
Inhibition of the RAF/MEK/ERK Signaling Cascade in Pancreatic Cancer: Recent Advances and Future Perspectives



Abstract
1. Introduction
2. RAF/MEK/ERK Signaling Pathway in Pancreatic Cancer
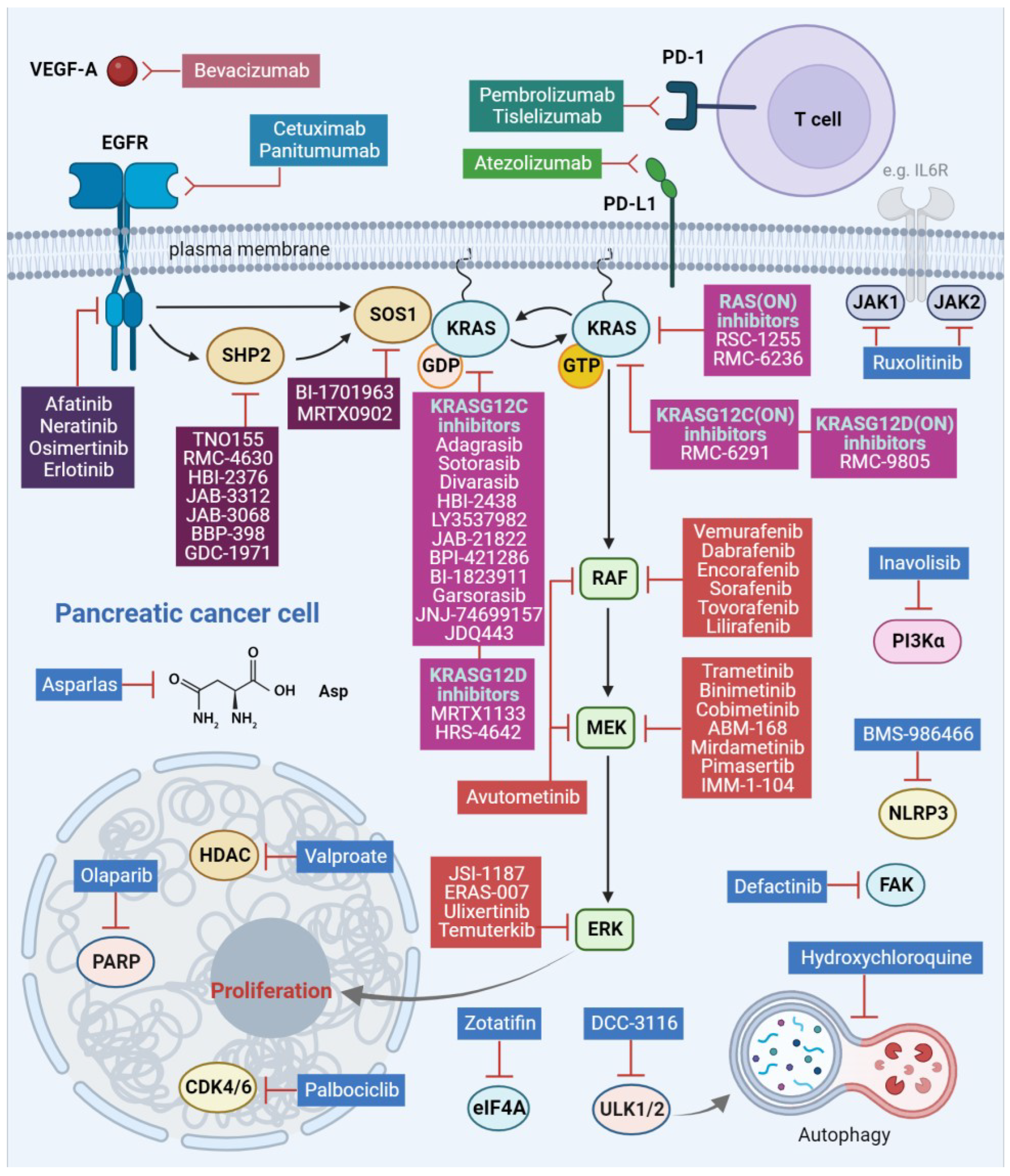
3. Targeting Strategies
3.1. EGFR Family Inhibition
3.2. RAF/MEK/ERK Pathway Component Inhibition
3.2.1. RAF Inhibition
3.2.2. MEK Inhibition
3.2.3. ERK Inhibition
3.3. RAF/MEK/ERK Pathway Regulator Inhibition
3.3.1. SHP2 Inhibition
3.3.2. SOS1 Inhibition
3.4. KRAS Inhibition
3.5. Toxicity Challenges
4. Discussion—Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2020, 17, 108–123. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Novel therapeutic approaches for pancreatic cancer by combined targeting of RAF→MEK→ERK signaling and autophagy survival response. Ann. Transl. Med. 2019, 7, S153. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Trejo, C.L.; Silva, J.M.; Gu, S.; Korkola, J.E.; Heiser, L.M.; Charles, R.P.; Rabinovich, B.A.; Hann, B.; Dankort, D.; et al. A central role for RAF→MEK→ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2012, 2, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Rudloff, U. Emerging kinase inhibitors for the treatment of pancreatic ductal adenocarcinoma. Expert Opin. Emerg. Drugs 2022, 27, 345–368. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 41, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.J.; O’Neil, B.H.; Berlin, J.; Ames, P.; McKinley, M.; Horan, J.; Catalano, P.M.; Davies, A.; Weekes, C.D.; Leichman, L. A phase 1b study of erlotinib in combination with gemcitabine and nab-paclitaxel in patients with previously untreated advanced pancreatic cancer: An Academic Oncology GI Cancer Consortium study. Cancer Chemother. Pharmacol. 2016, 77, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Benedetti, J.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; Flynn, P.J.; Rowland, K.M.; Atkins, J.N.; Mirtsching, B.C.; Rivkin, S.E.; et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J. Clin. Oncol. 2010, 28, 3605. [Google Scholar] [CrossRef] [PubMed]
- Safran, H.; Miner, T.; Bahary, N.; Whiting, S.; Lopez, C.D.; Sun, W.; Charpentier, K.; Shipley, J.; Anderson, E.; McNulty, B.; et al. Lapatinib and gemcitabine for metastatic pancreatic cancer. A phase II study. Am. J. Clin. Oncol. 2011, 34, 50–52. [Google Scholar] [CrossRef]
- Wu, Z.; Gabrielson, A.; Hwang, J.J.; Pishvaian, M.J.; Weiner, L.M.; Zhuang, T.; Ley, L.; Marshall, J.L.; He, A.R. Phase II study of lapatinib and capecitabine in second-line treatment for metastatic pancreatic cancer. Cancer Chemother. Pharmacol. 2015, 76, 1309–1314. [Google Scholar] [CrossRef]
- Haas, M.; Waldschmidt, D.T.; Stahl, M.; Reinacher-Schick, A.; Freiberg-Richter, J.; Fischer von Weikersthal, L.; Kaiser, F.; Kanzler, S.; Frickhofen, N.; Seufferlein, T.; et al. Afatinib plus gemcitabine versus gemcitabine alone as first-line treatment of metastatic pancreatic cancer: The randomised, open-label phase II ACCEPT study of the Arbeitsgemeinschaft Internistische Onkologie with an integrated analysis of the ‘burden of therapy’ method. Eur. J. Cancer 2021, 146, 95–106. [Google Scholar]
- Cardin, D.B.; Goff, L.; Li, C.I.; Shyr, Y.; Winkler, C.; DeVore, R.; Schlabach, L.; Holloway, M.; McClanahan, P.; Meyer, K.; et al. Phase II trial of sorafenib and erlotinib in advanced pancreatic cancer. Cancer Med. 2014, 3, 572–579. [Google Scholar] [CrossRef]
- Ko, A.H.; Bekaii-Saab, T.; Van Ziffle, J.; Mirzoeva, O.M.; Joseph, N.M.; Talasaz, A.; Kuhn, P.; Tempero, M.A.; Collisson, E.A.; Kelley, R.K.; et al. A Multicenter, Open-Label Phase II Clinical Trial of Combined MEK plus EGFR Inhibition for Chemotherapy-Refractory Advanced Pancreatic Adenocarcinoma. Clin. Cancer Res. 2016, 22, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Halfdanarson, T.R.; Foster, N.R.; Kim, G.P.; Meyers, J.P.; Smyrk, T.C.; McCullough, A.E.; Ames, M.M.; Jaffe, J.P.; Alberts, S.R. A Phase II Randomized Trial of Panitumumab, Erlotinib, and Gemcitabine versus Erlotinib and Gemcitabine in Patients with Untreated, Metastatic Pancreatic Adenocarcinoma: North Central Cancer Treatment Group Trial N064B. Oncologist 2019, 24, 589-e160. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.M.; Powell, E.D.; Asmis, T.R.; Jonker, D.J.; Hilton, J.F.; O’Callaghan, C.J.; Tu, D.; Parulekar, W.; Moore, M.J. Comorbidity, age and overall survival in patients with advanced pancreatic cancer—Results from NCIC CTG PA.3: A phase III trial of gemcitabine plus erlotinib or placebo. Eur. J. Cancer 2012, 48, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Dent, P.; Booth, L.; Roberts, J.L.; Liu, J.; Poklepovic, A.; Lalani, A.S.; Tuveson, D.; Martinez, J.; Hancock, J.F. Neratinib inhibits Hippo/YAP signaling, reduces mutant K-RAS expression, and kills pancreatic and blood cancer cells. Oncogene 2019, 38, 5890–5904. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, A.; Gilabert, M.; François, E.; Dahan, L.; Perrier, H.; Lamy, R.; Re, D.; Largillier, R.; Gasmi, M.; Tchiknavorian, X.; et al. BAYPAN study: A double-blind phase III randomized trial comparing gemcitabine plus sorafenib and gemcitabine plus placebo in patients with advanced pancreatic cancer. Ann. Oncol. 2012, 23, 2799–2805. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Karoulia, Z.; Wu, Y.; Ahmed, T.A.; Xin, Q.; Bollard, J.; Krepler, C.; Wu, X.; Zhang, C.; Bollag, G.; Herlyn, M.; et al. An Integrated Model of RAF Inhibitor Action Predicts Inhibitor Activity against Oncogenic BRAF Signaling. Cancer Cell 2016, 30, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.B.; Henry, J.R.; Kaufman, M.D.; Lu, W.P.; Smith, B.D.; Vogeti, S.; Rutkoski, T.J.; Wise, S.; Chun, L.; Zhang, Y.; et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 2015, 28, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Vakana, E.; Pratt, S.; Blosser, W.; Dowless, M.; Simpson, N.; Yuan, X.J.; Jaken, S.; Manro, J.; Stephens, J.; Zhang, Y.; et al. LY3009120, a panRAF inhibitor, has significant anti-tumor activity in BRAF and KRAS mutant preclinical models of colorectal cancer. Oncotarget 2017, 8, 9251–9266. [Google Scholar] [CrossRef] [PubMed]
- Desai, J.; Gan, H.; Barrow, C.; Jameson, M.; Atkinson, V.; Haydon, A.; Millward, M.; Begbie, S.; Brown, M.; Markman, B.; et al. Phase I, Open-Label, Dose-Escalation/Dose-Expansion Study of Lifirafenib (BGB-283), an RAF Family Kinase Inhibitor, in Patients with Solid Tumors. J. Clin. Oncol. 2020, 38, 2140–2150. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted with Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e4. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Nowak, J.A.; Camarda, N.D.; Moffitt, R.A.; Ghazani, A.A.; Hazar-Rethinam, M.; Raghavan, S.; Kim, J.; Brais, L.K.; Ragon, D.; et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 2018, 8, 1096–1111. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Zhang, Y.; Van Horn, R.D.; Yin, T.; Buchanan, S.; Yadav, V.; Mochalkin, I.; Wong, S.S.; Yue, Y.G.; Huber, L.; et al. Oncogenic BRAF Deletions That Function as Homodimers and Are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120. Cancer Discov. 2016, 6, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Igarashi, K.; Murakami, T.; Kiyuna, T.; Lwin, T.M.; Hwang, H.K.; Delong, J.C.; Clary, B.M.; Bouvet, M.; Unno, M.; et al. MEK inhibitors cobimetinib and trametinib, regressed a gemcitabine-resistant pancreatic-cancer patient-derived orthotopic xenograft (PDOX). Oncotarget 2017, 8, 47490–47496. [Google Scholar] [CrossRef]
- Seghers, A.K.; Cuyle, P.J.; Van Cutsem, E. Molecular Targeting of a BRAF Mutation in Pancreatic Ductal Adenocarcinoma: Case Report and Literature Review. Target. Oncol. 2020, 15, 407–410. [Google Scholar] [CrossRef]
- Li, H.S.; Yang, K.; Wang, Y. Remarkable response of BRAF (V600E)-mutated metastatic pancreatic cancer to BRAF/MEK inhibition: A case report. Gastroenterol. Rep. 2021, 10, goab031. [Google Scholar] [CrossRef]
- Grinshpun, A.; Zarbiv, Y.; Roszik, J.; Subbiah, V.; Hubert, A. Beyond KRAS: Practical molecular targets in pancreatic adenocarcinoma. Case Rep. Oncol. 2019, 12, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; He, D.; Chen, C.; Liu, X.; Ke, N. Vemurafenib Combined with Trametinib Significantly Benefits the Survival of a Patient with Stage IV Pancreatic Ductal Adenocarcinoma with BRAF V600E Mutation: A Case Report. Front. Oncol. 2022, 11, 801320. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Rana, T.; Kancharla, P.; Monga, D. Targeted Therapy for BRAF V600E Positive Pancreatic Adenocarcinoma: Two Case Reports. Cancer Genom. Proteom. 2023, 20, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Ardalan, B.; Azqueta, J.I.; England, J.; Eatz, T.A. Potential benefit of treatment with MEK inhibitors and chemotherapy in BRAF-mutated KRAS wild-type pancreatic ductal adenocarcinoma patients: A case report. Cold Spring Harb. Mol. Case Stud. 2021, 7, a006108. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Hidalgo, M.; Canon, J.L.; Macarulla, T.; Bazin, I.; Poddubskaya, E.; Manojlovic, N.; Radenkovic, D.; Verslype, C.; Raymond, E.; et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int. J. Cancer 2018, 143, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Van Laethem, J.L.; Riess, H.; Jassem, J.; Haas, M.; Martens, U.M.; Weekes, C.; Peeters, M.; Ross, P.; Bridgewater, J.; Melichar, B.; et al. Phase I/II Study of Refametinib (BAY 86-9766) in Combination with Gemcitabine in Advanced Pancreatic cancer. Target. Oncol. 2017, 12, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Bodoky, G.; Timcheva, C.; Spigel, D.R.; La Stella, P.J.; Ciuleanu, T.E.; Pover, G.; Tebbutt, N.C. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Investig. New Drugs 2012, 30, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Chung, V.; McDonough, S.; Philip, P.A.; Cardin, D.; Wang-Gillam, A.; Hui, L.; Tejani, M.A.; Seery, T.E.; Dy, I.A.; Al Baghdadi, T.; et al. Effect of Selumetinib and MK-2206 vs Oxaliplatin and Fluorouracil in Patients with Metastatic Pancreatic Cancer after Prior Therapy: SWOG S1115 Study Randomized Clinical Trial. JAMA Oncol. 2017, 3, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Bendell, J.C.; Papadopoulos, K.P.; Burris, H.A., 3rd; Patnaik, A.; Jones, S.F.; Rasco, D.; Cox, D.S.; Durante, M.; Bellew, K.M.; et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann. Oncol. 2015, 26, 58–64. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.; Fakih, M.; De Vos, F.Y.F.L.; Beck, J.T.; Merchan, J.; Shapiro, G.; Lin, C.-C.; Spratlin, J.; Cascella, T.; Sandalic, L.; et al. Phase Ib study of ribociclib (R) + trametinib (T) in patients (pts) with metastatic/advanced solid tumours. Ann. Oncol. 2020, 31, S484. [Google Scholar] [CrossRef]
- Rodon Ahnert, J.; Tan, D.S.; Garrido-Laguna, I.; Harb, W.; Bessudo, A.; Beck, J.T.; Rottey, S.; Bahary, N.; Kotecki, N.; Zhu, Z.; et al. Avelumab or talazoparib in combination with binimetinib in metastatic pancreatic ductal adenocarcinoma: Dose-finding results from phase Ib of the JAVELIN PARP MEKi trial. ESMO Open 2023, 8, 101584. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Baker, N.M.; Miermont, A.M.; Thurman, R.D.; Pierobon, M.; Tran, T.H.; Anderson, A.O.; Waters, A.M.; Diehl, J.N.; Papke, B.; et al. Atypical KRASG12R Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov. 2020, 10, 104–123. [Google Scholar] [CrossRef] [PubMed]
- Kenney, C.; Kunst, T.; Webb, S.; Christina, D., Jr.; Arrowood, C.; Steinberg, S.M.; Mettu, N.B.; Kim, E.J.; Rudloff, U. Phase II study of selumetinib, an orally active inhibitor of MEK1 and MEK2 kinases, in KRASG12R-mutant pancreatic ductal adenocarcinoma. Investig. New Drugs 2021, 39, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Ardalan, B.; Azqueta, J.; Sleeman, D. Cobimetinib Plus Gemcitabine: An Active Combination in KRAS G12R-Mutated Pancreatic Ductal Adenocarcinoma Patients in Previously Treated and Failed Multiple Chemotherapies. J. Pancreat. Cancer 2021, 7, 65–70. [Google Scholar] [CrossRef]
- Lopez, C.D.; Kardosh, A.; Chen, E.Y.; Pegna, G.J.; Goodyear, S.; Taber, E.; Rajagopalan, B.; Edmerson, E.; Vo, J.; Jackson, A.; et al. Casper: A phase I, open-label, dose finding study of calaspargase pegol-mnkl (cala) in combination with cobimetinib (cobi) in locally advanced or metastatic pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2023, 41, TPS772. [Google Scholar] [CrossRef]
- Grierson, P.M.; Tan, B.; Pedersen, K.S.; Park, H.; Suresh, R.; Amin, M.A.; Trikalinos, N.A.; Knoerzer, D.; Kreider, B.; Reddy, A.; et al. Phase Ib Study of Ulixertinib Plus Gemcitabine and Nab-Paclitaxel in Patients with Metastatic Pancreatic Adenocarcinoma. Oncologist 2023, 28, e115–e123. [Google Scholar] [CrossRef]
- Wang, J.; Johnson, M.; Barve, M.; Pelster, M.; Chen, X.; Li, Z.; Gordon, J.; Reiss, M.; Pai, S.; Falchook, G.; et al. Preliminary results from HERKULES-1: A phase 1b/2, open-label, multicenter study of ERAS-007, an oral ERK1/2 inhibitor, in patients with advanced or metastatic solid tumors. Eur. J. Cancer 2022, 174, S80–S81. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.; Witkiewicz, A.K.; Knudsen, E.S. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget 2014, 5, 6512–6525. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. 2016, 14, 979–990. [Google Scholar] [CrossRef]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Görgülü, K.; Dantes, Z.; Wörmann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef]
- Ahmed, T.A.; Adamopoulos, C.; Karoulia, Z.; Wu, X.; Sachidanandam, R.; Aaronson, S.A.; Poulikakos, P.I. SHP2 Drives Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent Tumors. Cell Rep. 2019, 26, 65–78.e5. [Google Scholar] [CrossRef] [PubMed]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.J.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G.; et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef]
- Mainardi, S.; Mulero-Sánchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; de Wit, N.; Gonçalves-Ribeiro, S.; et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.J.; Mulero-Sánchez, A.; Berninger, A.; Ruiz-Cañas, L.; Bosma, A.; Görgülü, K.; Wu, N.; Diakopoulos, K.N.; Kaya-Aksoy, E.; Ruess, D.A.; et al. Extensive preclinical validation of combined RMC-4550 and LY3214996 supports clinical investigation for KRAS mutant pancreatic cancer. Cell Rep. Med. 2022, 3, 100815. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRASG12C Inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Fedele, C.; Li, S.; Teng, K.W.; Foster, C.J.R.; Peng, D.; Ran, H.; Mita, P.; Geer, M.J.; Hattori, T.; Koide, A.; et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J. Exp. Med. 2021, 218, e20201414. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Jeng, H.H.; Taylor, L.J.; Bar-Sagi, D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat. Commun. 2012, 3, 1168. [Google Scholar] [CrossRef]
- Ma, Y.; Schulz, B.; Trakooljul, N.; Al Ammar, M.; Sekora, A.; Sender, S.; Hadlich, F.; Zechner, D.; Weiss, F.U.; Lerch, M.M.; et al. Inhibition of KRAS, MEK and PI3K Demonstrate Synergistic Anti-Tumor Effects in Pancreatic Ductal Adenocarcinoma Cell Lines. Cancers 2022, 14, 4467. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Gort, E.; Pant, S.; Lolkema, M.; Sebastian, M.; Scheffler, M.; Hwang, J.; Dünzinger, U.; Riemann, K.; Kitzing, T.; et al. 524P A phase I, open-label, dose-escalation trial of BI 1701963 in patients (pts) with KRAS mutated solid tumours: A snapshot analysis. Ann. Oncol. 2021, 32, S591–S592. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Murciano-Goroff, Y.R.; Suehnholz, S.P.; Drilon, A.; Chakravarty, D. Precision Oncology: 2023 in Review. Cancer Discov. 2023, 13, 2525–2531. [Google Scholar] [CrossRef]
- Sacher, A.; LoRusso, P.; Patel, M.R.; Miller, W.H., Jr.; Garralda, E.; Forster, M.D.; Santoro, A.; Falcon, A.; Kim, T.W.; Paz-Ares, L.; et al. Single-Agent Divarasib (GDC-6036) in Solid Tumors with a KRAS G12C Mutation. N. Engl. J. Med. 2023, 389, 710–721. [Google Scholar] [CrossRef]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRASG12D Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Herdeis, L.; Rudolph, D.; Zhao, Y.; Böttcher, J.; Vides, A.; Ayala-Santos, C.I.; Pourfarjam, Y.; Cuevas-Navarro, A.; Xue, J.Y.; et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature 2023, 619, 160–166. [Google Scholar] [CrossRef]
- Foote, J.B.; Mattox, T.E.; Keeton, A.B.; Purnachandra, G.N.; Maxuitenko, Y.; Chen, X.; Valiyaveettil, J.; Buchsbaum, D.J.; Piazza, G.A.; El-Rayes, B.F. Abstract 4140: Oncogenic KRAS inhibition with ADT-007 primes T cell responses in pancreatic ductal adenocarcinoma. Cancer Res. 2023, 83 (Suppl. 7), 4140. [Google Scholar] [CrossRef]
- Koltun, E.; Cregg, J.; Rice, M.A.; Whalen, D.M.; Freilich, R.; Jiang, J.; Hansen, R.; Bermingham, A.; Knox, D.; Dinglasan, J.; et al. Abstract 1260: First-in-class, orally bioavailable KRASG12V(ON) tri-complex inhibitors, as single agents and in combinations, drive profound anti-tumor activity in preclinical models of KRASG12V mutant cancers. Cancer Res. 2021, 13, 1260. [Google Scholar] [CrossRef]
- de Jesus, V.H.F.; Mathias-Machado, M.C.; de Farias, J.P.F.; Aruquipa, M.P.S.; Jácome, A.A.; Peixoto, R.D. Targeting KRAS in Pancreatic Ductal Adenocarcinoma: The Long Road to Cure. Cancers 2023, 15, 5015. [Google Scholar] [CrossRef] [PubMed]
- Koltun, E.S.; Rice, M.A.; Gustafson, W.C.; Wilds, D.; Jiang, J.; Lee, B.J.; Wang, Z.; Chang, S.; Flagella, M.; Mu, Y.; et al. Direct targeting of KRASG12X mutant cancers with RMC-6236, a first-in-class, RAS-selective, orally bioavailable, tri-complex RASMULTI(ON) inhibitor. Cancer Res. 2022, 82 (Suppl. S12), 3597. [Google Scholar] [CrossRef]
- Park, S.R.; Davis, M.; Doroshow, J.H.; Kummar, S. Safety and feasibility of targeted agent combinations in solid tumours. Nat. Rev. Clin. Oncol. 2013, 10, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lu, H.; Wang, H.; Loo, A.; Zhang, X.; Yang, G.; Kowal, C.; Delach, S.; Wang, Y.; Goldoni, S.; et al. Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling. Clin. Cancer Res. 2021, 27, 342–354. [Google Scholar] [CrossRef]
- Akhave, N.S.; Biter, A.B.; Hong, D.S. The Next Generation of KRAS Targeting: Reasons for Excitement and Concern. Mol. Cancer Ther. 2022, 21, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Li, B.T.; Falchook, G.S.; Durm, G.A.; Burns, T.F.; Skoulidis, F.; Ramalingam, S.S.; Spira, A.; Bestvina, C.M.; Goldberg, S.B.; Veluswamy, R.; et al. CodeBreaK 100/101: First report of safety/efficacy of sotorasib in combination with pembrolizumab or atezolizumab in advanced KRAS p.G12C NSCLC. J. Thorac. Oncol. 2022, 17, S10–S11. [Google Scholar] [CrossRef]
- Sacher, A.; Patel, M.R.; Miller, W.H., Jr.; Desai, J.; Garralda, E.; Bowyer, S.; Kim, T.W.; De Miguel, M.; Falcon, A.; Krebs, M.G.; et al. OA03.04 phase I A study to evaluate GDC-6036 monotherapy in patients with Non-small Cell Lung Cancer (NSCLC) with KRAS G12C mutation. J. Thorac. Oncol. 2022, 17, S8–S9. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRASG12C Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Darman, L.; Hassan, M.S.; Von Holzen, U.; Awasthi, N. Targeting KRAS for the potential treatment of pancreatic ductal adenocarcinoma: Recent advancements provide hope (Review). Oncol. Rep. 2023, 50, 206. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Azar, I.; Xiu, J.; Hall, M.J.; Hendifar, A.E.; Lou, E.; Hwang, J.J.; Gong, J.; Feldman, R.; Ellis, M.; et al. Molecular Characterization of KRAS Wild-type Tumors in Patients with Pancreatic Adenocarcinoma. Clin. Cancer Res. 2022, 28, 2704–2714. [Google Scholar] [CrossRef] [PubMed]
- Ardalan, B.; Ciner, A.; Baca, Y.; Darabi, S.; Kasi, A.; Lou, E.; Azqueta, J.I.; Xiu, J.; Nabhan, C.; Shields, A.F.; et al. Not all treated KRAS-mutant pancreatic adenocarcinomas are equal: KRAS G12D and survival outcome. J. Clin. Oncol. 2023, 41, 4020. [Google Scholar] [CrossRef]
- Ardalan, B.; Ciner, A.; Baca, Y.; Darabi, S.; Kasi, A.; Lou, E.; Azqueta, J.I.; Xiu, J.; Nabhan, C.; Shields, A.F.; et al. Prognostic indicators of KRAS G12X mutations in pancreatic cancer. J. Clin. Oncol. 2023, 41, 735. [Google Scholar] [CrossRef]
- Ciner, A.; Ardalan, B.; Baca, Y.; Darabi, S.; Kasi, A.; Lou, E.; Azqueta, J.I.; Xiu, J.; Nabhan, C.; Shields, A.F.; et al. KRAS G12C-mutated pancreatic cancer: Clinical outcomes based on chemotherapeutic regimen. J. Clin. Oncol. 2023, 41, 4150. [Google Scholar] [CrossRef]
- Suzuki, T.; Masugi, Y.; Inoue, Y.; Hamada, T.; Tanaka, M.; Takamatsu, M.; Arita, J.; Kato, T.; Kawaguchi, Y.; Kunita, A.; et al. KRAS variant allele frequency, but not mutation positivity, associates with survival of patients with pancreatic cancer. Cancer Sci. 2022, 113, 3097–3109. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Öllinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef]
- Adamopoulos, C.; Ahmed, T.A.; Tucker, M.R.; Ung, P.M.U.; Xiao, M.; Karoulia, Z.; Amabile, A.; Wu, X.; Aaronson, S.A.; Ang, C.; et al. Exploiting Allosteric Properties of RAF and MEK Inhibitors to Target Therapy-Resistant Tumors Driven by Oncogenic BRAF Signaling. Cancer Discov. 2021, 11, 1716–1735. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.B.; Cheng, N.; Markosyan, N.; Sor, R.; Kim, I.K.; Hallin, J.; Shoush, J.; Quinones, L.; Brown, N.V.; Bassett, J.B.; et al. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov. 2023, 13, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. The KRAS crowd targets its next cancer mutations. Nat. Rev. Drug Discov. 2023, 22, 167–171. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Drug(s) | Target(s) | Second Drug(s) | Second Target(s) | Phase | Clinical Study Code |
|---|---|---|---|---|---|
| Neratinib | EGFR, ERBB2/HER2 | Divalproex sodium (Valproate) | HDAC | I/II | NCT03919292 |
| Vemurafenib | BRAFV600E/K | Sorafenib | RAF | II | NCT05068752 |
| Lilirafenib | BRAF | Mirdametinib | MEK | I | NCT03905148 |
| Tovorafenib | RAF | Pimasertib | MEK | I/II | NCT04985604 |
| Avutometinib | MEK, RAF | Defactinib | FAK | I/II | NCT05669482 |
| ABM-168 | MEK | I | NCT05831995 | ||
| Binimetinib | MEK | Hydroxychloroquine | Autophagy | I | NCT04132505 |
| Binimetinib | MEK | Encorafenib | RAFV600E/K | II | NCT04390243 |
| Binimetinib | MEK | Palbociclib | CDK4/6 | II | NCT05554367 |
| Trametinib | MEK | Hydroxychloroquine | Autophagy | I | NCT03825289 |
| Trametinib | MEK | Ruxolitinib | JAK1/JAK2 | I | NCT04303403 |
| Cobimetinib | MEK | Calaspargase pegol-mnkl (Asparlas) | Asparagine | I | NCT05034627 |
| IMM-1-104 | MEK | I/II | NCT05585320 | ||
| Temuterkib | ERK | RMC-4630 | SHP2 | I | NCT04916236 |
| Temuterkib | ERK | Hydroxychloroquine sulfate | Autophagy | II | NCT04386057 |
| Ulixertinib | ERK | Palbociclib | CDK4/6 | I | NCT03454035 |
| ERAS-007 | ERK | Encorafenib Palbociclib Cetuximab * | BRAFV600E/K CDK4/6 EGFR | I/II | NCT05039177 |
| BI-1701963 | SOS1 | Adagrasib | KRASG12C | I | NCT04975256 |
| BI-1701963 | SOS1 | Trametinib | MEK | I | NCT04111458 |
| HBI-2376 | SHP2 | I | NCT05163028 | ||
| JAB-3068 | SHP2 | I/II | NCT03565003 | ||
| JAB-3312 | SHP2 | I | NCT04045496 | ||
| JAB-3312 | SHP2 | Binimetinib Pembrolizumab * Sotorasib Osimertinib | MEK PD-1 KRASG12C EGFR | I/II | NCT04720976 |
| BBP-398 | SHP2 | Sotorasib | KRASG12C | I | NCT05480865 |
| RMC-6291 | KRASG12C | I | NCT05462717 | ||
| RMC-6291 | KRASG12C | RMC-6236 | RAS (pan-mutant and wild-type) | I | NCT06128551 |
| HBI-2438 | KRASG12C | I | NCT05485974 | ||
| LY3537982 | KRASG12C | I | NCT04956640 | ||
| JAB-21822 | KRASG12C | II | NCT06008288 | ||
| JAB-21822 | KRASG12C | Cetuximab * | EGFR | I/II | NCT05002270 |
| JAB-21822 | KRASG12C | JAB-3312 | SHP2 | I/II | NCT05288205 |
| Adagrasib | KRASG12C | I | NCT05634525 | ||
| Adagrasib | KRASG12C | TNO155 | SHP2 | I/II | NCT04330664 |
| Adagrasib | KRASG12C | Afatinib Cetuximab * Pembrolizumab * | EGFR/HER2 EGFR PD-1 | I | NCT03785249 |
| Adagrasib | KRASG12C | Olaparib | PARP | I | NCT06130254 |
| Adagrasib | KRASG12C | BMS-986466 † −/+ cetuximab * | NLRP3 EGFR | I/II | NCT06024174 |
| Adagrasib | KRASG12C | MRTX0902 | SOS1 | I/II | NCT05578092 |
| BPI-421286 | KRASG12C | I | NCT05315180 | ||
| BI-1823911 | KRASG12C | BI-1701963 | SOS1 | I | NCT04973163 |
| Divarasib | KRASG12C | Atezolizumab * Cetuximab * Bevacizumab * Erlotinib GDC-1971 Inavolisib | PD-L1 EGFR VEGFA EGFR SHP2 PI3Kα | I | NCT04449874 |
| Garsorasib | KRASG12C | I | NCT04585035 | ||
| JNJ-74699157 | KRASG12C | I | NCT04006301 | ||
| JDQ443 | KRASG12C | TNO155 Tislelizumab * | SHP2 PD-1 | I/II | NCT04699188 |
| MK-1084 | KRASG12C | Pembrolizumab * | PD-1 | I | NCT05067283 |
| Sotorasib | KRASG12C | I/II | NCT03600883 | ||
| Sotorasib § | KRASG12C | II | NCT04185883 | ||
| Sotorasib | KRASG12C | I | NCT04380753 | ||
| Sotorasib | KRASG12C | Panitumumab * | EGFR | II | NCT05638295 |
| Sotorasib | KRASG12C | Panitumumab* | EGFR | II | NCT05993455 |
| Sotorasib | KRASG12C | DCC-3116 † | ULK1/2 | I/II | NCT04892017 |
| Sotorasib | KRASG12C | Zotatifin † | eIF4A | I/II | NCT04092673 |
| MRTX1133 | KRASG12D | I/II | NCT05737706 | ||
| RMC-9805 | KRASG12D | I | NCT06040541 | ||
| HRS-4642 | KRASG12D | I | NCT05533463 | ||
| RMC-6236 | RAS (pan-mutant and wild-type) | I | NCT05379985 | ||
| RSC-1255 | RAS (pan-mutant and wild-type) | I | NCT04678648 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamopoulos, C.; Cave, D.D.; Papavassiliou, A.G. Inhibition of the RAF/MEK/ERK Signaling Cascade in Pancreatic Cancer: Recent Advances and Future Perspectives. Int. J. Mol. Sci. 2024, 25, 1631. https://doi.org/10.3390/ijms25031631
Adamopoulos C, Cave DD, Papavassiliou AG. Inhibition of the RAF/MEK/ERK Signaling Cascade in Pancreatic Cancer: Recent Advances and Future Perspectives. International Journal of Molecular Sciences. 2024; 25(3):1631. https://doi.org/10.3390/ijms25031631
Chicago/Turabian StyleAdamopoulos, Christos, Donatella Delle Cave, and Athanasios G. Papavassiliou. 2024. "Inhibition of the RAF/MEK/ERK Signaling Cascade in Pancreatic Cancer: Recent Advances and Future Perspectives" International Journal of Molecular Sciences 25, no. 3: 1631. https://doi.org/10.3390/ijms25031631
APA StyleAdamopoulos, C., Cave, D. D., & Papavassiliou, A. G. (2024). Inhibition of the RAF/MEK/ERK Signaling Cascade in Pancreatic Cancer: Recent Advances and Future Perspectives. International Journal of Molecular Sciences, 25(3), 1631. https://doi.org/10.3390/ijms25031631