses. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 165539. [Google Scholar] [CrossRef]
- Pshezhetsky, A.V. Lysosomal Storage of Heparan Sulfate Causes Mitochondrial Defects, Altered Autophagy, and Neuronal Death in the Mouse Model of Mucopolysaccharidosis III Type C. Autophagy 2016, 12, 1059–1060. [Google Scholar] [CrossRef]
- Ballabio, A.; Gieselmann, V. Lysosomal Disorders: From Storage to Cellular Damage. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 684–696. [Google Scholar] [CrossRef]
- Holley, R.J.; Deligny, A.; Wei, W.; Watson, H.A.; Niñonuevo, M.R.; Dagälv, A.; Leary, J.A.; Bigger, B.W.; Kjellén, L.; Merry, C.L.R. Mucopolysaccharidosis Type I, Unique Structure of Accumulated Heparan Sulfate and Increased N-Sulfotransferase Activity in Mice Lacking α-l-Iduronidase. J. Biol. Chem. 2011, 286, 37515–37524. [Google Scholar] [CrossRef]
- McCarty, D.M.; DiRosario, J.; Gulaid, K.; Killedar, S.; Oosterhof, A.; van Kuppevelt, T.H.; Martin, P.T.; Fu, H. Differential Distribution of Heparan Sulfate Glycoforms and Elevated Expression of Heparan Sulfate Biosynthetic Enzyme Genes in the Brain of Mucopolysaccharidosis IIIB Mice. Metab. Brain Dis. 2011, 26, 9–19. [Google Scholar] [CrossRef]
- Bruyère, J.; Roy, E.; Ausseil, J.; Lemonnier, T.; Teyre, G.; Bohl, D.; Etienne-Manneville, S.; Lortat-Jacob, H.; Heard, J.M.; Vitry, S. Heparan Sulfate Saccharides Modify Focal Adhesions: Implication in Mucopolysaccharidosis Neuropathophysiology. J. Mol. Biol. 2015, 427, 775–791. [Google Scholar] [CrossRef]
- Ausseil, J.; Desmaris, N.; Bigou, S.; Attali, R.; Corbineau, S.; Vitry, S.; Parent, M.; Cheillan, D.; Fuller, M.; Maire, I.; et al. Early Neurodegeneration Progresses Independently of Microglial Activation by Heparan Sulfate in the Brain of Mucopolysaccharidosis IIIB Mice. PLoS ONE 2008, 3, e2296. [Google Scholar] [CrossRef]
- Bigger, B.W.; Begley, D.J.; Virgintino, D.; Pshezhetsky, A.V. Anatomical Changes and Pathophysiology of the Brain in Mucopolysaccharidosis Disorders. Mol. Genet. Metab. 2018, 125, 322–331. [Google Scholar] [CrossRef]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.A.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in Mouse Models of Mucopolysaccharidosis Type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef]
- Watson, H.A.; Holley, R.J.; Langford-Smith, K.J.; Wilkinson, F.L.; van Kuppevelt, T.H.; Wynn, R.F.; Wraith, J.E.; Merry, C.L.R.; Bigger, B.W. Heparan Sulfate Inhibits Hematopoietic Stem and Progenitor Cell Migration and Engraftment in Mucopolysaccharidosis I. J. Biol. Chem. 2014, 289, 36194–36203. [Google Scholar] [CrossRef]
- Pan, C.; Nelson, M.S.; Reyes, M.; Koodie, L.; Brazil, J.J.; Stephenson, E.J.; Zhao, R.C.; Peters, C.; Selleck, S.B.; Stringer, S.E.; et al. Functional Abnormalities of Heparan Sulfate in Mucopolysaccharidosis-I Are Associated with Defective Biologic Activity of FGF-2 on Human Multipotent Progenitor Cells. Blood 2005, 106, 1956–1964. [Google Scholar] [CrossRef]
- Walton, R.M.; Wolfe, J.H. Abnormalities in Neural Progenitor Cells in a Dog Model of Lysosomal Storage Disease. J. Neuropathol. Exp. Neurol. 2007, 66, 760–769. [Google Scholar] [CrossRef]
- Poli, E.F.; Zalfa, C.; D’Avanzo, F.; Tomanin, R.; Carlessi, L.; Bossi, M.; Nodari, L.R.; Binda, E.; Marmiroli, P.; Scarpa, M.; et al. Murine Neural Stem Cells Model Hunter Disease in Vitro: Glial Cell-Mediated Neurodegeneration as a Possible Mechanism Involved. Cell Death Dis. 2013, 4, e906. [Google Scholar] [CrossRef]
- Bellesso, S.; Salvalaio, M.; Lualdi, S.; Tognon, E.; Costa, R.; Braghetta, P.; Giraudo, C.; Stramare, R.; Rigon, L.; Filocamo, M.; et al. FGF Signaling Deregulation Is Associated with Early Developmental Skeletal Defects in Animal Models for Mucopolysaccharidosis Type II (MPSII). Hum. Mol. Genet. 2018, 27, 2262–2275. [Google Scholar] [CrossRef]
- Lamanna, W.C.; Lawrence, R.; Sarrazin, S.; Esko, J.D. Secondary Storage of Dermatan Sulfate in Sanfilippo Disease. J. Biol. Chem. 2011, 286, 6955–6962. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Oguma, T.; Dung, V.C.; Oikawa, H.; Gutiérrez, M.L.; Yamaguchi, S.; Suzuki, Y.; Fukushi, M.; Barrera, L.A.; et al. Validation of Disaccharide Compositions Derived from Dermatan Sulfate and Heparan Sulfate in Mucopolysaccharidoses and Mucolipidoses II and III by Tandem Mass Spectrometry. Mol. Genet. Metab. 2010, 99, 124–131. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. Int. J. Mol. Sci. 2020, 21, 2566. [Google Scholar] [CrossRef]
- Li, H.H.; Yu, W.-H.; Rozengurt, N.; Zhao, H.-Z.; Lyons, K.M.; Anagnostaras, S.; Fanselow, M.S.; Suzuki, K.; Vanier, M.T.; Neufeld, E.F. Mouse Model of Sanfilippo Syndrome Type B Produced by Targeted Disruption of the Gene Encoding α-N-Acetylglucosaminidase. Proc. Natl. Acad. Sci. USA 1999, 96, 14505–14510. [Google Scholar] [CrossRef]
- Derrick-Roberts, A.; Kaidonis, X.; Jackson, M.R.; Liaw, W.C.; Ding, X.; Ong, C.; Ranieri, E.; Sharp, P.; Fletcher, J.; Byers, S. Comparative Analysis of Brain Pathology in Heparan Sulphate Storing Mucopolysaccharidoses. Mol. Genet. Metab. 2020, 131, 197–205. [Google Scholar] [CrossRef]
- Baydakova, G.; Ilyushkina, A.; Gaffke, L.; Pierzynowska, K.; Bychkov, I.; Ługowska, A.; Wegrzyn, G.; Tylki-Szymanska, A.; Zakharova, E. Elevated LysoGb3 Concentration in the Neuronopathic Forms of Mucopolysaccharidoses. Diagnostics 2020, 10, 155. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D. Thematic Review Series: Brain Lipids. Cholesterol Metabolism in the Central Nervous System during Early Development and in the Mature Animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef]
- Ryazantsev, S.; Yu, W.-H.; Zhao, H.-Z.; Neufeld, E.F.; Ohmi, K. Lysosomal Accumulation of SCMAS (Subunit c of Mitochondrial ATP Synthase) in Neurons of the Mouse Model of Mucopolysaccharidosis III B. Mol. Genet. Metab. 2007, 90, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, K.; Kudo, L.C.; Ryazantsev, S.; Zhao, H.-Z.; Karsten, S.L.; Neufeld, E.F. Sanfilippo Syndrome Type B, a Lysosomal Storage Disease, Is Also a Tauopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 8332–8337. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Galvin, J.E.; Lee, V.M.-Y.; Rorke, L.B.; Dickson, D.W.; Wolfe, J.H.; Jones, M.Z.; Trojanowski, J.Q. Accumulation of Intracellular Amyloid-β Peptide (Aβ 1–40) in Mucopolysaccharidosis Brains. J. Neuropathol. Exp. Neurol. 1999, 58, 815–824. [Google Scholar] [CrossRef]
- Fraldi, A.; Klein, A.D.; Medina, D.L.; Settembre, C. Brain Disorders Due to Lysomal Dysfunction. Annu. Rev. Neurosci. 2015, 39, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Mahuran, D.J. Biochemical Consequences of Mutations Causing the GM2 Gangliosidoses. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1999, 1455, 105–138. [Google Scholar] [CrossRef]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated Globotriaosylsphingosine Is a Hallmark of Fabry Disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef]
- Pentchev, P.G.; Comly, M.E.; Kruth, H.S.; Vanier, M.T.; Wenger, D.A.; Patel, S.; Brady, R.O. A Defect in Cholesterol Esterification in Niemann-Pick Disease (Type C) Patients. Proc. Natl. Acad. Sci. USA 1985, 82, 8247–8251. [Google Scholar] [CrossRef]
- Black, M.M.; Aletta, J.M.; Greene, L.A. Regulation of Microtubule Composition and Stability during Nerve Growth Factor-Promoted Neurite Outgrowth. J. Cell Biol. 1986, 103, 545–557. [Google Scholar] [CrossRef]
- Drubin, D.G.; Kirschner, M.W. Tau Protein Function in Living Cells. J. Cell Biol. 1986, 103, 2739–2746. [Google Scholar] [CrossRef]
- Butler, D.; Brown, Q.B.; Chin, D.J.; Batey, L.; Karim, S.; Mutneja, M.S.; Karanian, D.A.; Bahr, B.A. Cellular Responses to Protein Accumulation Involve Autophagy and Lysosomal Enzyme Activation. Rejuvenation Res. 2005, 8, 227–237. [Google Scholar] [CrossRef]
- Monaco, A.; Fraldi, A. Protein Aggregation and Autophagy Dysfunction: New Lessons from Mucopolysaccharidoses. Autophagy 2021, 17, 3875–3876. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Q.; Li, S.; Li, X.-J.; Yang, W.; He, D. Microglial Autophagy in Alzheimer’s Disease and Parkinson’s Disease. Front. Aging Neurosci. 2023, 14, 1065183. [Google Scholar] [CrossRef] [PubMed]
- Minchev, D.; Kazakova, M.; Sarafian, V. Neuroinflammation and Autophagy in Parkinson’s Disease—Novel Perspectives. Int. J. Mol. Sci. 2022, 23, 14997. [Google Scholar] [CrossRef]
- Oh, Y.M.; Lee, S.W.; Kim, W.K.; Chen, S.; Church, V.A.; Cates, K.; Li, T.; Zhang, B.; Dolle, R.E.; Dahiya, S.; et al. Age-Related Huntington’s Disease Progression Modeled in Directly Reprogrammed Patient-Derived Striatal Neurons Highlights Impaired Autophagy. Nat. Neurosci. 2022, 25, 1420–1433. [Google Scholar] [CrossRef] [PubMed]
- Lazarev, V.F.; Dutysheva, E.A.; Kanunikov, I.E.; Guzhova, I.V.; Margulis, B.A. Protein Interactome of Amyloid-β as a Therapeutic Target. Pharmaceuticals 2023, 16, 312. [Google Scholar] [CrossRef]
- Khan, S.A.; Nelson, M.S.; Pan, C.; Gaffney, P.M.; Gupta, P. Endogenous Heparan Sulfate and Heparin Modulate Bone Morphogenetic Protein-4 Signaling and Activity. Am. J. Physiol.-Cell Physiol. 2008, 294, C1387–C1397. [Google Scholar] [CrossRef] [PubMed]
- Oussoren, E.; Brands, M.M.M.G.; Ruijter, G.J.G.; van der Ploeg, A.T.; Reuser, A.J.J. Bone, Joint and Tooth Development in Mucopolysaccharidoses: Relevance to Therapeutic Options. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 1542–1556. [Google Scholar] [CrossRef]
- Katagiri, T.; Watabe, T. Bone Morphogenetic Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021899. [Google Scholar] [CrossRef]
- Fisher, M.C.; Li, Y.; Seghatoleslami, M.R.; Dealy, C.N.; Kosher, R.A. Heparan Sulfate Proteoglycans Including Syndecan-3 Modulate BMP Activity during Limb Cartilage Differentiation. Matrix Biol. 2006, 25, 27–39. [Google Scholar] [CrossRef]
- Raman, R.; Sasisekharan, V.; Sasisekharan, R. Structural Insights into Biological Roles of Protein-Glycosaminoglycan Interactions. Chem. Biol. 2005, 12, 267–277. [Google Scholar] [CrossRef]
- Mende, M.; Bednarek, C.; Wawryszyn, M.; Sauter, P.; Biskup, M.B.; Schepers, U.; Bräse, S. Chemical Synthesis of Glycosaminoglycans. Chem. Rev. 2016, 116, 8193–8255. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.P.; Nader, H.B.; Mourão, P.A.S. Differentiation of Hunter’s and Hurler’s Syndromes by the Analysis of the Excreted Mucopolysaccharides. Biochem. Med. Metab. B 1973, 8, 371–379. [Google Scholar] [CrossRef]
- Mourão, P.A.S.; Rozenfeld, S.; Laredo, J.; Dietrich, C.P. The Distributions of Chondroitin Sulfates in Articular and Growth Cartilages of Human Bones. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1976, 428, 19–26. [Google Scholar] [CrossRef]
- Mourão, P.A.S. Distribution of Chondroitin 4–Sulfate and Chondroitin 6–Sulfate in Human Articular and Growth Cartilage. Arthritis Rheum. 1988, 31, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Leaver, A.G.; Triffitt, J.T.; Holbrook, I.B. Newer Knowledge of Non-Collagenous Protein in Dentin and Cortical Bone Matrix. Clin. Orthop. Relat. Res. 1975, 110, 269–292. [Google Scholar] [CrossRef]
- Hagiwara, H.; Aoki, T.; Yoshimi, T. Immunoelectron Microscopic Analysis of Chondroitin Sulfates during Calcification in the Rat Growth Plate Cartilage. Histochem. Cell Biol. 1995, 103, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Caterson, B.; Melrose, J. Keratan Sulfate, a Complex Glycosaminoglycan with Unique Functional Capability. Glycobiology 2018, 28, 182–206. [Google Scholar] [CrossRef]
- Meyer, K.; Linker, A.; Davidson, E.A.; Weissmann, B. The Mucopolysaccharides of Bovine Cornea. J. Biol. Chem. 1953, 205, 611–616. [Google Scholar] [CrossRef]
- Sajdera, S.W.; Hascall, V.C. Proteinpolysaccharide Complex from Bovine Nasal Cartilage A Comparison of Low and High Shear Extraction Procedures. J. Biol. Chem. 1969, 244, 77–87. [Google Scholar] [CrossRef]
- KIANI, C.; CHEN, L.; WU, Y.J.; YEE, A.J.; YANG, B.B. Structure and Function of Aggrecan. Cell Res. 2002, 12, 19–32. [Google Scholar] [CrossRef]
- Khan, S.; Alméciga-Díaz, C.J.; Sawamoto, K.; Mackenzie, W.G.; Theroux, M.C.; Pizarro, C.; Mason, R.W.; Orii, T.; Tomatsu, S. Mucopolysaccharidosis IVA and Glycosaminoglycans. Mol. Genet. Metab. 2017, 120, 78–95. [Google Scholar] [CrossRef]
- Sawamoto, K.; González, J.V.Á.; Piechnik, M.; Otero, F.J.; Couce, M.L.; Suzuki, Y.; Tomatsu, S. Mucopolysaccharidosis IVA: Diagnosis, Treatment, and Management. Int. J. Mol. Sci. 2020, 21, 1517. [Google Scholar] [CrossRef] [PubMed]
- Bishnoi, M.; Jain, A.; Hurkat, P.; Jain, S.K. Chondroitin Sulphate: A Focus on Osteoarthritis. Glycoconj. J. 2016, 33, 693–705. [Google Scholar] [CrossRef]
- Uwe, S. Anti-Inflammatory Interventions of NF-ΚB Signaling: Potential Applications and Risks. Biochem. Pharmacol. 2008, 75, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Fujitsuka, H.; Sawamoto, K.; Peracha, H.; Mason, R.W.; Mackenzie, W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Biomarkers in Patients with Mucopolysaccharidosis Type II and IV. Mol Genet. Metab. Rep. 2019, 19, 100455. [Google Scholar] [CrossRef]
- Franceschi, L.D.; Roseti, L.; Desando, G.; Facchini, A.; Grigolo, B. A Molecular and Histological Characterization of Cartilage from Patients with Morquio Syndrome. Osteoarthr. Cartil. 2007, 15, 1311–1317. [Google Scholar] [CrossRef][Green Version]
- Bank, R.A.; Groener, J.E.M.; van Gemund, J.J.; Maaswinkel, P.D.; Hoeben, K.A.; Schut, H.A.; Everts, V. Deficiency in N-Acetylgalactosamine-6-Sulfate Sulfatase Results in Collagen Perturbations in Cartilage of Morquio Syndrome A Patients. Mol. Genet. Metab. 2009, 97, 196–201. [Google Scholar] [CrossRef]
- Groebe, H.; Krins, M.; Schmidberger, H.; von Figura, K.; Harzer, K.; Kresse, H.; Paschke, E.; Sewell, A.; Ullrich, K. Morquio Syndrome (Mucopolysaccharidosis IV B) Associated with Beta-Galactosidase Deficiency. Report of Two Cases. Am. J. Hum. Genet. 1980, 32, 258–272. [Google Scholar]
- Huffman, F.G. Encyclopedia of Food Sciences and Nutrition, 2nd ed.; URONIC ACIDS; Elsevier Science B.V: Amsterdam, The Netherlands, 2003. [Google Scholar] [CrossRef]
- Bianco, P.; Fisher, L.W.; Young, M.F.; Termine, J.D.; Robey, P.G. Expression and Localization of the Two Small Proteoglycans Biglycan and Decorin in Developing Human Skeletal and Non-Skeletal Tissues. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1990, 38, 1549–1563. [Google Scholar] [CrossRef]
- Grande-Allen, K.J.; Griffin, B.P.; Ratliff, N.B.; Cosgrove, D.M.; Vesely, I. Glycosaminoglycan Profiles of Myxomatous Mitral Leaflets and Chordae Parallel the Severity of Mechanical Alterations. J. Am. Coll. Cardiol. 2003, 42, 271–277. [Google Scholar] [CrossRef]
- Latif, N.; Sarathchandra, P.; Taylor, P.M.; Antoniw, J.; Yacoub, M.H. Localization and Pattern of Expression of Extracellular Matrix Components in Human Heart Valves. J. Heart Valve Dis. 2005, 14, 218–227. [Google Scholar]
- Gupta, V.; Barzilla, J.E.; Mendez, J.S.; Stephens, E.H.; Lee, E.L.; Collard, C.D.; Laucirica, R.; Weigel, P.H.; Grande-Allen, K.J. Abundance and Location of Proteoglycans and Hyaluronan within Normal and Myxomatous Mitral Valves. Cardiovasc. Pathol. 2009, 18, 191–197. [Google Scholar] [CrossRef]
- Dangel, J.H. Cardiovascular Changes in Children with Mucopolysaccharide Storage Diseases and Related Disorders—Clinical and Echocardiographic Findings in 64 Patients. Eur. J. Pediatr. 1998, 157, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.N.; de Paula, A.C.; Leone, C.; Kim, C.A. Echocardiographic Study of Paediatric Patients with Mucopolysaccharidosis. Cardiol. Young 2010, 20, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kametani, K.; Koyama, Y.; Suzuki, D.; Imamura, Y.; Takehana, K.; Hiramatsu, K. Ring-Mesh Model of Proteoglycan Glycosaminoglycan Chains in Tendon Based on Three-Dimensional Reconstruction by Focused Ion Beam Scanning Electron Microscopy. J. Biol. Chem. 2016, 291, 23704–23708. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Takahashi, N.; Tangkawattana, P.; Minaguchi, J.; Mizumoto, S.; Yamada, S.; Miyake, N.; Hayashi, S.; Hatamochi, A.; Nakayama, J.; et al. Structural Alteration of Glycosaminoglycan Side Chains and Spatial Disorganization of Collagen Networks in the Skin of Patients with McEDS-CHST14. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2019, 1863, 623–631. [Google Scholar] [CrossRef]
- Toole, B.P.; Lowther, D.A. Dermatan Sulfate-Protein: Isolation from and Interaction with Collagen. Arch. Biochem. Biophys. 1968, 128, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Boskey, A.L.; Spevak, L.; Doty, S.B.; Rosenberg, L. Effects of Bone CS-Proteoglycans, DS-Decorin, and DS-Biglycan on Hydroxyapatite Formation in a Gelatin Gel. Calcif. Tissue Int. 1997, 61, 298–305. [Google Scholar] [CrossRef]
- Waddington, R.J.; Roberts, H.C.; Sugars, R.V.; Schönherr, E. Differential Roles for Small Leucine-Rich Proteoglycans in Bone Formation. Eur. Cells Mater. 2003, 6, 12–21. [Google Scholar] [CrossRef]
- Hua, R.; Ni, Q.; Eliason, T.D.; Han, Y.; Gu, S.; Nicolella, D.P.; Wang, X.; Jiang, J.X. Biglycan and Chondroitin Sulfate Play Pivotal Roles in Bone Toughness via Retaining Bound Water in Bone Mineral Matrix. Matrix Biol. 2020, 94, 95–109. [Google Scholar] [CrossRef]
- Fahnehjelm, K.T.; Ashworth, J.L.; Pitz, S.; Olsson, M.; Törnquist, A.L.; Lindahl, P.; Summers, C.G. Clinical Guidelines for Diagnosing and Managing Ocular Manifestations in Children with Mucopolysaccharidosis. Acta Ophthalmol. 2012, 90, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Pitz, S.; Hampel, U. Ophthalmological Findings in Mucopolysaccharidoses. J. Clin. Med. 2019, 8, 1467. [Google Scholar] [CrossRef]
- Dhamale, O.P.; Lawrence, R.; Wiegmann, E.M.; Shah, B.A.; Al-Mafraji, K.; Lamanna, W.C.; Lübke, T.; Dierks, T.; Boons, G.-J.; Esko, J.D. Arylsulfatase K Is the Lysosomal 2-Sulfoglucuronate Sulfatase. ACS Chem. Biol. 2017, 12, 367–373. [Google Scholar] [CrossRef]
- Trabszo, C.; Ramms, B.; Chopra, P.; Lüllmann-Rauch, R.; Stroobants, S.; Sproß, J.; Jeschke, A.; Schinke, T.; Boons, G.-J.; Esko, J.D.; et al. Arylsulfatase K Inactivation Causes Mucopolysaccharidosis Due to Deficient Glucuronate Desulfation of Heparan and Chondroitin Sulfate. Biochem. J. 2020, 477, 3433–3451. [Google Scholar] [CrossRef] [PubMed]
- Rustad, C.F.; Prescott, T.E.; Merckoll, E.; Kristensen, E.; Salvador, C.L.; Nordgarden, H.; Tveten, K. Phenotypic Expansion of ARSK-related Mucopolysaccharidosis. Am. J. Med. Genet. A 2022, 188, 3369–3373. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Chanmee, T.; Itano, N. Hyaluronan: Metabolism and Function. Biomolecules 2020, 10, 1525. [Google Scholar] [CrossRef]
- Tamer, T.M. Hyaluronan and Synovial Joint: Function, Distribution and Healing. Interdiscip. Toxicol. 2013, 6, 111–125. [Google Scholar] [CrossRef]
- LAURENT, T.C.; LAURENT, U.B.; FRASER, J.R.E. The Structure and Function of Hyaluronan: An Overview. Immunol. Cell Biol. 1996, 74, a1–a7. [Google Scholar] [CrossRef]
- Kreil, G. Hyaluronidases—A Group of Neglected Enzymes. Protein Sci. 1995, 4, 1666–1669. [Google Scholar] [CrossRef]
- Frost, G.I.; Csóka, A.B.; Wong, T.; Stern, R.; Csóka, T.B. Purification, Cloning, and Expression of Human Plasma Hyaluronidase. Biochem. Biophys. Res. Commun. 1997, 236, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Imundo, L.; LeDuc, C.A.; Guha, S.; Brown, M.; Perino, G.; Gushulak, L.; Triggs-Raine, B.; Chung, W.K. A Complete Deficiency of Hyaluronoglucosaminidase 1 (HYAL1) Presenting as Familial Juvenile Idiopathic Arthritis. J. Inherit. Metab. Dis. 2011, 34, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.C.; Atmuri, V.; Hemming, R.J.; Farley, J.; Mort, J.S.; Byers, S.; Hombach-Klonisch, S.; Csoka, A.B.; Stern, R.; Triggs-Raine, B.L. A Mouse Model of Human Mucopolysaccharidosis IX Exhibits Osteoarthritis. Hum. Mol. Genet. 2008, 17, 1904–1915. [Google Scholar] [CrossRef]
- VanTeeffelen, J.W.; Brands, J.; Stroes, E.S.; Vink, H. Endothelial Glycocalyx: Sweet Shield of Blood Vessels. Trends Cardiovasc. Med. 2007, 17, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Morishita, K.; Petty, R.E. Musculoskeletal Manifestations of Mucopolysaccharidoses. Rheumatology 2011, 50 (Suppl. S5), v19–v25. [Google Scholar] [CrossRef]
- Peracha, H.; Sawamoto, K.; Averill, L.; Kecskemethy, H.; Theroux, M.; Thacker, M.; Nagao, K.; Pizarro, C.; Mackenzie, W.; Kobayashi, H.; et al. Molecular Genetics and Metabolism, Special Edition: Diagnosis, Diagnosis and Prognosis of Mucopolysaccharidosis IVA. Mol. Genet. Metab. 2018, 125, 18–37. [Google Scholar] [CrossRef]
- Tomatsu, S.; Alméciga-Díaz, C.J.; Montaño, A.M.; Yabe, H.; Tanaka, A.; Dung, V.C.; Giugliani, R.; Kubaski, F.; Mason, R.W.; Yasuda, E.; et al. Therapies for the Bone in Mucopolysaccharidoses. Mol. Genet. Metab. 2015, 114, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.; Muenzer, J. The Metabolic and Molecular Bases of Inherited Disease; Scriver, C., Beaudet, A.L., Valle, D., Sly, W., Eds.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Pereira, V.G.; Gazarini, M.L.; Rodrigues, L.C.; da Silva, F.H.; Han, S.W.; Martins, A.M.; Tersariol, I.L.S.; D’Almeida, V. Evidence of Lysosomal Membrane Permeabilization in Mucopolysaccharidosis Type I: Rupture of Calcium and Proton Homeostasis. J. Cell. Physiol. 2010, 223, 335–342. [Google Scholar] [CrossRef]
- Micsenyi, M.C.; Sikora, J.; Stephney, G.; Dobrenis, K.; Walkley, S.U. Lysosomal Membrane Permeability Stimulates Protein Aggregate Formation in Neurons of a Lysosomal Disease. J. Neurosci. 2013, 33, 10815–10827. [Google Scholar] [CrossRef]
- Kendall, R.L.; Holian, A. The Role of Lysosomal Ion Channels in Lysosome Dysfunction. Inhal. Toxicol. 2021, 33, 41–54. [Google Scholar] [CrossRef]
- Xu, J.; Núñez, G. The NLRP3 Inflammasome: Activation and Regulation. Trends Biochem. Sci. 2023, 48, 331–344. [Google Scholar] [CrossRef]
- Parker, H.; Bigger, B.W. The Role of Innate Immunity in Mucopolysaccharide Diseases. J. Neurochem. 2019, 148, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. Lysosomal Storage Disorders: The Cellular Impact of Lysosomal Dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy Controls IL-1β Secretion by Targeting Pro-IL-1β for Degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, M.T.; Moro, E.; Erickson, R.P. The Pathogenesis of Lysosomal Storage Disorders: Beyond the Engorgement of Lysosomes to Abnormal Development and Neuroinflammation. Hum. Mol. Genet. 2018, 27, R119–R129. [Google Scholar] [CrossRef]
- Tillo, M.; Lamanna, W.C.; Dwyer, C.A.; Sandoval, D.R.; Pessentheiner, A.R.; Al-Azzam, N.; Sarrazin, S.; Gonzales, J.C.; Kan, S.-H.; Andreyev, A.Y.; et al. Impaired Mitophagy in Sanfilippo a Mice Causes Hypertriglyceridemia and Brown Adipose Tissue Activation. J. Biol. Chem. 2022, 298, 102159. [Google Scholar] [CrossRef] [PubMed]
- Hashem, S.I.; Murphy, A.N.; Divakaruni, A.S.; Klos, M.L.; Nelson, B.C.; Gault, E.C.; Rowland, T.J.; Perry, C.N.; Gu, Y.; Dalton, N.D.; et al. Impaired Mitophagy Facilitates Mitochondrial Damage in Danon Disease. J. Mol. Cell. Cardiol. 2017, 108, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Stepien, K.M.; Roncaroli, F.; Turton, N.; Hendriksz, C.J.; Roberts, M.; Heaton, R.A.; Hargreaves, I. Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review. J. Clin. Med. 2020, 9, 2596. [Google Scholar] [CrossRef]
- Liedtke, M.; Völkner, C.; Hermann, A.; Frech, M.J. Impact of Organelle Transport Deficits on Mitophagy and Autophagy in Niemann–Pick Disease Type C. Cells 2022, 11, 507. [Google Scholar] [CrossRef]
- Stepien, K.M.; Cufflin, N.; Donald, A.; Jones, S.; Church, H.; Hargreaves, I.P. Secondary Mitochondrial Dysfunction as a Cause of Neurodegenerative Dysfunction in Lysosomal Storage Diseases and an Overview of Potential Therapies. Int. J. Mol. Sci. 2022, 23, 10573. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-Interacting Protein Links Oxidative Stress to Inflammasome Activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Liu, J.; Han, X.; Zhang, T.; Tian, K.; Li, Z.; Luo, F. Reactive Oxygen Species (ROS) Scavenging Biomaterials for Anti-Inflammatory Diseases: From Mechanism to Therapy. J. Hematol. Oncol. 2023, 16, 116. [Google Scholar] [CrossRef] [PubMed]
- Srinivasula, S.M.; Poyet, J.-L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD Protein ASC Is an Activating Adaptor for Caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar] [CrossRef] [PubMed]
- Goodall, K.J.; Poon, I.K.H.; Phipps, S.; Hulett, M.D. Soluble Heparan Sulfate Fragments Generated by Heparanase Trigger the Release of Pro-Inflammatory Cytokines through TLR-4. PLoS ONE 2014, 9, e109596. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.; Means, T.K.; Heine, H.; Yoshimura, A.; Kusumoto, S.; Fukase, K.; Fenton, M.J.; Oikawa, M.; Qureshi, N.; Monks, B.; et al. Toll-like Receptor 4 Imparts Ligand-Specific Recognition of Bacterial Lipopolysaccharide. J. Clin. Investig. 2000, 105, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.R.; Yamasaki, K.; Radek, K.A.; Nardo, A.D.; Goodarzi, H.; Golenbock, D.; Beutler, B.; Gallo, R.L. Recognition of Hyaluronan Released in Sterile Injury Involves a Unique Receptor Complex Dependent on Toll-like Receptor 4, CD44, and MD-2. J. Biol. Chem. 2007, 282, 18265–18275. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.R.; Kessler, S.P.; Rho, H.K.; Cowman, M.K.; Motte, C.A. de la. Specific-Sized Hyaluronan Fragments Promote Expression of Human β-Defensin 2 in Intestinal Epithelium. J. Biol. Chem. 2012, 287, 30610–30624. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Bremer, L.; Aulin, C.; Harris, H.E. Fragmented Hyaluronan Has No Alarmin Function Assessed in Arthritis Synovial Fibroblast and Chondrocyte Cultures. Innate Immun. 2018, 24, 131–141. [Google Scholar] [CrossRef]
- Cowman, M.K.; Shortt, C.; Arora, S.; Fu, Y.; Villavieja, J.; Rathore, J.; Huang, X.; Rakshit, T.; Jung, G.I.; Kirsch, T. Role of Hyaluronan in Inflammatory Effects on Human Articular Chondrocytes. Inflammation 2019, 42, 1808–1820. [Google Scholar] [CrossRef]
- Altman, R.; Bedi, A.; Manjoo, A.; Niazi, F.; Shaw, P.; Mease, P. Anti-Inflammatory Effects of Intra-Articular Hyaluronic Acid: A Systematic Review. Cartilage 2019, 10, 43–52. [Google Scholar] [CrossRef]
- Jolly, R.D.; Allan, F.J.; Collett, M.G.; Rozaklis, T.; Muller, V.J.; Hopwood, J.J. Mucopolysaccharidosis IIIA (Sanfilippo Syndrome) in a New Zealand Huntaway Dog with Ataxia. N. Z. Vet. J. 2000, 48, 144–148. [Google Scholar] [CrossRef]
- McGlynn, R.; Dobrenis, K.; Walkley, S.U. Differential Subcellular Localization of Cholesterol, Gangliosides, and Glycosaminoglycans in Murine Models of Mucopolysaccharide Storage Disorders. J. Comp. Neurol. 2004, 480, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Jou, I.; Lee, J.H.; Park, S.Y.; Yoon, H.J.; Joe, E.-H.; Park, E.J. Gangliosides Trigger Inflammatory Responses via TLR4 in Brain Glia. Am. J. Pathol. 2006, 168, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Ellison, S.M.; Holley, R.J.; O’Leary, C.; Liao, A.; Asadi, J.; Glover, E.; Ghosh, A.; Jones, S.; Wilkinson, F.L.; et al. Haematopoietic Stem Cell Gene Therapy with IL-1Ra Rescues Cognitive Loss in Mucopolysaccharidosis IIIA. EMBO Mol. Med. 2020, 12, e11185. [Google Scholar] [CrossRef] [PubMed]
- Raymond, G.V.; Pasquali, M.; Polgreen, L.E.; Dickson, P.I.; Miller, W.P.; Orchard, P.J.; Lund, T.C. Elevated Cerebral Spinal Fluid Biomarkers in Children with Mucopolysaccharidosis I-H. Sci. Rep. 2016, 6, 38305. [Google Scholar] [CrossRef] [PubMed]
- Khalid, O.; Vera, M.U.; Gordts, P.L.; Ellinwood, N.M.; Schwartz, P.H.; Dickson, P.I.; Esko, J.D.; Wang, R.Y. Immune-Mediated Inflammation May Contribute to the Pathogenesis of Cardiovascular Disease in Mucopolysaccharidosis Type I. PLoS ONE 2016, 11, e0150850. [Google Scholar] [CrossRef]
- Bigg, P.W.; Baldo, G.; Sleeper, M.M.; O’Donnell, P.A.; Bai, H.; Rokkam, V.R.P.; Liu, Y.; Wu, S.; Giugliani, R.; Casal, M.L.; et al. Pathogenesis of Mitral Valve Disease in Mucopolysaccharidosis VII Dogs. Mol. Genet. Metab. 2013, 110, 319–328. [Google Scholar] [CrossRef][Green Version]
- Opoka-Winiarska, V.; Jurecka, A.; Emeryk, A.; Tylki-Szymańska, A. Osteoimmunology in Mucopolysaccharidoses Type I, II, VI and VII. Immunological Regulation of the Osteoarticular System in the Course of Metabolic Inflammation. Osteoarthr. Cartil. 2013, 21, 1813–1823. [Google Scholar] [CrossRef]
- Simonaro, C.M.; Ge, Y.; Eliyahu, E.; He, X.; Jepsen, K.J.; Schuchman, E.H. Involvement of the Toll-like Receptor 4 Pathway and Use of TNF-α Antagonists for Treatment of the Mucopolysaccharidoses. Proc. Natl. Acad. Sci. USA 2010, 107, 222–227. [Google Scholar] [CrossRef]
- Xing, E.M.; Wu, S.; Ponder, K.P. The Effect of Tlr4 and/or C3 Deficiency and of Neonatal Gene Therapy on Skeletal Disease in Mucopolysaccharidosis VII Mice. Mol. Genet. Metab. 2015, 114, 209–216. [Google Scholar] [CrossRef]
- Manohar, K.; Hosfield, B.D.; Mesfin, F.M.; Colgate, C.; Shelley, W.C.; Liu, J.; Zeng, L.; Brokaw, J.P.; Markel, T.A. Chondroitin Sulfate Supplementation Improves Clinical Outcomes in a Murine Model of Necrotizing Enterocolitis. Physiol. Rep. 2023, 11, e15819. [Google Scholar] [CrossRef]
- Vallières, M.; Souich, P.D. Modulation of Inflammation by Chondroitin Sulfate. Osteoarthr. Cartil. 2010, 18, S1–S6. [Google Scholar] [CrossRef] [PubMed]
- du Souich, P.; García, A.G.; Vergés, J.; Montell, E. Immunomodulatory and Anti-inflammatory Effects of Chondroitin Sulphate. J. Cell. Mol. Med. 2009, 13, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.-K.; Tabata, Y. Chondroitin-6-Sulfate Attenuates Inflammatory Responses in Murine Macrophages via Suppression of NF-ΚB Nuclear Translocation. Acta Biomater. 2014, 10, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Donida, B.; Marchetti, D.P.; Biancini, G.B.; Deon, M.; Manini, P.R.; da Rosa, H.T.; Moura, D.J.; Saffi, J.; Bender, F.; Burin, M.G.; et al. Oxidative Stress and Inflammation in Mucopolysaccharidosis Type IVA Patients Treated with Enzyme Replacement Therapy. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Donida, B.; Marchetti, D.P.; Jacques, C.E.D.; Ribas, G.; Deon, M.; Manini, P.; da Rosa, H.T.; Moura, D.J.; Saffi, J.; Giugliani, R.; et al. Oxidative Profile Exhibited by Mucopolysaccharidosis Type IVA Patients at Diagnosis: Increased Keratan Urinary Levels. Mol. Genet. Metab. Rep. 2017, 11, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Song, H.; Xiao, X.; Yu, J.; Yu, J.; Yang, Y.; Huang, Q.; Zang, L.; Han, T.; Zhang, D.; et al. Tectoridin Alleviates Lipopolysaccharide-Induced Inflammation via Inhibiting TLR4-NF-ΚB/NLRP3 Signaling In Vivo and In Vitro. Immunopharmacol. Immunotoxicol. 2022, 44, 641–655. [Google Scholar] [CrossRef]
- Xiang, P.; Chen, T.; Mou, Y.; Wu, H.; Xie, P.; Lu, G.; Gong, X.; Hu, Q.; Zhang, Y.; Ji, H. NZ Suppresses TLR4/NF-ΚB Signalings and NLRP3 Inflammasome Activation in LPS-Induced RAW264.7 Macrophages. Inflamm. Res. 2015, 64, 799–808. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-Mediated Neurotoxicity: Uncovering the Molecular Mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Burm, S.M.; Zuiderwijk-Sick, E.A.; Jong, A.E.J.‘t; van der Putten, C.; Veth, J.; Kondova, I.; Bajramovic, J.J. Inflammasome-Induced IL-1β Secretion in Microglia Is Characterized by Delayed Kinetics and Is Only Partially Dependent on Inflammatory Caspases. J. Neurosci. 2015, 35, 678–687. [Google Scholar] [CrossRef]
- Chen, A.-Q.; Fang, Z.; Chen, X.-L.; Yang, S.; Zhou, Y.-F.; Mao, L.; Xia, Y.-P.; Jin, H.-J.; Li, Y.-N.; You, M.-F.; et al. Microglia-Derived TNF-α Mediates Endothelial Necroptosis Aggravating Blood Brain–Barrier Disruption after Ischemic Stroke. Cell Death Dis. 2019, 10, 487. [Google Scholar] [CrossRef]
- Gong, Q.; Lin, Y.; Lu, Z.; Xiao, Z. Microglia-Astrocyte Cross Talk through IL-18/IL-18R Signaling Modulates Migraine-like Behavior in Experimental Models of Migraine. Neuroscience 2020, 451, 207–215. [Google Scholar] [CrossRef]
- Semino, C.; Carta, S.; Gattorno, M.; Sitia, R.; Rubartelli, A. Progressive Waves of IL-1β Release by Primary Human Monocytes via Sequential Activation of Vesicular and Gasdermin D-Mediated Secretory Pathways. Cell Death Dis. 2018, 9, 1088. [Google Scholar] [CrossRef]
- Navarro, S.; Debili, N.; Bernaudin, J.F.; Vainchenker, W.; Doly, J. Regulation of the Expression of IL-6 in Human Monocytes. J. Immunol. 1989, 142, 4339–4345. [Google Scholar] [CrossRef]
- Schildberger, A.; Rossmanith, E.; Eichhorn, T.; Strassl, K.; Weber, V. Monocytes, Peripheral Blood Mononuclear Cells, and THP-1 Cells Exhibit Different Cytokine Expression Patterns Following Stimulation with Lipopolysaccharide. Mediat. Inflamm. 2013, 2013, 697972. [Google Scholar] [CrossRef]
- Mussbacher, M.; Derler, M.; Basílio, J.; Schmid, J.A. NF-ΚB in Monocytes and Macrophages—An Inflammatory Master Regulator in Multitalented Immune Cells. Front. Immunol. 2023, 14, 1134661. [Google Scholar] [CrossRef]
- Könnecke, H.; Bechmann, I. The Role of Microglia and Matrix Metalloproteinases Involvement in Neuroinflammation and Gliomas. Clin. Dev. Immunol. 2013, 2013, 914104. [Google Scholar] [CrossRef]
- Albornoz, E.A.; Amarilla, A.A.; Modhiran, N.; Parker, S.; Li, X.X.; Wijesundara, D.K.; Aguado, J.; Zamora, A.P.; McMillan, C.L.D.; Liang, B.; et al. SARS-CoV-2 Drives NLRP3 Inflammasome Activation in Human Microglia through Spike Protein. Mol. Psychiatry 2023, 28, 2878–2893. [Google Scholar] [CrossRef]
- Nolan, R.A.; Reeb, K.L.; Rong, Y.; Matt, S.M.; Johnson, H.S.; Runner, K.; Gaskill, P.J. Dopamine Activates NF-ΚB and Primes the NLRP3 Inflammasome in Primary Human Macrophages. Brain Behav. Immun.-Health 2020, 2, 100030. [Google Scholar] [CrossRef]
- Newby, A.C. Metalloproteinase Expression in Monocytes and Macrophages and Its Relationship to Atherosclerotic Plaque Instability. Arter. Thromb. Vasc. Biol. 2008, 28, 2108–2114. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-ΚB Pathway for the Therapy of Diseases: Mechanism and Clinical Study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Qin, J.; Ma, Z.; Chen, X.; Shu, S. Microglia Activation in Central Nervous System Disorders: A Review of Recent Mechanistic Investigations and Development Efforts. Front. Neurol. 2023, 14, 1103416. [Google Scholar] [CrossRef]
- Visse, R.; Nagase, H. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef]
- Simonaro, C.M.; D’Angelo, M.; Haskins, M.E.; Schuchman, E.H. Joint and Bone Disease in Mucopolysaccharidoses VI and VII: Identification of New Therapeutic Targets and BioMarkers Using Animal Models. Pediatr. Res. 2005, 57 Pt 1, 701–707. [Google Scholar] [CrossRef]
- Reddy, V.S.; Prabhu, S.D.; Mummidi, S.; Valente, A.J.; Venkatesan, B.; Shanmugam, P.; Delafontaine, P.; Chandrasekar, B. Interleukin-18 Induces EMMPRIN Expression in Primary Cardiomyocytes via JNK/Sp1 Signaling and MMP-9 in Part via EMMPRIN and through AP-1 and NF-ΚB Activation. Am. J. Physiol.-Hear. Circ. Physiol. 2010, 299, H1242–H1254. [Google Scholar] [CrossRef]
- Li, W.; Li, H.; Bocking, A.D.; Challis, J.R.G. Tumor Necrosis Factor Stimulates Matrix Metalloproteinase 9 Secretion from Cultured Human Chorionic Trophoblast Cells Through TNF Receptor 1 Signaling to IKBKB-NFKB and MAPK1/3 Pathway. Biol. Reprod. 2010, 83, 481–487. [Google Scholar] [CrossRef]
- Suzuki, M.; Hashizume, M.; Yoshida, H.; Shiina, M.; Mihara, M. IL-6 and IL-1 Synergistically Enhanced the Production of MMPs from Synovial Cells by up-Regulating IL-6 Production and IL-1 Receptor I Expression. Cytokine 2010, 51, 178–183. [Google Scholar] [CrossRef]
- Goto, H.; Ishihara, Y.; Kikuchi, T.; Izawa, A.; Ozeki, N.; Okabe, E.; Kamiya, Y.; Ozawa, Y.; Mizutani, H.; Yamamoto, G.; et al. Interleukin-1 Receptor Antagonist Has a Novel Function in the Regulation of Matrix Metalloproteinase-13 Expression. PLoS ONE 2015, 10, e0140942. [Google Scholar] [CrossRef]
- Wang, F.; Guan, M.; Wei, L.; Yan, H. IL-18 Promotes the Secretion of Matrix Metalloproteinases in Human Periodontal Ligament Fibroblasts by Activating NF-ΚB Signaling. Mol. Med. Rep. 2019, 19, 703–710. [Google Scholar] [CrossRef]
- Mirastschijski, U.; Lupše, B.; Maedler, K.; Sarma, B.; Radtke, A.; Belge, G.; Dorsch, M.; Wedekind, D.; McCawley, L.J.; Boehm, G.; et al. Matrix Metalloproteinase-3 Is Key Effector of TNF-α-Induced Collagen Degradation in Skin. Int. J. Mol. Sci. 2019, 20, 5234. [Google Scholar] [CrossRef]
- Duan, L.; Ma, B.; Liang, Y.; Chen, J.; Zhu, W.; Li, M.; Wang, D. Cytokine Networking of Chondrocyte Dedifferentiation in Vitro and Its Implications for Cell-Based Cartilage Therapy. Am. J. Transl. Res. 2014, 7, 194–208. [Google Scholar]
- Zhu, H.; Yan, X.; Zhang, M.; Ji, F.; Wang, S. MiR-21-5p Protects IL-1β-Induced Human Chondrocytes from Degradation. J. Orthop. Surg. Res. 2019, 14, 118. [Google Scholar] [CrossRef]
- Buffolo, F.; Petrosino, V.; Albini, M.; Moschetta, M.; Carlini, F.; Floss, T.; de Rosbo, N.K.; Cesca, F.; Rocchi, A.; Uccelli, A.; et al. Neuroinflammation Induces Synaptic Scaling through IL-1β-Mediated Activation of the Transcriptional Repressor REST/NRSF. Cell Death Dis. 2021, 12, 180. [Google Scholar] [CrossRef]
- van Kralingen, C.; Kho, D.T.; Costa, J.; Angel, C.E.; Graham, E.S. Exposure to Inflammatory Cytokines IL-1β and TNFα Induces Compromise and Death of Astrocytes; Implications for Chronic Neuroinflammation. PLoS ONE 2013, 8, e84269. [Google Scholar] [CrossRef]
- Nakamura, S.; Otani, T.; Okura, R.; Ijiri, Y.; Motoda, R.; Kurimoto, M.; Orita, K. Expression and Responsiveness of Human Interleukin-18 Receptor (IL-18R) on Hematopoietic Cell Lines. Leukemia 2000, 14, 1052–1059. [Google Scholar] [CrossRef]
- Schneider, R.; Leven, P.; Mallesh, S.; Breßer, M.; Schneider, L.; Mazzotta, E.; Fadda, P.; Glowka, T.; Vilz, T.O.; Lingohr, P.; et al. IL-1-Dependent Enteric Gliosis Guides Intestinal Inflammation and Dysmotility and Modulates Macrophage Function. Commun. Biol. 2022, 5, 811. [Google Scholar] [CrossRef]
- Gerdes, N.; Sukhova, G.K.; Libby, P.; Reynolds, R.S.; Young, J.L.; Schönbeck, U. Expression of Interleukin (IL)-18 and Functional IL-18 Receptor on Human Vascular Endothelial Cells, Smooth Muscle Cells, and Macrophages. J. Exp. Med. 2002, 195, 245–257. [Google Scholar] [CrossRef]
- Prinz, M.; Hanisch, U. Murine Microglial Cells Produce and Respond to Interleukin-18. J. Neurochem. 1999, 72, 2215–2218. [Google Scholar] [CrossRef]
- Zhao, J.-F.; Ren, T.; Li, X.-Y.; Guo, T.-L.; Liu, C.-H.; Wang, X. Research Progress on the Role of Microglia Membrane Proteins or Receptors in Neuroinflammation and Degeneration. Front. Cell. Neurosci. 2022, 16, 831977. [Google Scholar] [CrossRef]
- Rex, D.A.B.; Agarwal, N.; Prasad, T.S.K.; Kandasamy, R.K.; Subbannayya, Y.; Pinto, S.M. A Comprehensive Pathway Map of IL-18-Mediated Signalling. J. Cell Commun. Signal. 2020, 14, 257–266. [Google Scholar] [CrossRef]
- Hirooka, Y.; Nozaki, Y. Interleukin-18 in Inflammatory Kidney Disease. Front. Med. 2021, 8, 639103. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.-K. An Update on the Regulatory Mechanisms of NLRP3 Inflammasome Activation. Cell. Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer’s Disease. Neurochem. Res. 2020, 45, 2560–2572. [Google Scholar] [CrossRef]
- Eglitis, M.A.; Mezey, É. Hematopoietic Cells Differentiate into Both Microglia and Macroglia in the Brains of Adult Mice. Proc. Natl. Acad. Sci. USA 1997, 94, 4080–4085. [Google Scholar] [CrossRef]
- Ono, K.; Takii, T.; Onozaki, K.; Ikawa, M.; Okabe, M.; Sawada, M. Migration of Exogenous Immature Hematopoietic Cells into Adult Mouse Brain Parenchyma under GFP-Expressing Bone Marrow Chimera. Biochem. Biophys. Res. Commun. 1999, 262, 610–614. [Google Scholar] [CrossRef]
- Asheuer, M.; Pflumio, F.; Benhamida, S.; Dubart-Kupperschmitt, A.; Fouquet, F.; Imai, Y.; Aubourg, P.; Cartier, N. Human CD34+ Cells Differentiate into Microglia and Express Recombinant Therapeutic Protein. Proc. Natl. Acad. Sci. USA 2004, 101, 3557–3562. [Google Scholar] [CrossRef]
- Cogle, C.R.; Yachnis, A.T.; Laywell, E.D.; Zander, D.S.; Wingard, J.R.; Steindler, D.A.; Scott, E.W. Bone Marrow Transdifferentiation in Brain after Transplantation: A Retrospective Study. Lancet 2004, 363, 1432–1437. [Google Scholar] [CrossRef]
- Araya, K.; Sakai, N.; Mohri, I.; Kagitani-Shimono, K.; Okinaga, T.; Hashii, Y.; Ohta, H.; Nakamichi, I.; Aozasa, K.; Taniike, M.; et al. Localized Donor Cells in Brain of a Hunter Disease Patient after Cord Blood Stem Cell Transplantation. Mol. Genet. Metab. 2009, 98, 255–263. [Google Scholar] [CrossRef]
- Tanaka, A.; Okuyama, T.; Suzuki, Y.; Sakai, N.; Takakura, H.; Sawada, T.; Tanaka, T.; Otomo, T.; Ohashi, T.; Ishige-Wada, M.; et al. Long-Term Efficacy of Hematopoietic Stem Cell Transplantation on Brain Involvement in Patients with Mucopolysaccharidosis Type II: A Nationwide Survey in Japan. Mol. Genet. Metab. 2012, 107, 513–520. [Google Scholar] [CrossRef]
- Yasuda, E.; Mackenzie, W.G.; Ruhnke, K.D.; Shimada, T.; Mason, R.W.; Zustin, J.; Martin, P.L.; Thacker, M.M.; Orii, T.; Sai, Y.; et al. Long-Term Follow-up of Post Hematopoietic Stem Cell Transplantation for Hurler Syndrome: Clinical, Biochemical, and Pathological Improvements. Mol. Genet. Metab. Rep. 2015, 2, 65–76. [Google Scholar] [CrossRef]
- Aldenhoven, M.; Wynn, R.F.; Orchard, P.J.; O’Meara, A.; Veys, P.; Fischer, A.; Valayannopoulos, V.; Neven, B.; Rovelli, A.; Prasad, V.K.; et al. Long-Term Outcome of Hurler Syndrome Patients after Hematopoietic Cell Transplantation: An International Multicenter Study. Blood 2015, 125, 2164–2172. [Google Scholar] [CrossRef]
- Shapiro, E.G.; Lockman, L.A.; Balthazor, M.; Krivit, W. Neuropsychological Outcomes of Several Storage Diseases with and without Bone Marrow Transplantation. J. Inherit. Metab. Dis. 1995, 18, 413–429. [Google Scholar] [CrossRef]
- Sivakumur, P.; Wraith, J.E. Bone Marrow Transplantation in Mucopolysaccharidosis Type IIIA: A Comparison of an Early Treated Patient with His Untreated Sibling. J. Inherit. Metab. Dis. 1999, 22, 849–850. [Google Scholar] [CrossRef]
- Pardridge, W.M. Recent Developments in Peptide Drug Delivery to the Brain. Pharmacol. Toxicol. 1992, 71, 3–10. [Google Scholar] [CrossRef]
- Pardridge, W.M. Neurotrophins, Neuroprotection and the Blood-Brain Barrier. Curr. Opin. Investig. Drugs 2002, 3, 1753–1757. [Google Scholar]
- Zhao, P.; Zhang, N.; An, Z. Engineering Antibody and Protein Therapeutics to Cross the Blood–Brain Barrier. Antib. Ther. 2022, 5, 311–331. [Google Scholar] [CrossRef]
- Giugliani, R.; Giugliani, L.; Poswar, F.d.O.; Donis, K.C.; Corte, A.D.; Schmidt, M.; Boado, R.J.; Nestrasil, I.; Nguyen, C.; Chen, S.; et al. Neurocognitive and Somatic Stabilization in Pediatric Patients with Severe Mucopolysaccharidosis Type I after 52 Weeks of Intravenous Brain-Penetrating Insulin Receptor Antibody-Iduronidase Fusion Protein (Valanafusp Alpha): An Open Label Phase 1-2 Trial. Orphanet J. Rare Dis. 2018, 13, 110. [Google Scholar] [CrossRef]
- Cheng, R.; Dhorajia, V.V.; Kim, J.; Kim, Y. Mitochondrial Iron Metabolism and Neurodegenerative Diseases. Neurotoxicology 2022, 88, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, T.; Eto, Y.; Sakai, N.; Minami, K.; Yamamoto, T.; Sonoda, H.; Yamaoka, M.; Tachibana, K.; Hirato, T.; Sato, Y. Iduronate-2-Sulfatase with Anti-Human Transferrin Receptor Antibody for Neuropathic Mucopolysaccharidosis II: A Phase 1/2 Trial. Mol. Ther. 2019, 27, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, T.; Eto, Y.; Sakai, N.; Nakamura, K.; Yamamoto, T.; Yamaoka, M.; Ikeda, T.; So, S.; Tanizawa, K.; Sonoda, H.; et al. A Phase 2/3 Trial of Pabinafusp Alfa, IDS Fused with Anti-Human Transferrin Receptor Antibody, Targeting Neurodegeneration in MPS-II. Mol. Ther. 2021, 29, 671–679. [Google Scholar] [CrossRef]
- Giugliani, R.; Martins, A.M.; Okuyama, T.; Eto, Y.; Sakai, N.; Nakamura, K.; Morimoto, H.; Minami, K.; Yamamoto, T.; Yamaoka, M.; et al. Enzyme Replacement Therapy with Pabinafusp Alfa for Neuronopathic Mucopolysaccharidosis II: An Integrated Analysis of Preclinical and Clinical Data. Int. J. Mol. Sci. 2021, 22, 10938. [Google Scholar] [CrossRef] [PubMed]
- Tjakra, M.; Wang, Y.; Vania, V.; Hou, Z.; Durkan, C.; Wang, N.; Wang, G. Overview of Crosstalk between Multiple Factor of Transcytosis in Blood Brain Barrier. Front. Neurosci. 2020, 13, 1436. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Chou, T. Mathematical Models of Blood-Brain Barrier Transport of Monoclonal Antibodies Targeting the Transferrin Receptor and the Insulin Receptor. Pharmaceuticals 2021, 14, 535. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-Brain Barrier Delivery for Lysosomal Storage Disorders with IgG-Lysosomal Enzyme Fusion Proteins. Adv. Drug Deliv. Rev. 2022, 184, 114234. [Google Scholar] [CrossRef] [PubMed]
- Arguello, A.; Mahon, C.S.; Calvert, M.E.K.; Chan, D.; Dugas, J.C.; Pizzo, M.E.; Thomsen, E.R.; Chau, R.; Damo, L.A.; Duque, J.; et al. Molecular Architecture Determines Brain Delivery of a Transferrin Receptor–Targeted Lysosomal Enzyme. J. Exp. Med. 2022, 219, e20211057. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Yoden, E.; Tanaka, N.; Kinoshita, M.; Imakiire, A.; Hirato, T.; Minami, K. Nonclinical Safety Evaluation of Pabinafusp Alfa, an Anti-Human Transferrin Receptor Antibody and Iduronate-2-Sulfatase Fusion Protein, for the Treatment of Neuronopathic Mucopolysaccharidosis Type II. Mol. Genet. Metab. Rep. 2021, 27, 100758. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Tomatsu, S.; Gutierrez, M.A.; Miyamoto, K.; Trandafirescu, G.G.; Lopez, P.L.C.; Grubb, J.H.; Kanai, R.; Kobayashi, H.; Yamaguchi, S.; et al. Enhancement of Drug Delivery to Bone: Characterization of Human Tissue-Nonspecific Alkaline Phosphatase Tagged with an Acidic Oligopeptide. Mol. Genet. Metab. 2006, 88, 244–255. [Google Scholar] [CrossRef]
- Montaño, A.M.; Oikawa, H.; Tomatsu, S.; Nishioka, T.; Vogler, C.; Gutierrez, M.A.; Oguma, T.; Tan, Y.; Grubb, J.H.; Dung, V.C.; et al. Acidic Amino Acid Tag Enhances Response to Enzyme Replacement in Mucopolysaccharidosis Type VII Mice. Mol. Genet. Metab. 2008, 94, 178–189. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Dung, V.C.; Ohashi, A.; Oikawa, H.; Oguma, T.; Orii, T.; Barrera, L.; Sly, W.S. Enhancement of Drug Delivery: Enzyme-Replacement Therapy for Murine Morquio A Syndrome. Mol. Ther. 2010, 18, 1094–1102. [Google Scholar] [CrossRef]
- Sawamoto, K.; Karumuthil-Melethil, S.; Khan, S.; Stapleton, M.; Bruder, J.T.; Danos, O.; Tomatsu, S. Liver-Targeted AAV8 Gene Therapy Ameliorates Skeletal and Cardiovascular Pathology in a Mucopolysaccharidosis IVA Murine Model. Mol. Ther.-Methods Clin. Dev. 2020, 18, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-Viral Vectors for Gene-Based Therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef]
- Hastie, E.; Samulski, R.J. Adeno-Associated Virus at 50: A Golden Anniversary of Discovery, Research, and Gene Therapy Success—A Personal Perspective. Hum. Gene Ther. 2015, 26, 257–265. [Google Scholar] [CrossRef]
- Rose, J.A.; Hoggan, M.D.; Shatkin, A.J. Nucleic Acid from an Adeno-Associated Virus: Chemical and Physical Studies. Proc. Natl. Acad. Sci. USA 1966, 56, 86–92. [Google Scholar] [CrossRef]
- Zhang, R.; Cao, L.; Cui, M.; Sun, Z.; Hu, M.; Zhang, R.; Stuart, W.; Zhao, X.; Yang, Z.; Li, X.; et al. Adeno-Associated Virus 2 Bound to Its Cellular Receptor AAVR. Nat. Microbiol. 2019, 4, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yang, H.; Colosi, P. Effect of Genome Size on AAV Vector Packaging. Mol. Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef]
- Berns, K.I.; Linden, R.M. The Cryptic Life Style of Adenoassociated Virus. Bioessays 1995, 17, 237–245. [Google Scholar] [CrossRef]
- Choi, V.W.; McCarty, D.M.; Samulski, R.J. Host Cell DNA Repair Pathways in Adeno-Associated Viral Genome Processing. J. Virol. 2006, 80, 10346–10356. [Google Scholar] [CrossRef]
- Nakai, H.; Yant, S.R.; Storm, T.A.; Fuess, S.; Meuse, L.; Kay, M.A. Extrachromosomal Recombinant Adeno-Associated Virus Vector Genomes Are Primarily Responsible for Stable Liver Transduction In Vivo. J. Virol. 2001, 75, 6969–6976. [Google Scholar] [CrossRef] [PubMed]
- Gil-Farina, I.; Fronza, R.; Kaeppel, C.; Lopez-Franco, E.; Ferreira, V.; D’Avola, D.; Benito, A.; Prieto, J.; Petry, H.; Gonzalez-Aseguinolaza, G.; et al. Recombinant AAV Integration Is Not Associated with Hepatic Genotoxicity in Nonhuman Primates and Patients. Mol. Ther. 2016, 24, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Conese, M.; Auriche, C.; Ascenzioni, F. Gene Therapy Progress and Prospects: Episomally Maintained Self-Replicating Systems. Gene Ther. 2004, 11, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Donsante, A.; Miller, D.G.; Li, Y.; Vogler, C.; Brunt, E.M.; Russell, D.W.; Sands, M.S. AAV Vector Integration Sites in Mouse Hepatocellular Carcinoma. Science 2007, 317, 477. [Google Scholar] [CrossRef] [PubMed]
- Bell, P.; Wang, L.; Lebherz, C.; Flieder, D.B.; Bove, M.S.; Wu, D.; Gao, G.P.; Wilson, J.M.; Wivel, N.A. No Evidence for Tumorigenesis of AAV Vectors in a Large-Scale Study in Mice. Mol. Ther. 2005, 12, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Bell, P.; Moscioni, A.D.; McCarter, R.J.; Wu, D.; Gao, G.; Hoang, A.; Sanmiguel, J.C.; Sun, X.; Wivel, N.A.; Raper, S.E.; et al. Analysis of Tumors Arising in Male B6C3F1 Mice with and without AAV Vector Delivery to Liver. Mol. Ther. 2006, 14, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Rosas, L.E.; Grieves, J.L.; Zaraspe, K.; Perle, K.M.L.; Fu, H.; McCarty, D.M. Patterns of ScAAV Vector Insertion Associated with Oncogenic Events in a Mouse Model for Genotoxicity. Mol. Ther. 2012, 20, 2098–2110. [Google Scholar] [CrossRef]
- Walia, J.S.; Altaleb, N.; Bello, A.; Kruck, C.; LaFave, M.C.; Varshney, G.K.; Burgess, S.M.; Chowdhury, B.; Hurlbut, D.; Hemming, R.; et al. Long-Term Correction of Sandhoff Disease Following Intravenous Delivery of RAAV9 to Mouse Neonates. Mol. Ther. 2015, 23, 414–422. [Google Scholar] [CrossRef]
- Kao, C.-Y.; Yang, S.-J.; Tao, M.-H.; Jeng, Y.-M.; Yu, I.-S.; Lin, S.-W. Incorporation of the Factor IX Padua Mutation into FIX-Triple Improves Clotting Activity in Vitro and in Vivo. Thromb. Haemost. 2013, 110, 244–256. [Google Scholar] [CrossRef]
- Chandler, R.J.; LaFave, M.C.; Varshney, G.K.; Trivedi, N.S.; Carrillo-Carrasco, N.; Senac, J.S.; Wu, W.; Hoffmann, V.; Elkahloun, A.G.; Burgess, S.M.; et al. Vector Design Influences Hepatic Genotoxicity after Adeno-Associated Virus Gene Therapy. J. Clin. Investig. 2015, 125, 870–880. [Google Scholar] [CrossRef]
- Li, H.; Malani, N.; Hamilton, S.R.; Schlachterman, A.; Bussadori, G.; Edmonson, S.E.; Shah, R.; Arruda, V.R.; Mingozzi, F.; Wright, J.F.; et al. Assessing the Potential for AAV Vector Genotoxicity in a Murine Model. Blood 2011, 117, 3311–3319. [Google Scholar] [CrossRef] [PubMed]
- Fraldi, A.; Serafini, M.; Sorrentino, N.C.; Gentner, B.; Aiuti, A.; Bernardo, M.E. Gene Therapy for Mucopolysaccharidoses: In Vivo and Ex Vivo Approaches. Ital. J. Pediatr. 2018, 44 (Suppl. S2), 130. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, K.; Chen, H.-H.; Alméciga-Díaz, C.J.; Mason, R.W.; Tomatsu, S. Gene Therapy for Mucopolysaccharidoses. Mol. Genet. Metab. 2017, 123, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, F.K.; Samulski, T.; Shenk, T.; Samulski, R.J. Second-Strand Synthesis Is a Rate-Limiting Step for Efficient Transduction by Recombinant Adeno-Associated Virus Vectors. J. Virol. 1996, 70, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- McCarty, D.; Monahan, P.; Samulski, R. Self-Complementary Recombinant Adeno-Associated Virus (ScAAV) Vectors Promote Efficient Transduction Independently of DNA Synthesis. Gene Ther. 2001, 8, 1248–1254. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhao, W.; Zhong, L.; Han, Z.; Li, B.; Ma, W.; Weigel-Kelley, K.A.; Warrington, K.H.; Srivastava, A. Self-Complementary Recombinant Adeno-Associated Viral Vectors Packaging Capacity And The Role of Rep Proteins in Vector Purity. Hum. Gene Ther. 2007, 18, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Duncan, F.J.; Naughton, B.J.; Zaraspe, K.; Murrey, D.A.; Meadows, A.S.; Clark, K.R.; Newsom, D.E.; White, P.; Fu, H.; McCarty, D.M. Broad Functional Correction of Molecular Impairments by Systemic Delivery of ScAAVrh74-HSGSH Gene Delivery in MPS IIIA Mice. Mol. Ther. 2015, 23, 638–647. [Google Scholar] [CrossRef]
- Fu, H.; Cataldi, M.P.; Ware, T.A.; Zaraspe, K.; Meadows, A.S.; Murrey, D.A.; McCarty, D.M. Functional Correction of Neurological and Somatic Disorders at Later Stages of Disease in MPS IIIA Mice by Systemic ScAAV9-HSGSH Gene Delivery. Mol. Ther.-Methods Clin. Dev. 2016, 3, 16036. [Google Scholar] [CrossRef]
- Bobo, T.A.; Samowitz, P.N.; Robinson, M.I.; Fu, H. Targeting the Root Cause of Mucopolysaccharidosis IIIA with a New ScAAV9 Gene Replacement Vector. Mol. Ther.-Methods Clin. Dev. 2020, 19, 474–485. [Google Scholar] [CrossRef]
- Saville, J.T.; Derrick-Roberts, A.L.K.; McIntyre, C.; Fuller, M. Systemic ScAAV9.U1a.HSGSH Delivery Corrects Brain Biochemistry in Mucopolysaccharidosis Type IIIA at Early and Later Stages of Disease. Hum. Gene Ther. 2021, 32, 420–430. [Google Scholar] [CrossRef]
- Mori, S.; Wang, L.; Takeuchi, T.; Kanda, T. Two Novel Adeno-Associated Viruses from Cynomolgus Monkey: Pseudotyping Characterization of Capsid Protein. Virology 2004, 330, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Michelfelder, S.; Trepel, M. Chapter 2 Adeno-Associated Viral Vectors and Their Redirection to Cell-Type Specific Receptors. Adv. Genet. 2009, 67, 29–60. [Google Scholar] [CrossRef]
- Kern, A.; Schmidt, K.; Leder, C.; Müller, O.J.; Wobus, C.E.; Bettinger, K.; der Lieth, C.W.V.; King, J.A.; Kleinschmidt, J.A. Identification of a Heparin-Binding Motif on Adeno-Associated Virus Type 2 Capsids. J. Virol. 2003, 77, 11072–11081. [Google Scholar] [CrossRef]
- Boyle, M.P.; Enke, R.A.; Reynolds, J.B.; Mogayzel, P.J.; Guggino, W.B.; Zeitlin, P.L. Membrane-Associated Heparan Sulfate Is Not Required for RAAV-2 Infection of Human Respiratory Epithelia. Virol. J. 2006, 3, 29. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Handa, A.; Muramatsu, S.; Qiu, J.; Mizukami, H.; Brown, K.E. Adeno-Associated Virus (AAV)-3-Based Vectors Transduce Haematopoietic Cells Not Susceptible to Transduction with AAV-2-Based Vectors. J. Gen. Virol. 2000, 81, 2077–2084. [Google Scholar] [CrossRef] [PubMed]
- Asokan, A.; Schaffer, D.V.; Samulski, R.J. The AAV Vector Toolkit: Poised at the Clinical Crossroads. Mol. Ther. 2012, 20, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Alméciga-Díaz, C.J.; Montaño, A.M.; Barrera, L.A.; Tomatsu, S. Tailoring the AAV2 Capsid Vector for Bone-Targeting. Pediatr. Res. 2018, 84, 545–551. [Google Scholar] [CrossRef]
- Fu, H.; Meadows, A.S.; Pineda, R.J.; Kunkler, K.L.; Truxal, K.V.; McBride, K.L.; Flanigan, K.M.; McCarty, D.M. Differential Prevalence of Antibodies Against Adeno-Associated Virus in Healthy Children and Patients with Mucopolysaccharidosis III: Perspective for AAV-Mediated Gene Therapy. Hum. Gene Ther. Clin. Dev. 2017, 28, 187–196. [Google Scholar] [CrossRef]
- Mingozzi, F.; High, K.A. Immune Responses to AAV Vectors: Overcoming Barriers to Successful Gene Therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Halbert, C.L.; Standaert, T.A.; Aitken, M.L.; Alexander, I.E.; Russell, D.W.; Miller, A.D. Transduction by Adeno-Associated Virus Vectors in the Rabbit Airway: Efficiency, Persistence, and Readministration. J. Virol. 1997, 71, 5932–5941. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, T.; Qiao, C.; Zhou, L.; Wang, B.; Zhang, J.; Chen, C.; Li, J.; Xiao, X. Adeno-Associated Virus Serotype 8 Efficiently Delivers Genes to Muscle and Heart. Nat. Biotechnol. 2005, 23, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, S.C.; Dane, A.P.; Spinoulas, A.; Logan, G.J.; Alexander, I.E. Gene Delivery to the Juvenile Mouse Liver Using AAV2/8 Vectors. Mol. Ther. 2008, 16, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, V.M.; Meadows, A.S.; Pineda, R.J.; Camboni, M.; McCarty, D.M.; Fu, H. Effective Depletion of Pre-Existing Anti-AAV Antibodies Requires Broad Immune Targeting. Mol. Ther.-Methods Clin. Dev. 2017, 4, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Meliani, A.; Boisgerault, F.; Hardet, R.; Marmier, S.; Collaud, F.; Ronzitti, G.; Leborgne, C.; Verdera, H.C.; Sola, M.S.; Charles, S.; et al. Antigen-Selective Modulation of AAV Immunogenicity with Tolerogenic Rapamycin Nanoparticles Enables Successful Vector Re-Administration. Nat. Commun. 2018, 9, 4098. [Google Scholar] [CrossRef] [PubMed]
- Retroviruses; Coffin, J., Hughes, S., Varmus, H., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Blaese, R.M.; Culver, K.W.; Miller, A.D.; Carter, C.S.; Fleisher, T.; Clerici, M.; Shearer, G.; Chang, L.; Chiang, Y.; Tolstoshev, P.; et al. T Lymphocyte-Directed Gene Therapy for ADA− SCID: Initial Trial Results After 4 Years. Science 1995, 270, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana, M.; Six, E.; Lagresle-Peyrou, C.; André-Schmutz, I.; Hacein-Bey-Abina, S. Gene Therapy for X-Linked Severe Combined Immunodeficiency: Where Do We Stand? Hum. Gene Ther. 2016, 27, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional Mutagenesis Combined with Acquired Somatic Mutations Causes Leukemogenesis Following Gene Therapy of SCID-X1 Patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional Oncogenesis in 4 Patients after Retrovirus-Mediated Gene Therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The Genotoxic Potential of Retroviral Vectors Is Strongly Modulated by Vector Design and Integration Site Selection in a Mouse Model of HSC Gene Therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef]
- Zychlinski, D.; Schambach, A.; Modlich, U.; Maetzig, T.; Meyer, J.; Grassman, E.; Mishra, A.; Baum, C. Physiological Promoters Reduce the Genotoxic Risk of Integrating Gene Vectors. Mol. Ther. 2008, 16, 718–725. [Google Scholar] [CrossRef]
- Thornhill, S.I.; Schambach, A.; Howe, S.J.; Ulaganathan, M.; Grassman, E.; Williams, D.; Schiedlmeier, B.; Sebire, N.J.; Gaspar, H.B.; Kinnon, C.; et al. Self-Inactivating Gammaretroviral Vectors for Gene Therapy of X-Linked Severe Combined Immunodeficiency. Mol. Ther. 2008, 16, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Throm, R.E.; Ouma, A.A.; Zhou, S.; Chandrasekaran, A.; Lockey, T.; Greene, M.; Ravin, S.S.D.; Moayeri, M.; Malech, H.L.; Sorrentino, B.P.; et al. Efficient Construction of Producer Cell Lines for a SIN Lentiviral Vector for SCID-X1 Gene Therapy by Concatemeric Array Transfection. Blood 2009, 113, 5104–5110. [Google Scholar] [CrossRef]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In Vivo Gene Delivery and Stable Transduction of Nondividing Cells by a Lentiviral Vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Matta, H.; Hozayev, B.; Tomar, R.; Chugh, P.; Chaudhary, P.M. Use of Lentiviral Vectors for Delivery of Small Interfering RNA. Cancer Biol. Ther. 2003, 2, 206–210. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Delenda, C. Lentiviral Vectors: Optimization of Packaging, Transduction and Gene Expression. J. Gene Med. 2004, 6 (Suppl. S1), S125–S138. [Google Scholar] [CrossRef] [PubMed]
- Anson, D.S.; Bielicki, J.; Hopwood, J.J. Correction of Mucopolysaccharidosis Type I Fibroblasts by Retroviral-Mediated Transfer of the Human α-l-Iduronidase Gene. Hum. Gene Ther. 1992, 3, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Rintz, E.; Higuchi, T.; Kobayashi, H.; Galileo, D.S.; Wegrzyn, G.; Tomatsu, S. Promoter Considerations in the Design of Lentiviral Vectors for Use in Treating Lysosomal Storage Diseases. Mol. Ther.-Methods Clin. Dev. 2022, 24, 71–87. [Google Scholar] [CrossRef]
- Gentner, B.; Tucci, F.; Galimberti, S.; Fumagalli, F.; Pellegrin, M.D.; Silvani, P.; Camesasca, C.; Pontesilli, S.; Darin, S.; Ciotti, F.; et al. Hematopoietic Stem- and Progenitor-Cell Gene Therapy for Hurler Syndrome. N. Engl. J. Med. 2021, 385, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten Things You Should Know about Transposable Elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Hou, X.; Du, Y.; Deng, Y.; Wu, J.; Cao, G. Sleeping Beauty Transposon System for Genetic Etiological Research and Gene Therapy of Cancers. Cancer Biol. Ther. 2015, 16, 8–16. [Google Scholar] [CrossRef][Green Version]
- Kebriaei, P.; Izsvák, Z.; Narayanavari, S.A.; Singh, H.; Ivics, Z. Gene Therapy with the Sleeping Beauty Transposon System. Trends Genet. 2017, 33, 852–870. [Google Scholar] [CrossRef]
- Ye, L.; Lam, S.Z.; Yang, L.; Suzuki, K.; Zou, Y.; Lin, Q.; Zhang, Y.; Clark, P.; Peng, L.; Chen, S. AAV-Mediated Delivery of a Sleeping Beauty Transposon and an MRNA-Encoded Transposase for the Engineering of Therapeutic Immune Cells. Nat. Biomed. Eng. 2023. [Google Scholar] [CrossRef] [PubMed]
- Harmening, N.; Johnen, S.; Izsvák, Z.; Ivics, Z.; Kropp, M.; Bascuas, T.; Walter, P.; Kreis, A.; Pajic, B.; Thumann, G. Enhanced Biosafety of the Sleeping Beauty Transposon System by Using MRNA as Source of Transposase to Efficiently and Stably Transfect Retinal Pigment Epithelial Cells. Biomolecules 2023, 13, 658. [Google Scholar] [CrossRef] [PubMed]
- Mattern, L.; Otten, K.; Miskey, C.; Fuest, M.; Izsvák, Z.; Ivics, Z.; Walter, P.; Thumann, G.; Johnen, S. Molecular and Functional Characterization of BDNF-Overexpressing Human Retinal Pigment Epithelial Cells Established by Sleeping Beauty Transposon-Mediated Gene Transfer. Int. J. Mol. Sci. 2022, 23, 12982. [Google Scholar] [CrossRef]
- Aronovich, E.L.; Bell, J.B.; Belur, L.R.; Gunther, R.; Koniar, B.; Erickson, D.C.C.; Schachern, P.A.; Matise, I.; McIvor, R.S.; Whitley, C.B.; et al. Prolonged Expression of a Lysosomal Enzyme in Mouse Liver after Sleeping Beauty Transposon-mediated Gene Delivery: Implications for Non-viral Gene Therapy of Mucopolysaccharidoses. J. Gene Med. 2007, 9, 403–415. [Google Scholar] [CrossRef]
- Aronovich, E.L.; Bell, J.B.; Khan, S.A.; Belur, L.R.; Gunther, R.; Koniar, B.; Schachern, P.A.; Parker, J.B.; Carlson, C.S.; Whitley, C.B.; et al. Systemic Correction of Storage Disease in MPS I NOD/SCID Mice Using the Sleeping Beauty Transposon System. Mol. Ther. 2009, 17, 1136–1144. [Google Scholar] [CrossRef]
- Aronovich, E.L.; Hall, B.C.; Bell, J.B.; McIvor, R.S.; Hackett, P.B. Quantitative Analysis of α-L-Iduronidase Expression in Immunocompetent Mice Treated with the Sleeping Beauty Transposon System. PLoS ONE 2013, 8, e78161. [Google Scholar] [CrossRef] [PubMed]
- Aronovich, E.L.; Hyland, K.A.; Hall, B.C.; Bell, J.B.; Olson, E.R.; Rusten, M.U.; Hunter, D.W.; Ellinwood, N.M.; McIvor, R.S.; Hackett, P.B. Prolonged Expression of Secreted Enzymes in Dogs After Liver-Directed Delivery of Sleeping Beauty Transposons: Implications for Non-Viral Gene Therapy of Systemic Disease. Hum. Gene Ther. 2017, 28, 551–564. [Google Scholar] [CrossRef]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S.; et al. Phase I Trials Using Sleeping Beauty to Generate CD19-Specific CAR T Cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef]
- Chicaybam, L.; Abdo, L.; Viegas, M.; Marques, L.V.C.; de Sousa, P.; Batista-Silva, L.R.; Alves-Monteiro, V.; Bonecker, S.; Monte-Mór, B.; Bonamino, M.H. Transposon-Mediated Generation of CAR-T Cells Shows Efficient Anti B-Cell Leukemia Response after Ex Vivo Expansion. Gene Ther. 2020, 27, 85–95. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and Function of NF-ΚB Transcription Factors in the Immune System. Immunology 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology, 10th ed.; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Eliyahu, E.; Wolfson, T.; Ge, Y.; Jepsen, K.J.; Schuchman, E.H.; Simonaro, C.M. Anti-TNF-Alpha Therapy Enhances the Effects of Enzyme Replacement Therapy in Rats with Mucopolysaccharidosis Type VI. PLoS ONE 2011, 6, e22447. [Google Scholar] [CrossRef] [PubMed]
- Polgreen, L.E.; Kunin-Batson, A.; Rudser, K.; Vehe, R.K.; Utz, J.J.; Whitley, C.B.; Dickson, P. Pilot Study of the Safety and Effect of Adalimumab on Pain, Physical Function, and Musculoskeletal Disease in Mucopolysaccharidosis Types I and II. Mol. Genet. Metab. Rep. 2017, 10, 75–80. [Google Scholar] [CrossRef]
- Müller-Miny, L.; Heming, M.; Lautwein, T.; Ruck, T.; Lu, I.-N.; Wiendl, H.; Hörste, G.M.Z. Alemtuzumab Treatment Exemplifies Discordant Immune Effects of Blood and Cerebrospinal Fluid in Multiple Sclerosis. J. Neuroimmunol. 2023, 378, 578088. [Google Scholar] [CrossRef]
- Torres-Acosta, N.; O’Keefe, J.H.; O’Keefe, E.L.; Isaacson, R.; Small, G. Therapeutic Potential of TNF-α Inhibition for Alzheimer’s Disease Prevention. J. Alzheimer’s Dis. 2020, 78, 619–626. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Broadwell, R.D. Passage of Cytokines across the Blood-Brain Barrier. Neuroimmunomodulation 1995, 2, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Sjöström, E.O.; Culot, M.; Leickt, L.; Åstrand, M.; Nordling, E.; Gosselet, F.; Kaiser, C. Transport Study of Interleukin-1 Inhibitors Using a Human in Vitro Model of the Blood-Brain Barrier. Brain Behav. Immun.-Heal. 2021, 16, 100307. [Google Scholar] [CrossRef]
- Cliteur, M.; van der Kolk, A.; Hannink, G.; Hofmeijer, J.; Jolink, W.; Klijn, C.; Schreuder, F. Anakinra in Cerebral Haemorrhage to Target Secondary Injury Resulting from Neuroinflammation (ACTION): Study Protocol of a Phase II Randomised Clinical Trial. Eur. Stroke J. 2023, 23969873231200690. [Google Scholar] [CrossRef]
- Rintz, E.; Pierzynowska, K.; Podlacha, M.; Węgrzyn, G. Has Resveratrol a Potential for Mucopolysaccharidosis Treatment? Eur. J. Pharmacol. 2020, 888, 173534. [Google Scholar] [CrossRef]
- Bar, S.; Prasad, M.; Datta, R. Neuromuscular Degeneration and Locomotor Deficit in a Drosophila Model of Mucopolysaccharidosis VII Is Attenuated by Treatment with Resveratrol. Dis. Model. Mech. 2018, 11, dmm036954. [Google Scholar] [CrossRef]
- Rintz, E.; Podlacha, M.; Cyske, Z.; Pierzynowska, K.; Węgrzyn, G.; Gaffke, L. Activities of (Poly)Phenolic Antioxidants and Other Natural Autophagy Modulators in the Treatment of Sanfilippo Disease: Remarkable Efficacy of Resveratrol in Cellular and Animal Models. Neurotherapeutics 2023, 20, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Montero, R.; Yubero, D.; Salgado, M.C.; González, M.J.; Campistol, J.; O’Callaghan, M.d.M.; Pineda, M.; Delgadillo, V.; Maynou, J.; Fernandez, G.; et al. Plasma Coenzyme Q10 Status Is Impaired in Selected Genetic Conditions. Sci. Rep. 2019, 9, 793. [Google Scholar] [CrossRef] [PubMed]
- Jacques, C.E.D.; Lopes, F.F.; Poletto, E.; Vera, L.N.P.; Vianna, P.; Reinhardt, L.S.; Baldo, G.; Vargas, C.R. Evaluation of Oxidative Stress and Mitochondrial Function in a Type II Mucopolysaccharidosis Cellular Model: In Vitro Effects of Genistein and Coenzyme Q10. Metab. Brain Dis. 2023, 38, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Gabandé-Rodríguez, E.; Pérez-Cañamás, A.; Soto-Huelin, B.; Mitroi, D.N.; Sánchez-Redondo, S.; Martínez-Sáez, E.; Venero, C.; Peinado, H.; Ledesma, M.D. Lipid-induced Lysosomal Damage after Demyelination Corrupts Microglia Protective Function in Lysosomal Storage Disorders. EMBO J. 2019, 38, e99553. [Google Scholar] [CrossRef]
- David, A.; Chazeirat, T.; Saidi, A.; Lalmanach, G.; Lecaille, F. The Interplay of Glycosaminoglycans and Cysteine Cathepsins in Mucopolysaccharidosis. Biomedicines 2023, 11, 810. [Google Scholar] [CrossRef]
- Baldo, G.; Tavares, A.M.V.; Gonzalez, E.; Poletto, E.; Mayer, F.Q.; Matte, U.d.S.; Giugliani, R. Progressive Heart Disease in Mucopolysaccharidosis Type I Mice May Be Mediated by Increased Cathepsin B Activity. Cardiovasc. Pathol. 2017, 27, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.A.; Martins, G.R.; Tavares, A.M.V.; Viegas, M.; Poletto, E.; Giugliani, R.; Matte, U.; Baldo, G. Cathepsin B Inhibition Attenuates Cardiovascular Pathology in Mucopolysaccharidosis I Mice. Life Sci. 2018, 196, 102–109. [Google Scholar] [CrossRef]
- Ghosh, P.; Wu, J.; Shimmon, S.; Zannettino, A.C.; Gronthos, S.; Itescu, S. Pentosan Polysulfate Promotes Proliferation and Chondrogenic Differentiation of Adult Human Bone Marrow-Derived Mesenchymal Precursor Cells. Arthritis Res. Ther. 2010, 12, R28. [Google Scholar] [CrossRef]
- Smith, M.M.; Melrose, J. Pentosan Polysulfate Affords Pleotropic Protection to Multiple Cells and Tissues. Pharmaceuticals 2023, 16, 437. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; DeAngelis, V.; Zhu, C.; Schuchman, E.H.; Simonaro, C.M. JIMD Reports, Volume 43. JIMD Rep. 2018, 43, 37–52. [Google Scholar] [CrossRef]
- Frohbergh, M.; Ge, Y.; Meng, F.; Karabul, N.; Solyom, A.; Lai, A.; Iatridis, J.; Schuchman, E.H.; Simonaro, C.M. Dose Responsive Effects of Subcutaneous Pentosan Polysulfate Injection in Mucopolysaccharidosis Type VI Rats and Comparison to Oral Treatment. PLoS ONE 2014, 9, e100882. [Google Scholar] [CrossRef] [PubMed]
- Simonaro, C.M.; Tomatsu, S.; Sikora, T.; Kubaski, F.; Frohbergh, M.; Guevara, J.M.; Wang, R.Y.; Vera, M.; Kang, J.L.; Smith, L.J.; et al. Pentosan Polysulfate: Oral Versus Subcutaneous Injection in Mucopolysaccharidosis Type I Dogs. PLoS ONE 2016, 11, e0153136. [Google Scholar] [CrossRef] [PubMed]
- Hennermann, J.B.; Gökce, S.; Solyom, A.; Mengel, E.; Schuchman, E.H.; Simonaro, C.M. Treatment with Pentosan Polysulphate in Patients with MPS I: Results from an Open Label, Randomized, Monocentric Phase II Study. J. Inherit. Metab. Dis. 2016, 39, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Orii, K.; Lim, A.; Tomatsu, S.; Stapleton, M.; Suzuki, Y.; Simonaro, C.M.; Schuchman, E.H.; Fukao, T.; Matsumoto, T. Safety Study of Sodium Pentosan Polysulfate for Adult Patients with Mucopolysaccharidosis Type II. Diagnostics 2019, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Guo, Q.; Zhu, Q.; Tan, R.; Bai, D.; Bu, X.; Lin, B.; Zhao, K.; Pan, C.; Chen, H.; et al. Flavonoid VI-16 Protects against DSS-Induced Colitis by Inhibiting Txnip-Dependent NLRP3 Inflammasome Activation in Macrophages via Reducing Oxidative Stress. Mucosal Immunol. 2019, 12, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Leon-Oliva, D.D.; García-Montero, C.; Fraile-Martinez, O.; Boaru, D.L.; de Castro, A.V.; Saez, M.A.; Lopez-Gonzalez, L.; Bujan, J.; Alvarez-Mon, M.A.; et al. Reframing the Link between Metabolism and NLRP3 Inflammasome: Therapeutic Opportunities. Front. Immunol. 2023, 14, 1232629. [Google Scholar] [CrossRef] [PubMed]
- Ghayur, T.; Banerjee, S.; Hugunin, M.; Butler, D.; Herzog, L.; Carter, A.; Quintal, L.; Sekut, L.; Talanian, R.; Paskind, M.; et al. Caspase-1 Processes IFN-γ-Inducing Factor and Regulates LPS-Induced IFN-γ Production. Nature 1997, 386, 619–623. [Google Scholar] [CrossRef]
- Amand, M.; Adams, P.; Schober, R.; Iserentant, G.; Servais, J.-Y.; Moutschen, M.; Seguin-Devaux, C. The Anti-Caspase 1 Inhibitor VX-765 Reduces Immune Activation, CD4+ T Cell Depletion, Viral Load, and Total HIV-1 DNA in HIV-1 Infected Humanized Mice. eLife 2023, 12, e83207. [Google Scholar] [CrossRef]
- Jiao, M.; Wang, J.; Liu, W.; Zhao, X.; Qin, Y.; Zhang, C.; Yin, H.; Zhao, C. VX-765 Inhibits Pyroptosis and Reduces Inflammation to Prevent Acute Liver Failure by Upregulating PPARα Expression. Ann. Hepatol. 2023, 28, 101082. [Google Scholar] [CrossRef]
- Qian, L.; Zhang, C.; Wu, J.; Yao, S.Q. Fused Bicyclic Caspase-1 Inhibitors Assembled by Copper-Free Strain-Promoted Alkyne–Azide Cycloaddition (SPAAC). Chem. A Eur. J. 2017, 23, 360–369. [Google Scholar] [CrossRef]
- Dong, L.; Qiao, H.; Zhang, X.; Zhang, X.; Wang, C.; Wang, L.; Cui, L.; Zhao, J.; Xing, Y.; Li, Y.; et al. Parthenolide Is Neuroprotective in Rat Experimental Stroke Model: Downregulating NF-κB, Phospho-P38MAPK, and Caspase-1 and Ameliorating BBB Permeability. Mediat. Inflamm. 2013, 2013, 370804. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Sun, Q.; Zhong, X.; Zeng, M.; Zeng, H.; Shi, X.; Li, Z.; Wang, Y.; Zhao, Q.; Shao, F.; et al. Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell 2020, 180, 941–955.e20. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Sun, Y.; Liu, S.; Yang, S.; Chen, C.; Zhang, Z.; Chu, S.; Yang, Y.; Pei, G.; Lin, M.; et al. Targeting Pyroptosis as a Preventive and Therapeutic Approach for Stroke. Cell Death Discov. 2023, 9, 155. [Google Scholar] [CrossRef]
- Jacobsen, E.; Martensen-Larsen, O. Treatment of alcoholism with tetraethylthiuram disulfide (Antabus®). J. Am. Med. Assoc. 1949, 139, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, J.; Zhao, J.; Chen, S.; Zhou, T.; Xu, J. Treatment of Severe Acute Pancreatitis and Related Lung Injury by Targeting Gasdermin D-Mediated Pyroptosis. Front. Cell Dev. Biol. 2021, 9, 780142. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Chen, S.; Li, C.; Xu, J.; Wang, L. Application of Disulfiram and Its Metabolites in Treatment of Inflammatory Disorders. Front. Pharmacol. 2022, 12, 795078. [Google Scholar] [CrossRef] [PubMed]
- Takashima, K.; Matsunaga, N.; Yoshimatsu, M.; Hazeki, K.; Kaisho, T.; Uekata, M.; Hazeki, O.; Akira, S.; Iizawa, Y.; Ii, M. Analysis of Binding Site for the Novel Small-molecule TLR4 Signal Transduction Inhibitor TAK-242 and Its Therapeutic Effect on Mouse Sepsis Model. Br. J. Pharmacol. 2009, 157, 1250–1262. [Google Scholar] [CrossRef]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (Resatorvid), a Small-Molecule Inhibitor of Toll-Like Receptor (TLR) 4 Signaling, Binds Selectively to TLR4 and Interferes with Interactions between TLR4 and Its Adaptor Molecules. Mol. Pharmacol. 2011, 79, 34–41. [Google Scholar] [CrossRef]
- Yu, R.; Ma, C.; Li, G.; Xu, J.; Feng, D.; Lan, X. Inhibition of Toll-Like Receptor 4 Signaling Pathway Accelerates the Repair of Avascular Necrosis of Femoral Epiphysis through Regulating Macrophage Polarization in Perthes Disease. Tissue Eng. Regen. Med. 2023, 20, 489–501. [Google Scholar] [CrossRef]
- Feng, Y.; Ju, Y.; Wu, Q.; Sun, G.; Yan, Z. TAK-242, a Toll-like Receptor 4 Antagonist, against Brain Injury by Alleviates Autophagy and Inflammation in Rats. Open Life Sci. 2023, 18, 20220662. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, Q.; Luo, Q.; Wang, L.; Chen, J.; Xiong, Y.; Wu, G.; Chang, L.; Liu, P.; Shu, H. A Single Dose of Ketamine Relieves Fentanyl-Induced-Hyperalgesia by Reducing Inflammation Initiated by the TLR4/NF-ΚB Pathway in Rat Spinal Cord Neurons. Drug Discov. Ther. 2023, 17, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Rice, T.W.; Wheeler, A.P.; Bernard, G.R.; Vincent, J.-L.; Angus, D.C.; Aikawa, N.; Demeyer, I.; Sainati, S.; Amlot, N.; Cao, C.; et al. A Randomized, Double-Blind, Placebo-Controlled Trial of TAK-242 for the Treatment of Severe Sepsis. Crit. Care Med. 2010, 38, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-L.; Yin, H.-R.; He, Q.-Y.; Wang, Y. Targeting the NLRP3 Inflammasome as New Therapeutic Avenue for Inflammatory Bowel Disease. Biomed. Pharmacother. 2021, 138, 111442. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.; Billinton, A.; Bock, M.G.; Doedens, J.R.; Gabel, C.A.; Holloway, M.K.; Porter, R.A.; Reader, V.; Scanlon, J.; Schooley, K.; et al. Discovery of Clinical Candidate NT-0796, a Brain-Penetrant and Highly Potent NLRP3 Inflammasome Inhibitor for Neuroinflammatory Disorders. J. Med. Chem. 2023, 66, 14897–14911. [Google Scholar] [CrossRef]
- Hou, B.; Zhang, Y.; Liang, P.; He, Y.; Peng, B.; Liu, W.; Han, S.; Yin, J.; He, X. Inhibition of the NLRP3-Inflammasome Prevents Cognitive Deficits in Experimental Autoimmune Encephalomyelitis Mice via the Alteration of Astrocyte Phenotype. Cell Death Dis. 2020, 11, 377. [Google Scholar] [CrossRef] [PubMed]
- Haddad, J.J.; Abdel-Karim, N.E. NF-ΚB Cellular and Molecular Regulatory Mechanisms and Pathways: Therapeutic Pattern or Pseudoregulation? Cell. Immunol. 2011, 271, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting Edge: NF-ΚB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Pierce, J.W.; Schoenleber, R.; Jesmok, G.; Best, J.; Moore, S.A.; Collins, T.; Gerritsen, M.E. Novel Inhibitors of Cytokine-Induced IκBα Phosphorylation and Endothelial Cell Adhesion Molecule Expression Show Anti-Inflammatory Effects in Vivo. J. Biol. Chem. 1997, 272, 21096–21103. [Google Scholar] [CrossRef]
- Mori, N.; Yamada, Y.; Ikeda, S.; Yamasaki, Y.; Tsukasaki, K.; Tanaka, Y.; Tomonaga, M.; Yamamoto, N.; Fujii, M. Bay 11-7082 Inhibits Transcription Factor NF-ΚB and Induces Apoptosis of HTLV-I–Infected T-Cell Lines and Primary Adult T-Cell Leukemia Cells. Blood 2002, 100, 1828–1834. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.-W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-Inflammatory Compounds Parthenolide and Bay 11-7082 Are Direct Inhibitors of the Inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Li, X.; Wang, C.; Gu, S.; Dai, L.; Zhang, J.; Fan, Y.; Wu, J. Repeated Neonatal Sevoflurane Induced Neurocognitive Impairment through NF-ΚB-Mediated Pyroptosis. J. Neuroinflamm. 2021, 18, 180. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, S.A.; Casili, G.; Basilotta, R.; Lanza, M.; Filippone, A.; Raciti, G.; Puliafito, I.; Colarossi, L.; Esposito, E.; Paterniti, I. NLRP3 Inflammasome Inhibitor BAY-117082 Reduces Oral Squamous Cell Carcinoma Progression. Int. J. Mol. Sci. 2021, 22, 11108. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.; Shi, M.; Lang, Y.; Chao, Z.; Jin, T.; Cui, L.; Zhu, J. Adoptive Transfer of Immunomodulatory M2 Macrophages Suppresses Experimental Autoimmune Encephalomyelitis in C57BL/6 Mice via Blockading NF-κB Pathway. Clin. Exp. Immunol. 2021, 204, 199–211. [Google Scholar] [CrossRef]
- Teng, X.; Hayashida, T.; Murata, T.; Nagayama, A.; Seki, T.; Takahashi, M.; Kitagawa, Y. A Transposon Screen Identifies Enhancement of NF-ΚB Pathway as a Mechanism of Resistance to Eribulin. Breast Cancer 2021, 28, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, A.R.; Tong, X.Y.; Ruiz-Cordero, R.; Bregy, A.; Bethea, J.R.; Bramlett, H.M.; Norenberg, M.D. Activation of NF-ΚB Mediates Astrocyte Swelling and Brain Edema in Traumatic Brain Injury. J. Neurotrauma 2014, 31, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Guffon, N.; Bin-Dorel, S.; Decullier, E.; Paillet, C.; Guitton, J.; Fouilhoux, A. Evaluation of Miglustat Treatment in Patients with Type III Mucopolysaccharidosis: A Randomized, Double-Blind, Placebo-Controlled Study. J. Pediatr. 2011, 159, 838–844.e1. [Google Scholar] [CrossRef]
- Entchev, E.; Jantzen, I.; Masson, P.; Bocart, S.; Bournique, B.; Luccarini, J.-M.; Bouchot, A.; Lacombe, O.; Junien, J.-L.; Broqua, P.; et al. Odiparcil, a Potential Glycosaminoglycans Clearance Therapy in Mucopolysaccharidosis VI—Evidence from in Vitro and in Vivo Models. PLoS ONE 2020, 15, e0233032. [Google Scholar] [CrossRef]
- Schwartz, N.B.; Rodén, L.; Dorfman, A. Biosynthesis of Chondroitin Sulfate: Interaction between Xylosyltransferase and Galactosyltransferase. Biochem. Biophys. Res. Commun. 1974, 56, 717–724. [Google Scholar] [CrossRef]
- Almeida, R.; Levery, S.B.; Mandel, U.; Kresse, H.; Schwientek, T.; Bennett, E.P.; Clausen, H. Cloning and expression of a proteoglycan UDP-galactose: β-xylose β1, 4-galactosyltransferase I: A seventh member of the human β4-galactosyltransferase gene family. J. Biol. Chem. 1999, 274, 26165–26171. [Google Scholar] [CrossRef]
- Mizumoto, S.; Yamada, S. Congenital Disorders of Deficiency in Glycosaminoglycan Biosynthesis. Front. Genet. 2021, 12, 717535. [Google Scholar] [CrossRef] [PubMed]
- Galligani, L.; Hopwood, J.; Schwartz, N.B.; Dorfman, A. Stimulation of Synthesis of Free Chondroitin Sulfate Chains by Beta-D-Xylosides in Cultured Cells. J. Biol. Chem. 1975, 250, 5400–5406. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, N.B. Regulation of Chondroitin Sulfate Synthesis. Effect of Beta-Xylosides on Synthesis of Chondroitin Sulfate Proteoglycan, Chondroitin Sulfate Chains, and Core Protein. J. Biol. Chem. 1977, 252, 6316–6321. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.J.; Coup, D.; Millet, J.; Brown, N.L. The Effect of the β-D-Xyloside Naroparcil on Circulating Plasma Glycosaminoglycans: An explanation for its known antithrombotic activity in the rabbit. J. Biol. Chem. 1995, 270, 2662–2668. [Google Scholar] [CrossRef]
- Upreti, V.V.; Khurana, M.; Cox, D.S.; Eddington, N.D. Determination of Endogenous Glycosaminoglycans Derived Disaccharides in Human Plasma by HPLC: Validation and Application in a Clinical Study. J. Chromatogr. B 2006, 831, 156–162. [Google Scholar] [CrossRef]
- Myers, A.L.; Upreti, V.V.; Khurana, M.; Eddington, N.D. Characterization of Total Plasma Glycosaminoglycan Levels in Healthy Volunteers Following Oral Administration of a Novel Antithrombotic Odiparcil with Aspirin or Enoxaparin. J. Clin. Pharmacol. 2008, 48, 1158–1170. [Google Scholar] [CrossRef]
- Guffon, N.; Chowdary, P.; Teles, E.L.; Hughes, D.; Hennermann, J.B.; Huot-Marchand, P.; Faudot-Vernier, E.; Lacombe, O.; Fiquet, A.; Richard, M.; et al. Oral Treatment for Mucopolysaccharidosis VI: Outcomes of the First Phase IIa Study with Odiparcil. J. Inherit. Metab. Dis. 2022, 45, 340–352. [Google Scholar] [CrossRef]




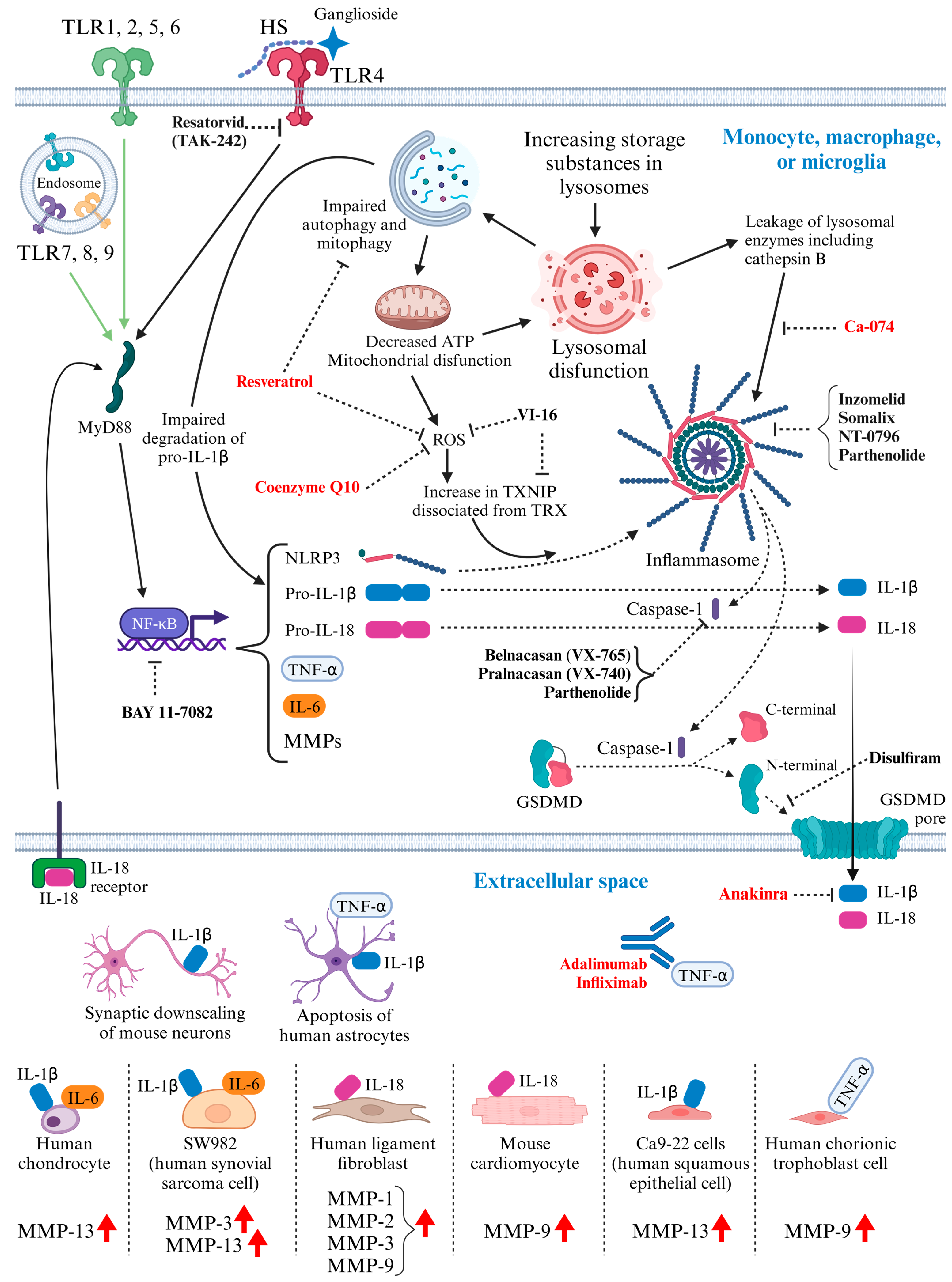{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Impaired Enzyme | Gene | Primary Storage Material | Symptoms |
|---|---|---|---|---|
| Skeletal disease, soft tissue storage, and CNS symptoms | ||||
| MPS I (Hurler syndrome) | α-L-Iduronidase | IDUA | DS, HS | Dysostosis multiplex, short stature, short neck and trunk, kyphoscoliosis, rigidity of joints, micrognathia, coarse facial features, macroglossia, retinal degeneration, corneal clouding, cardiac valvular disease, cardiomyopathy, hepatosplenomegaly, cognitive impairment, developmental delay |
| MPS II (Hunter syndrome) | Iduronate-2-sulfatase | IDS | DS, HS | Dysostosis multiplex, short stature, short neck and trunk, kyphoscoliosis, rigidity of joints, coarse facial features, cardiac valvular disease, no corneal clouding, retinal degeneration, hepatosplenomegaly, cognitive impairment, developmental delay |
| MPS VII (Sly syndrome) | β-Glucuronidase | GUSB | HS, C4S and C6S DS | Dysostosis multiplex, short stature, short neck and trunk, kyphoscoliosis, rigidity of joints, coarse facial features, hydrops fetalis, hepatosplenomegaly, corneal clouding, cognitive impairment, developmental delay |
| Mainly CNS involvement with less skeletal and soft-tissue disease | ||||
| MPS IIIA (Sanfilippo syndrome A) | N-sulfoglucosamine sulfohydrolase | SGSH | HS | Developmental delay, cognitive impairment, hyperactivity, spasticity, motor dysfunction |
| MPS IIIB (Sanfilippo syndrome B) | N-acetyl-α-glucosaminidase | NAGLU | HS | |
| MPS IIIC (Sanfilippo syndrome C) | Acetyl-CoA:α-glucosaminide N-acetyltransferase | HGSNAT | HS | |
| MPS IIID (Sanfilippo syndrome D) | Glucosamine (N-acetyl)-6-sulfatase | GNS | HS | |
| Mainly skeletal, cartilage, and ligament disease; no CNS symptoms | ||||
| MPS IVA (Morquio syndrome A) | Galactose/N-acetylgalactosamine-6-sulfatase | GALNS | KS, C6S | Dysostosis multiplex, short stature, short neck and trunk, pectus carinatum, kyphoscoliosis, genu valgum, motor dysfunction, hyperlaxity of joints, cardiac valvular disease, corneal clouding, narrowing trachea: MPS IVA has more severe symptoms. |
| MPS IVB (Morquio syndrome B) | β-Galactosidase | GLB1 | KS | |
| Skeletal disease and soft tissue storage; no CNS symptoms | ||||
| MPS VI (Maroteaux–Lamy syndrome) | N-acetylgalactosamine-4-sulfatase | ARSB | DS, C4S | Dysostosis multiplex, short stature, short neck and trunk, kyphoscoliosis, motor dysfunction, cardiac valvular disease, corneal clouding |
| MPS X | Arylsulfatase K | ARSK | DS, HS, KS, CS | Dysostosis multiplex, short stature, coarse facial features, mild lens and vitreous opacity, cardiac valvular disease |
| Mainly soft tissues around joints, with no CNS symptoms | ||||
| MPS IX (Natowicz syndrome) | Hyaluronidase | HYAL1 | HA | Large-joint effusion, periarticular soft-tissue masses, mild facial changes, short stature |
| Unclassified | ||||
| MPS-plus syndrome | Target protein: vacuolar protein sorting-associated protein 33A | VPS33A | DS, HS | Increased prenatal thickness of the fetus, coarse facial features, dysostosis multiplex, rigidity of joints, congenital heart defects, anemia, hepatomegaly, nephromegaly, proteinuria, psychomotor delay |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ago, Y.; Rintz, E.; Musini, K.S.; Ma, Z.; Tomatsu, S. Molecular Mechanisms in Pathophysiology of Mucopolysaccharidosis and Prospects for Innovative Therapy. Int. J. Mol. Sci. 2024, 25, 1113. https://doi.org/10.3390/ijms25021113
Ago Y, Rintz E, Musini KS, Ma Z, Tomatsu S. Molecular Mechanisms in Pathophysiology of Mucopolysaccharidosis and Prospects for Innovative Therapy. International Journal of Molecular Sciences. 2024; 25(2):1113. https://doi.org/10.3390/ijms25021113
Chicago/Turabian StyleAgo, Yasuhiko, Estera Rintz, Krishna Sai Musini, Zhengyu Ma, and Shunji Tomatsu. 2024. "Molecular Mechanisms in Pathophysiology of Mucopolysaccharidosis and Prospects for Innovative Therapy" International Journal of Molecular Sciences 25, no. 2: 1113. https://doi.org/10.3390/ijms25021113
APA StyleAgo, Y., Rintz, E., Musini, K. S., Ma, Z., & Tomatsu, S. (2024). Molecular Mechanisms in Pathophysiology of Mucopolysaccharidosis and Prospects for Innovative Therapy. International Journal of Molecular Sciences, 25(2), 1113. https://doi.org/10.3390/ijms25021113