

Copy Number Variations in Pancreatic Cancer: From Biological Significance to Clinical Utility


{kind=link}
{kind=link}
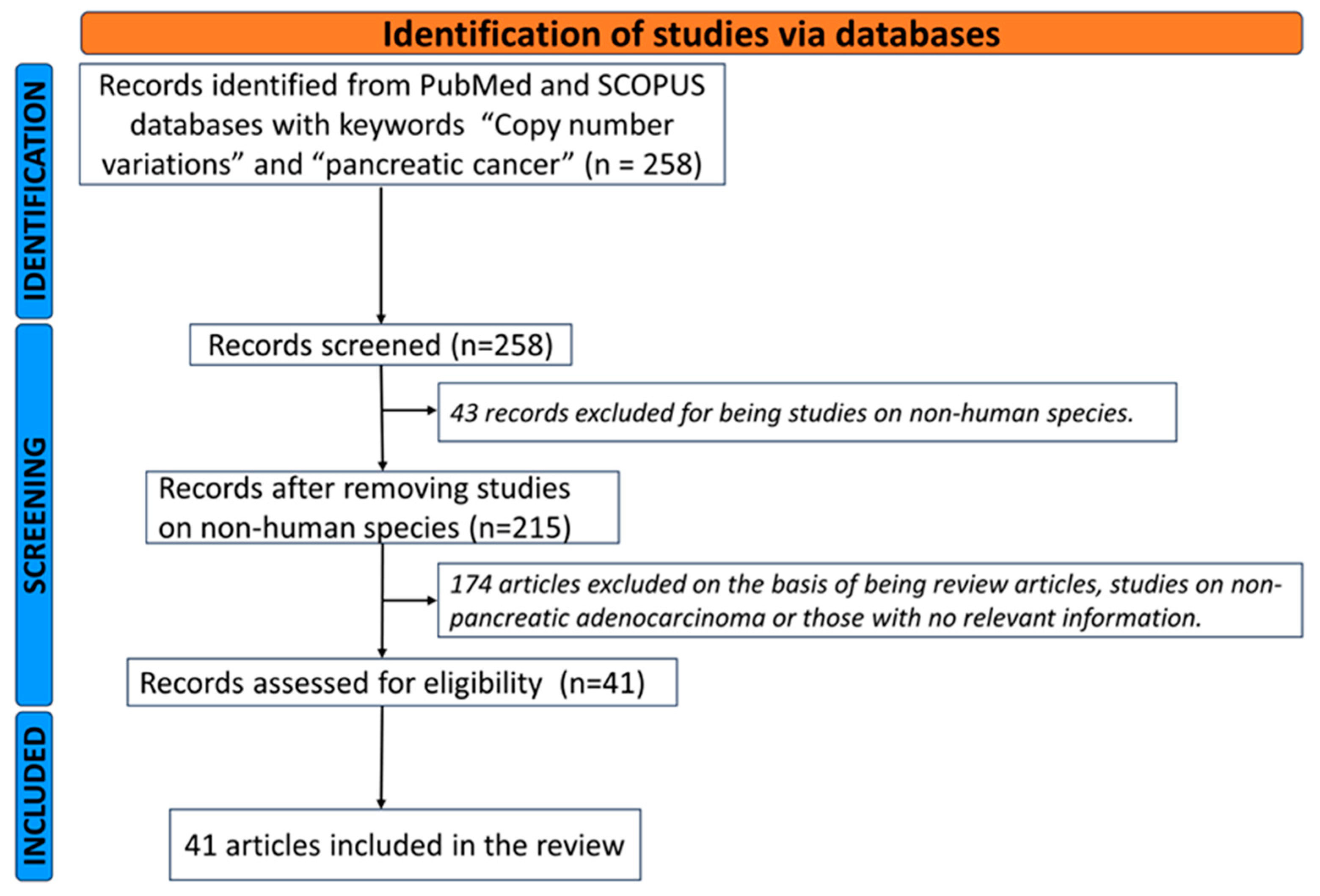{kind=link}
Abstract
1. Introduction
1.1. Classification of CNVs
1.2. Mechanisms of CNV Formation
1.2.1. Genomic Factors and Molecular Mechanisms of CNV Formation
1.2.2. Environmental Factors in CNV Formation
1.3. Distribution of CNVs
1.4. Identification and Detection of CNVs
1.4.1. “Whole” Approaches
1.4.2. “Targeted” Approaches
1.5. Implications of CNVs
- (i)
- direct influence on the expression of a gene product, giving rise to changing levels of a protein. For example, Miller et al. [53] demonstrated an almost perfect correlation between the α-synuclein (SNCA) gene dosage and its mRNA and protein levels in Parkinson disease. SNCA triplication resulted in a doubling in the effective load of the normal gene and increased deposition of aggregated forms of the protein level in the brain into insoluble fractions.
- (ii)
- alteration of regulatory regions due to CNVs on non-coding sequences. This directly influences the levels and timing of expression and the cellular localization of the related protein. For example, the regulation of SOX9 gene expression in the testis is governed by a set of regulatory elements (RevSex and XYSR) located upstream of its promoter [54,55]. Loss of one or both of these regions in an XY individual results in a loss of SOX9 expression and male-to-female sex reversal [55], while duplication of the RevSex region in an XX individual could increase SOX9 expression and lead to female-to-male sex reversal [56,57,58,59].
- (iii)
- recombination of functional domains of different genes, leading to the formation of modified or new products with newly acquired functions, as seen in the example of glucocorticoid-remediable aldosteronism (GRA). Some researchers have shown that it is caused by a chimeric 11 β-hydroxylase (CYP11B1)/aldosterone synthase (CYP11B2) gene formed when a gene duplication resulting from unequal crossing over fuses the 5′ regulatory region of 11/β-hydroxylase to the coding sequences of aldosterone synthase [60]. The ectopic expression of CYP11B2 in the adrenal zona fasciculata may be responsible for these abnormalities because the gene is normally only expressed in the adrenal zona glomerulosa [35,60].
1.6. CNVs and Cancer
2. CNVs and Pancreatic Ductal Adenocarcinoma
2.1. Mechanisms of Pancreatic Cancer Pathogenesis
2.2. Identification and Analysis of CNVs in PDAC
2.3. CNV-Based Classifications of PDAC
2.3.1. Structural Variation Profiles
2.3.2. Molecular Subtypes
3. CNV Studies in PDAC
3.1. Literature Review
3.2. CNVs in PDAC Stages and Grades
3.3. CNVs in PDAC Chemoresistance
4. Challenges and Limitations in Clinical Application
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hruban, R.H.; Gaida, M.M.; Thompson, E.; Hong, S.M.; Noe, M.; Brosens, L.A.; Jongepier, M.; Offerhaus, G.J.A.; Wood, L.D. Why is pancreatic cancer so deadly? The pathologist’s view. J. Pathol. 2019, 248, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Shlien, A.; Malkin, D. Copy number variations and cancer. Genome Med. 2009, 1, 62. [Google Scholar] [CrossRef] [PubMed]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Nowakowska, B. Clinical interpretation of copy number variants in the human genome. J. Appl. Genet. 2017, 58, 449–457. [Google Scholar] [CrossRef]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genom. Hum. Genet. 2009, 10, 451–481. [Google Scholar] [CrossRef]
- Nguyen, D.Q.; Webber, C.; Ponting, C.P. Bias of selection on human copy-number variants. PLoS Genet. 2006, 2, e20. [Google Scholar] [CrossRef]
- Choy, K.W.; Setlur, S.R.; Lee, C.; Lau, T.K. The impact of human copy number variation on a new era of genetic testing. BJOG 2010, 117, 391–398. [Google Scholar] [CrossRef]
- Bruder, C.E.; Piotrowski, A.; Gijsbers, A.A.; Andersson, R.; Erickson, S.; Diaz de Stahl, T.; Menzel, U.; Sandgren, J.; von Tell, D.; Poplawski, A.; et al. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am. J. Hum. Genet. 2008, 82, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, A.; Bruder, C.E.; Andersson, R.; Diaz de Stahl, T.; Menzel, U.; Sandgren, J.; Poplawski, A.; von Tell, D.; Crasto, C.; Bogdan, A.; et al. Somatic mosaicism for copy number variation in differentiated human tissues. Hum. Mutat. 2008, 29, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, L.A.; Rasi, C.; Razzaghian, H.R.; Pakalapati, G.; Waite, L.; Thilbeault, K.S.; Ronowicz, A.; Wineinger, N.E.; Tiwari, H.K.; Boomsma, D.; et al. Age-related somatic structural changes in the nuclear genome of human blood cells. Am. J. Hum. Genet. 2012, 90, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, M.C.; Agrawal, A.; Cole, J.W.; Hansel, N.N.; Barnes, K.C.; Beaty, T.H.; Bennett, S.N.; Bierut, L.J.; Boerwinkle, E.; Doheny, K.F.; et al. The Gene, Environment Association Studies consortium (GENEVA): Maximizing the knowledge obtained from GWAS by collaboration across studies of multiple conditions. Genet. Epidemiol. 2010, 34, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Laurie, C.C.; Laurie, C.A.; Rice, K.; Doheny, K.F.; Zelnick, L.R.; McHugh, C.P.; Ling, H.; Hetrick, K.N.; Pugh, E.W.; Amos, C.; et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 2012, 44, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Pos, O.; Radvanszky, J.; Buglyo, G.; Pos, Z.; Rusnakova, D.; Nagy, B.; Szemes, T. DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomed. J. 2021, 44, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Zhang, F.; Lupski, J.R. Mechanisms for human genomic rearrangements. Pathogenetics 2008, 1, 4. [Google Scholar] [CrossRef]
- Lupski, J.R. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998, 14, 417–422. [Google Scholar] [CrossRef]
- Shaw, C.J.; Lupski, J.R. Implications of human genome architecture for rearrangement-based disorders: The genomic basis of disease. Hum. Mol. Genet. 2004, 13 (Suppl. S1), R57–R64. [Google Scholar] [CrossRef]
- Hastings, P.J.; Lupski, J.R.; Rosenberg, S.M.; Ira, G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009, 10, 551–564. [Google Scholar] [CrossRef]
- Hastings, P.J.; Ira, G.; Lupski, J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009, 5, e1000327. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.H.; Schild, D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res. 2001, 477, 131–153. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Ozdemir, A.C.; Birkeland, S.R.; Wilson, T.E.; Glover, T.W. Hydroxyurea induces de novo copy number variants in human cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17360–17365. [Google Scholar] [CrossRef] [PubMed]
- Mullenders, L.H.F. Solar UV damage to cellular DNA: From mechanisms to biological effects. Photochem. Photobiol. Sci. 2018, 17, 1842–1852. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Wilson, T.E.; Glover, T.W. Replication stress and mechanisms of CNV formation. Curr. Opin. Genet. Dev. 2012, 22, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.O.A.; Pinto, I.P.; Goncalves, M.W.; da Silva, J.F.; Oliveira, L.G.; da Cruz, A.S.; Silva, D.M.E.; da Silva, C.C.; Pereira, R.W.; da Cruz, A.D. Small de novo CNVs as biomarkers of parental exposure to low doses of ionizing radiation of caesium-137. Sci. Rep. 2018, 8, 5914. [Google Scholar] [CrossRef] [PubMed]
- Zitzelsberger, H.; Unger, K. DNA copy number alterations in radiation-induced thyroid cancer. Clin. Oncol. 2011, 23, 289–296. [Google Scholar] [CrossRef]
- Zitzelsberger, H.; Lehmann, L.; Hieber, L.; Weier, H.U.; Janish, C.; Fung, J.; Negele, T.; Spelsberg, F.; Lengfelder, E.; Demidchik, E.P.; et al. Cytogenetic changes in radiation-induced tumors of the thyroid. Cancer Res. 1999, 59, 135–140. [Google Scholar]
- Hovhannisyan, G.; Harutyunyan, T.; Aroutiounian, R.; Liehr, T. DNA Copy Number Variations as Markers of Mutagenic Impact. Int. J. Mol. Sci. 2019, 20, 4723. [Google Scholar] [CrossRef]
- Kim, P.M.; Korbel, J.O.; Gerstein, M.B. Positive selection at the protein network periphery: Evaluation in terms of structural constraints and cellular context. Proc. Natl. Acad. Sci. USA 2007, 104, 20274–20279. [Google Scholar] [CrossRef] [PubMed]
- Yatsenko, S.A.; Brundage, E.K.; Roney, E.K.; Cheung, S.W.; Chinault, A.C.; Lupski, J.R. Molecular mechanisms for subtelomeric rearrangements associated with the 9q34.3 microdeletion syndrome. Hum. Mol. Genet. 2009, 18, 1924–1936. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Shaw, C.A.; Lu, X.Y.; Sahoo, T.; Bacino, C.A.; Lalani, S.R.; Stankiewicz, P.; Yatsenko, S.A.; Li, Y.; Neill, S.; et al. Identification of chromosome abnormalities in subtelomeric regions by microarray analysis: A study of 5380 cases. Am. J. Med. Genet. A 2008, 146A, 2242–2251. [Google Scholar] [CrossRef] [PubMed]
- She, X.; Horvath, J.E.; Jiang, Z.; Liu, G.; Furey, T.S.; Christ, L.; Clark, R.; Graves, T.; Gulden, C.L.; Alkan, C.; et al. The structure and evolution of centromeric transition regions within the human genome. Nature 2004, 430, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Agiannitopoulos, K.; Pepe, G.; Tsaousis, G.N.; Potska, K.; Bouzarelou, D.; Katseli, A.; Ntogka, C.; Meintani, A.; Tsoulos, N.; Giassas, S.; et al. Copy Number Variations (CNVs) Account for 10.8% of Pathogenic Variants in Patients Referred for Hereditary Cancer Testing. Cancer Genom. Proteom. 2023, 20, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.J.; Locke, D.P.; McGrath, S.D.; Cheng, Z.; Bailey, J.A.; Vallente, R.U.; Pertz, L.M.; Clark, R.A.; Schwartz, S.; Segraves, R.; et al. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005, 77, 78–88. [Google Scholar] [CrossRef]
- McCarroll, S.A.; Altshuler, D.M. Copy-number variation and association studies of human disease. Nat. Genet. 2007, 39 (Suppl. S7), S37–S42. [Google Scholar] [CrossRef] [PubMed]
- Weckselblatt, B.; Rudd, M.K. Human Structural Variation: Mechanisms of Chromosome Rearrangements. Trends Genet. 2015, 31, 587–599. [Google Scholar] [CrossRef]
- Cantsilieris, S.; Baird, P.N.; White, S.J. Molecular methods for genotyping complex copy number polymorphisms. Genomics 2013, 101, 86–93. [Google Scholar] [CrossRef]
- Singh, A.K.; Olsen, M.F.; Lavik, L.A.S.; Vold, T.; Drablos, F.; Sjursen, W. Detecting copy number variation in next generation sequencing data from diagnostic gene panels. BMC Med. Genom. 2021, 14, 214. [Google Scholar] [CrossRef]
- Medvedev, P.; Stanciu, M.; Brudno, M. Computational methods for discovering structural variation with next-generation sequencing. Nat. Methods 2009, 6 (Suppl. S11), S13–S20. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.E.; Walter, K.; Stewart, C.; Handsaker, R.E.; Chen, K.; Alkan, C.; Abyzov, A.; Yoon, S.C.; Ye, K.; Cheetham, R.K.; et al. Mapping copy number variation by population-scale genome sequencing. Nature 2011, 470, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Kidd, J.M.; Marques-Bonet, T.; Aksay, G.; Antonacci, F.; Hormozdiari, F.; Kitzman, J.O.; Baker, C.; Malig, M.; Mutlu, O.; et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009, 41, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Xuan, Z.; Makarov, V.; Ye, K.; Sebat, J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009, 19, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, M.; Goes, F.S.; Zandi, P.P. Whole-genome CNV analysis: Advances in computational approaches. Front. Genet. 2015, 6, 138. [Google Scholar] [CrossRef] [PubMed]
- Mellars, G.; Gomez, K. Mutation detection by Southern blotting. Methods Mol. Biol. 2011, 688, 281–291. [Google Scholar] [PubMed]
- Higuchi, R.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR analysis: Real-time monitoring of DNA amplification reactions. Biotechnology 1993, 11, 1026–1030. [Google Scholar] [CrossRef]
- Sellner, L.N.; Taylor, G.R. MLPA and MAPH: New techniques for detection of gene deletions. Hum. Mutat. 2004, 23, 413–419. [Google Scholar] [CrossRef]
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002, 30, e57. [Google Scholar] [CrossRef]
- Jakobsson, M.; Scholz, S.W.; Scheet, P.; Gibbs, J.R.; VanLiere, J.M.; Fung, H.C.; Szpiech, Z.A.; Degnan, J.H.; Wang, K.; Guerreiro, R.; et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature 2008, 451, 998–1003. [Google Scholar] [CrossRef]
- Janiak, M.C. Of starch and spit. elife 2019, 8, e47523. [Google Scholar] [CrossRef] [PubMed]
- Hardy, K.; Brand-Miller, J.; Brown, K.D.; Thomas, M.G.; Copeland, L. The Importance of Dietary Carbohydrate in Human Evolution. Q. Rev. Biol. 2015, 90, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.W.; Hague, S.M.; Clarimon, J.; Baptista, M.; Gwinn-Hardy, K.; Cookson, M.R.; Singleton, A.B. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 2004, 62, 1835–1838. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Ji, X.; Xing, Y.; Chen, Y.W.; Tao, J. A rare case of 46, XX SRY-negative male with approximately 74-kb duplication in a region upstream of SOX9. Eur. J. Med. Genet. 2013, 56, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.J.; Sock, E.; Buchberger, A.; Just, W.; Denzer, F.; Hoepffner, W.; German, J.; Cole, T.; Mann, J.; Seguin, J.H.; et al. Copy number variation of two separate regulatory regions upstream of SOX9 causes isolated 46,XY or 46,XX disorder of sex development. J. Med. Genet. 2015, 52, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Benko, S.; Gordon, C.T.; Mallet, D.; Sreenivasan, R.; Thauvin-Robinet, C.; Brendehaug, A.; Thomas, S.; Bruland, O.; David, M.; Nicolino, M.; et al. Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. J. Med. Genet. 2011, 48, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.J.; Willatt, L.; Homfray, T.; Woods, C.G. A SOX9 duplication and familial 46,XX developmental testicular disorder. N. Engl. J. Med. 2011, 364, 91–93. [Google Scholar] [CrossRef]
- Hyon, C.; Chantot-Bastaraud, S.; Harbuz, R.; Bhouri, R.; Perrot, N.; Peycelon, M.; Sibony, M.; Rojo, S.; Piguel, X.; Bilan, F.; et al. Refining the regulatory region upstream of SOX9 associated with 46,XX testicular disorders of Sex Development (DSD). Am. J. Med. Genet. A 2015, 167A, 1851–1858. [Google Scholar] [CrossRef]
- Vetro, A.; Dehghani, M.R.; Kraoua, L.; Giorda, R.; Beri, S.; Cardarelli, L.; Merico, M.; Manolakos, E.; Parada-Bustamante, A.; Castro, A.; et al. Testis development in the absence of SRY: Chromosomal rearrangements at SOX9 and SOX3. Eur. J. Hum. Genet. 2015, 23, 1025–1032. [Google Scholar] [CrossRef]
- Deng, N.; Goh, L.K.; Wang, H.; Das, K.; Tao, J.; Tan, I.B.; Zhang, S.; Lee, M.; Wu, J.; Lim, K.H.; et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 2012, 61, 673–684. [Google Scholar] [CrossRef]
- Brown, J.S.; Amend, S.R.; Austin, R.H.; Gatenby, R.A.; Hammarlund, E.U.; Pienta, K.J. Updating the Definition of Cancer. Mol. Cancer Res. 2023, 21, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Sun, J.; Li, G.; Zhu, Y.; Zhang, S.; Kim, S.T.; Sun, J.; Wiklund, F.; Wiley, K.; Isaacs, S.D.; et al. Association of a germ-line copy number variation at 2p24.3 and risk for aggressive prostate cancer. Cancer Res. 2009, 69, 2176–2179. [Google Scholar] [CrossRef] [PubMed]
- Fanciulli, M.; Petretto, E.; Aitman, T.J. Gene copy number variation and common human disease. Clin. Genet. 2010, 77, 201–213. [Google Scholar] [CrossRef]
- Shao, X.; Lv, N.; Liao, J.; Long, J.; Xue, R.; Ai, N.; Xu, D.; Fan, X. Copy number variation is highly correlated with differential gene expression: A pan-cancer study. BMC Med. Genet. 2019, 20, 175. [Google Scholar] [CrossRef] [PubMed]
- Higgins, M.E.; Claremont, M.; Major, J.E.; Sander, C.; Lash, A.E. CancerGenes: A gene selection resource for cancer genome projects. Nucleic Acids Res. 2007, 35, D721–D726. [Google Scholar] [CrossRef] [PubMed]
- Park, R.W.; Kim, T.M.; Kasif, S.; Park, P.J. Identification of rare germline copy number variations over-represented in five human cancer types. Mol. Cancer 2015, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Nagy, R.; Sweet, K.; Eng, C. Highly penetrant hereditary cancer syndromes. Oncogene 2004, 23, 6445–6470. [Google Scholar] [CrossRef]
- Thompson, E.D.; Roberts, N.J.; Wood, L.D.; Eshleman, J.R.; Goggins, M.G.; Kern, S.E.; Klein, A.P.; Hruban, R.H. The genetics of ductal adenocarcinoma of the pancreas in the year 2020: Dramatic progress, but far to go. Mod. Pathol. 2020, 33, 2544–2563. [Google Scholar] [CrossRef]
- Wood, L.D.; Canto, M.I.; Jaffee, E.M.; Simeone, D.M. Pancreatic Cancer: Pathogenesis, Screening, Diagnosis, and Treatment. Gastroenterology 2022, 163, 386–402.e1. [Google Scholar] [CrossRef]
- Morani, A.C.; Hanafy, A.K.; Ramani, N.S.; Katabathina, V.S.; Yedururi, S.; Dasyam, A.K.; Prasad, S.R. Hereditary and Sporadic Pancreatic Ductal Adenocarcinoma: Current Update on Genetics and Imaging. Radiol. Imaging Cancer 2020, 2, e190020. [Google Scholar] [CrossRef]
- Willis, J.A.; Mukherjee, S.; Orlow, I.; Viale, A.; Offit, K.; Kurtz, R.C.; Olson, S.H.; Klein, R.J. Genome-wide analysis of the role of copy-number variation in pancreatic cancer risk. Front. Genet. 2014, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yu, D.; Wu, C.; Zhai, K.; Jiang, G.; Cao, G.; Wang, C.; Liu, Y.; Sun, M.; Li, Z.; et al. Copy number variation at 6q13 functions as a long-range regulator and is associated with pancreatic cancer risk. Carcinogenesis 2012, 33, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Al-Sukhni, W.; Joe, S.; Lionel, A.C.; Zwingerman, N.; Zogopoulos, G.; Marshall, C.R.; Borgida, A.; Holter, S.; Gropper, A.; Moore, S.; et al. Identification of germline genomic copy number variation in familial pancreatic cancer. Hum. Genet. 2012, 131, 1481–1494. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Chelala, C.; Bhakta, V.; Chaplin, T.; Caulee, K.; Baril, P.; Young, B.D.; Lemoine, N.R. Genome-wide DNA copy number analysis in pancreatic cancer using high-density single nucleotide polymorphism arrays. Oncogene 2008, 27, 1951–1960. [Google Scholar] [CrossRef]
- Lucito, R.; Suresh, S.; Walter, K.; Pandey, A.; Lakshmi, B.; Krasnitz, A.; Sebat, J.; Wigler, M.; Klein, A.P.; Brune, K.; et al. Copy-number variants in patients with a strong family history of pancreatic cancer. Cancer Biol. Ther. 2007, 6, 1592–1599. [Google Scholar] [CrossRef]
- Harada, T.; Baril, P.; Gangeswaran, R.; Kelly, G.; Chelala, C.; Bhakta, V.; Caulee, K.; Mahon, P.C.; Lemoine, N.R. Identification of genetic alterations in pancreatic cancer by the combined use of tissue microdissection and array-based comparative genomic hybridisation. Br. J. Cancer 2007, 96, 373–382. [Google Scholar] [CrossRef]
- Fujita, M.; Sugama, S.; Nakai, M.; Takenouchi, T.; Wei, J.; Urano, T.; Inoue, S.; Hashimoto, M. alpha-Synuclein stimulates differentiation of osteosarcoma cells: Relevance to down-regulation of proteasome activity. J. Biol. Chem. 2007, 282, 5736–5748. [Google Scholar] [CrossRef]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef]
- Fu, B.; Luo, M.; Lakkur, S.; Lucito, R.; Iacobuzio-Donahue, C.A. Frequent genomic copy number gain and overexpression of GATA-6 in pancreatic carcinoma. Cancer Biol. Ther. 2008, 7, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, Z.; Fu, B.; Pan, F.; Yachida, S.; Dhara, M.; Albesiano, E.; Li, L.; Naito, Y.; Vilardell, F.; et al. GATA6 activates Wnt signaling in pancreatic cancer by negatively regulating the Wnt antagonist Dickkopf-1. PLoS ONE 2011, 6, e22129. [Google Scholar] [CrossRef]
- O’Kane, G.M.; Grunwald, B.T.; Jang, G.H.; Masoomian, M.; Picardo, S.; Grant, R.C.; Denroche, R.E.; Zhang, A.; Wang, Y.; Lam, B.; et al. GATA6 Expression Distinguishes Classical and Basal-like Subtypes in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4901–4910. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.B.; Chadwick, D.; Zhang, A.; et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin. Cancer Res. 2018, 24, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Wirth, M.; Keller, U.; Saur, D. Rationale for MYC imaging and targeting in pancreatic cancer. EJNMMI Res. 2021, 11, 104. [Google Scholar] [CrossRef]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Beaulieu, M.E.; Jauset, T.; Masso-Valles, D.; Martinez-Martin, S.; Rahl, P.; Maltais, L.; Zacarias-Fluck, M.F.; Casacuberta-Serra, S.; Serrano Del Pozo, E.; Fiore, C.; et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med. 2019, 11, eaar5012. [Google Scholar] [CrossRef]
- Hayashi, A.; Fan, J.; Chen, R.; Ho, Y.J.; Makohon-Moore, A.P.; Lecomte, N.; Zhong, Y.; Hong, J.; Huang, J.; Sakamoto, H.; et al. A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma. Nat. Cancer 2020, 1, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental Determinants of Pancreatic Cancer. Physiol. Rev. 2020, 100, 1707–1751. [Google Scholar] [CrossRef] [PubMed]
- Sodir, N.M.; Kortlever, R.M.; Barthet, V.J.A.; Campos, T.; Pellegrinet, L.; Kupczak, S.; Anastasiou, P.; Swigart, L.B.; Soucek, L.; Arends, M.J.; et al. MYC Instructs and Maintains Pancreatic Adenocarcinoma Phenotype. Cancer Discov. 2020, 10, 588–607. [Google Scholar] [CrossRef] [PubMed]
- Ischenko, I.; D’Amico, S.; Rao, M.; Li, J.; Hayman, M.J.; Powers, S.; Petrenko, O.; Reich, N.C. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat. Commun. 2021, 12, 1482. [Google Scholar] [CrossRef] [PubMed]
- Maddipati, R.; Norgard, R.J.; Baslan, T.; Rathi, K.S.; Zhang, A.; Saeid, A.; Higashihara, T.; Wu, F.; Kumar, A.; Annamalai, V.; et al. MYC Levels Regulate Metastatic Heterogeneity in Pancreatic Adenocarcinoma. Cancer Discov. 2022, 12, 542–561. [Google Scholar] [CrossRef]
- Hassan, Z.; Schneeweis, C.; Wirth, M.; Veltkamp, C.; Dantes, Z.; Feuerecker, B.; Ceyhan, G.O.; Knauer, S.K.; Weichert, W.; Schmid, R.M.; et al. MTOR inhibitor-based combination therapies for pancreatic cancer. Br. J. Cancer 2018, 118, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Herrmann, D.; Evans, T.J.; Morton, J.P.; Timpson, P. Combating pancreatic cancer with PI3K pathway inhibitors in the era of personalised medicine. Gut 2019, 68, 742–758. [Google Scholar] [CrossRef]
- Driscoll, D.R.; Karim, S.A.; Sano, M.; Gay, D.M.; Jacob, W.; Yu, J.; Mizukami, Y.; Gopinathan, A.; Jodrell, D.I.; Evans, T.R.; et al. mTORC2 Signaling Drives the Development and Progression of Pancreatic Cancer. Cancer Res. 2016, 76, 6911–6923. [Google Scholar] [CrossRef]
- Morran, D.C.; Wu, J.; Jamieson, N.B.; Mrowinska, A.; Kalna, G.; Karim, S.A.; Au, A.Y.; Scarlett, C.J.; Chang, D.K.; Pajak, M.Z.; et al. Targeting mTOR dependency in pancreatic cancer. Gut 2014, 63, 1481–1489. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Kumarasamy, V.; Ruiz, A.; Sivinski, J.; Chung, S.; Grant, A.; Vail, P.; Chauhan, S.S.; Jie, T.; Riall, T.S.; et al. Cell cycle plasticity driven by MTOR signaling: Integral resistance to CDK4/6 inhibition in patient-derived models of pancreatic cancer. Oncogene 2019, 38, 3355–3370. [Google Scholar] [CrossRef]
- Allen-Petersen, B.L.; Risom, T.; Feng, Z.; Wang, Z.; Jenny, Z.P.; Thoma, M.C.; Pelz, K.R.; Morton, J.P.; Sansom, O.J.; Lopez, C.D.; et al. Activation of PP2A and Inhibition of mTOR Synergistically Reduce MYC Signaling and Decrease Tumor Growth in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2019, 79, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Stranger, B.E.; Forrest, M.S.; Dunning, M.; Ingle, C.E.; Beazley, C.; Thorne, N.; Redon, R.; Bird, C.P.; de Grassi, A.; Lee, C.; et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 2007, 315, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Krimpenfort, P.; Ijpenberg, A.; Song, J.Y.; van der Valk, M.; Nawijn, M.; Zevenhoven, J.; Berns, A. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature 2007, 448, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Roussel, M.F. The INK4 family of cell cycle inhibitors in cancer. Oncogene 1999, 18, 5311–5317. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.L.; Wu, S.F.; Huang, X.; Zhou, S. Integrated Analysis of ECT2 and COL17A1 as Potential Biomarkers for Pancreatic Cancer. Dis. Markers 2022, 2022, 9453549. [Google Scholar] [CrossRef] [PubMed]
- Kozawa, K.; Sekai, M.; Ohba, K.; Ito, S.; Sako, H.; Maruyama, T.; Kakeno, M.; Shirai, T.; Kuromiya, K.; Kamasaki, T.; et al. The CD44/COL17A1 pathway promotes the formation of multilayered, transformed epithelia. Curr. Biol. 2021, 31, 3086–3097.e7. [Google Scholar] [CrossRef]
- Ho, T.T.B.; Nasti, A.; Seki, A.; Komura, T.; Inui, H.; Kozaka, T.; Kitamura, Y.; Shiba, K.; Yamashita, T.; Yamashita, T.; et al. Combination of gemcitabine and anti-PD-1 antibody enhances the anticancer effect of M1 macrophages and the Th1 response in a murine model of pancreatic cancer liver metastasis. J. Immunother. Cancer 2020, 8, e001367. [Google Scholar] [CrossRef]
- Pu, N.; Gao, S.; Yin, H.; Li, J.A.; Wu, W.; Fang, Y.; Zhang, L.; Rong, Y.; Xu, X.; Wang, D.; et al. Cell-intrinsic PD-1 promotes proliferation in pancreatic cancer by targeting CYR61/CTGF via the hippo pathway. Cancer Lett. 2019, 460, 42–53. [Google Scholar] [CrossRef]
- Hayashi, H.; Higashi, T.; Miyata, T.; Yamashita, Y.I.; Baba, H. Recent advances in precision medicine for pancreatic ductal adenocarcinoma. Ann. Gastroenterol. Surg. 2021, 5, 457–466. [Google Scholar] [CrossRef]
- Zhan, Q.; Wen, C.; Zhao, Y.; Fang, L.; Jin, Y.; Zhang, Z.; Zou, S.; Li, F.; Yang, Y.; Wu, L.; et al. Identification of copy number variation-driven molecular subtypes informative for prognosis and treatment in pancreatic adenocarcinoma of a Chinese cohort. eBioMedicine 2021, 74, 103716. [Google Scholar] [CrossRef]
- Ben-David, U.; Amon, A. Context is everything: Aneuploidy in cancer. Nat. Rev. Genet. 2020, 21, 44–62. [Google Scholar] [CrossRef]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e3. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Balli, D.; Rech, A.J.; Stanger, B.Z.; Vonderheide, R.H. Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 3129–3138. [Google Scholar] [CrossRef] [PubMed]
- Petersson, A.; Andersson, N.; Hau, S.O.; Eberhard, J.; Karlsson, J.; Chattopadhyay, S.; Valind, A.; Elebro, J.; Nodin, B.; Leandersson, K.; et al. Branching Copy-Number Evolution and Parallel Immune Profiles across the Regional Tumor Space of Resected Pancreatic Cancer. Mol. Cancer Res. 2022, 20, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, Y.; Liu, X.; Zhou, K.; Wu, J.; Chen, J.; Chen, C.; Chen, L.; Zheng, J. Development and clinical validation of a novel 9-gene prognostic model based on multi-omics in pancreatic adenocarcinoma. Pharmacol. Res. 2021, 164, 105370. [Google Scholar] [CrossRef]
- Chen, S.; Auletta, T.; Dovirak, O.; Hutter, C.; Kuntz, K.; El-ftesi, S.; Kendall, J.; Han, H.; Von Hoff, D.D.; Ashfaq, R.; et al. Copy number alterations in pancreatic cancer identify recurrent PAK4 amplification. Cancer Biol. Ther. 2008, 7, 1793–1802. [Google Scholar] [CrossRef]
- Zheng, Q.; Yu, X.; Zhang, Q.; He, Y.; Guo, W. Genetic characteristics and prognostic implications of m1A regulators in pancreatic cancer. Biosci. Rep. 2021, 41, BSR20210337. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhang, J.; Xu, C.; Yang, J.; Zhang, Y.; Liu, M.; Shi, X.; Li, X.; Zhan, H.; Chen, W.; et al. An integrated model of N6-methyladenosine regulators to predict tumor aggressiveness and immune evasion in pancreatic cancer. eBioMedicine 2021, 65, 103271. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.L.; Wu, Z.; Li, R.K.; Xing, X.; Jiang, Y.S.; Li, J.; Wang, Y.H.; Hu, L.P.; Wang, X.; Qin, W.T.; et al. Deciphering the genomic and lncRNA landscapes of aerobic glycolysis identifies potential therapeutic targets in pancreatic cancer. Int. J. Biol. Sci. 2021, 17, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.W.; Shi, Z.Z.; Shen, T.Y.; Che, X.; Wang, Z.; Shi, S.S.; Xu, X.; Cai, Y.; Zhao, P.; Wang, C.F.; et al. Identification of genomic alterations in pancreatic cancer using array-based comparative genomic hybridization. PLoS ONE 2014, 9, e114616. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Ashida, R.; Ohshima, K.; Uesaka, K.; Sugiura, T.; Okamura, Y.; Ohgi, K.; Ohnami, S.; Nagashima, T.; Yamaguchi, K. Genomic landscape of pancreatic cancer in the Japanese version of the Cancer Genome Atlas. Ann. Gastroenterol. Surg. 2023, 7, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Huang, C.; Cui Zhou, D.; Hu, Y.; Lih, T.M.; Savage, S.R.; Krug, K.; Clark, D.J.; Schnaubelt, M.; Chen, L.; et al. Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 2021, 184, 5031–5052.e26. [Google Scholar] [CrossRef]
- Zhang, X.; Mao, T.; Zhang, B.; Xu, H.; Cui, J.; Jiao, F.; Chen, D.; Wang, Y.; Hu, J.; Xia, Q.; et al. Characterization of the genomic landscape in large-scale Chinese patients with pancreatic cancer. eBioMedicine 2022, 77, 103897. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ding, Y.; Gong, Y.; Zhao, S.; Li, M.; Li, X.; Song, G.; Zhai, B.; Liu, J.; Shao, Y.; et al. The genetic landscape of pancreatic head ductal adenocarcinoma in China and prognosis stratification. BMC Cancer 2022, 22, 186. [Google Scholar] [CrossRef]
- Detlefsen, S.; Boldt, H.B.; Burton, M.; Thomsen, M.M.; Rasmussen, L.G.; Orbeck, S.V.; Pfeiffer, P.; Mortensen, M.B.; de Stricker, K. High overall copy number variation burden by genome-wide methylation profiling holds negative prognostic value in surgically treated pancreatic ductal adenocarcinoma. Hum. Pathol. 2023, 142, 68–80. [Google Scholar] [CrossRef]
- Kwon, M.J.; Jeon, J.Y.; Park, H.R.; Nam, E.S.; Cho, S.J.; Shin, H.S.; Kwon, J.H.; Kim, J.S.; Han, B.; Kim, D.H.; et al. Low frequency of KRAS mutation in pancreatic ductal adenocarcinomas in Korean patients and its prognostic value. Pancreas 2015, 44, 484–492. [Google Scholar] [CrossRef]
- Shin, S.H.; Kim, S.C.; Hong, S.M.; Kim, Y.H.; Song, K.B.; Park, K.M.; Lee, Y.J. Genetic alterations of K-ras, p53, c-erbB-2, and DPC4 in pancreatic ductal adenocarcinoma and their correlation with patient survival. Pancreas 2013, 42, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Giovannetti, E.; Hwang, J.H.; Petrini, I.; Wang, Q.; Voortman, J.; Wang, Y.; Steinberg, S.M.; Funel, N.; Meltzer, P.S.; et al. Loss of 18q22.3 involving the carboxypeptidase of glutamate-like gene is associated with poor prognosis in resected pancreatic cancer. Clin. Cancer Res. 2012, 18, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.T.; Deiotte, R.; Lenkiewicz, E.; Malasi, S.; Holley, T.; Evers, L.; Posner, R.G.; Jones, T.; Han, H.; Sausen, M.; et al. Clinical study of genomic drivers in pancreatic ductal adenocarcinoma. Br. J. Cancer 2017, 117, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Shi, X.; Gao, S.; Hou, Q.; Jiang, L.; Li, B.; Shen, J.; Wang, H.; Shen, S.; Zhang, G.; et al. The Landscape of Genetic Alterations Stratified Prognosis in Oriental Pancreatic Cancer Patients. Front. Oncol. 2021, 11, 717989. [Google Scholar] [CrossRef] [PubMed]
- Fujii, A.; Masuda, T.; Iwata, M.; Tobo, T.; Wakiyama, H.; Koike, K.; Kosai, K.; Nakano, T.; Kuramitsu, S.; Kitagawa, A.; et al. The novel driver gene ASAP2 is a potential druggable target in pancreatic cancer. Cancer Sci. 2021, 112, 1655–1668. [Google Scholar] [CrossRef]
- Birnbaum, D.J.; Bertucci, F.; Finetti, P.; Adelaide, J.; Giovannini, M.; Turrini, O.; Delpero, J.R.; Raoul, J.L.; Chaffanet, M.; Moutardier, V.; et al. Expression of Genes with Copy Number Alterations and Survival of Patients with Pancreatic Adenocarcinoma. Cancer Genom. Proteom. 2016, 13, 191–200. [Google Scholar]
- van Hattem, W.A.; Carvalho, R.; Li, A.; Offerhaus, G.J.; Goggins, M. Amplification of EMSY gene in a subset of sporadic pancreatic adenocarcinomas. Int. J. Clin. Exp. Pathol. 2008, 1, 343–351. [Google Scholar]
- Huang, L.; Yuan, X.; Zhao, L.; Han, Q.; Yan, H.; Yuan, J.; Guan, S.; Xu, X.; Dai, G.; Wang, J.; et al. Gene signature developed for predicting early relapse and survival in early-stage pancreatic cancer. BJS Open 2023, 7, zrad031. [Google Scholar] [CrossRef]
- Kalimuthu, S.N.; Wilson, G.W.; Grant, R.C.; Seto, M.; O’Kane, G.; Vajpeyi, R.; Notta, F.; Gallinger, S.; Chetty, R. Morphological classification of pancreatic ductal adenocarcinoma that predicts molecular subtypes and correlates with clinical outcome. Gut 2020, 69, 317–328. [Google Scholar] [CrossRef]
- Thomsen, M.M.; Larsen, M.H.; Di Caterino, T.; Hedegaard Jensen, G.; Mortensen, M.B.; Detlefsen, S. Accuracy and clinical outcomes of pancreatic EUS-guided fine-needle biopsy in a consecutive series of 852 specimens. Endosc. Ultrasound 2022, 11, 306–318. [Google Scholar]
- Fitzpatrick, M.J.; Hernandez-Barco, Y.G.; Krishnan, K.; Casey, B.; Pitman, M.B. Evaluating triage protocols for endoscopic ultrasound-guided fine needle biopsies of the pancreas. J. Am. Soc. Cytopathol. 2020, 9, 396–404. [Google Scholar] [CrossRef]
- Tibiletti, M.G.; Carnevali, I.; Pensotti, V.; Chiaravalli, A.M.; Facchi, S.; Volorio, S.; Mariette, F.; Mariani, P.; Fortuzzi, S.; Pierotti, M.A.; et al. OncoPan((R)): An NGS-Based Screening Methodology to Identify Molecular Markers for Therapy and Risk Assessment in Pancreatic Ductal Adenocarcinoma. Biomedicines 2022, 10, 1208. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, Y.S.; Iwata, T.; Okugawa, Y.; Tanaka, K.; Sakurai, H.; Watanabe, M. HER2-positive adenocarcinoma arising from heterotopic pancreas tissue in the duodenum: A case report. World J. Gastroenterol. 2021, 27, 4738–4745. [Google Scholar] [CrossRef] [PubMed]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Goldberg, S.B.; Narayan, A.; Kole, A.J.; Decker, R.H.; Teysir, J.; Carriero, N.J.; Lee, A.; Nemati, R.; Nath, S.K.; Mane, S.M.; et al. Early Assessment of Lung Cancer Immunotherapy Response via Circulating Tumor DNA. Clin. Cancer Res. 2018, 24, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Lu, L.; Zhou, Z.; Liu, J.; Zhang, D.; Nan, K.; Zhao, X.; Li, F.; Tian, L.; Dong, H.; et al. CNV Detection from Circulating Tumor DNA in Late Stage Non-Small Cell Lung Cancer Patients. Genes 2019, 10, 926. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ma, R.; Luo, C.; Xie, Q.; Ning, X.; Sun, K.; Meng, F.; Zhou, M.; Sun, J. Noninvasive early differential diagnosis and progression monitoring of ovarian cancer using the copy number alterations of plasma cell-free DNA. Transl. Res. 2023, 262, 12–24. [Google Scholar] [CrossRef]
- Kuuselo, R.; Simon, R.; Karhu, R.; Tennstedt, P.; Marx, A.H.; Izbicki, J.R.; Yekebas, E.; Sauter, G.; Kallioniemi, A. 19q13 amplification is associated with high grade and stage in pancreatic cancer. Genes Chromosomes Cancer 2010, 49, 569–575. [Google Scholar] [CrossRef]
- Gutierrez, M.L.; Munoz-Bellvis, L.; Abad Mdel, M.; Bengoechea, O.; Gonzalez-Gonzalez, M.; Orfao, A.; Sayagues, J.M. Association between genetic subgroups of pancreatic ductal adenocarcinoma defined by high density 500 K SNP-arrays and tumor histopathology. PLoS ONE 2011, 6, e22315. [Google Scholar] [CrossRef]
- Sung, H.Y.; Wu, H.G.; Ahn, J.H.; Park, W.Y. Dcr3 inhibit p53-dependent apoptosis in gamma-irradiated lung cancer cells. Int. J. Radiat. Biol. 2010, 86, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, C.; Zhuang, G.; Luo, H.; Su, J.; Yin, P.; Wang, J. Decoy receptor 3 overexpression and immunologic tolerance in hepatocellular carcinoma (HCC) development. Cancer Investig. 2008, 26, 965–974. [Google Scholar] [CrossRef]
- Ho, C.H.; Chen, C.L.; Li, W.Y.; Chen, C.J. Decoy receptor 3, upregulated by Epstein-Barr virus latent membrane protein 1, enhances nasopharyngeal carcinoma cell migration and invasion. Carcinogenesis 2009, 30, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Luo, D. Over-expression of decoy receptor 3 in gastric precancerous lesions and carcinoma. Ups. J. Med. Sci. 2008, 113, 297–304. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsuji, S.; Hosotani, R.; Yonehara, S.; Masui, T.; Tulachan, S.S.; Nakajima, S.; Kobayashi, H.; Koizumi, M.; Toyoda, E.; Ito, D.; et al. Endogenous decoy receptor 3 blocks the growth inhibition signals mediated by Fas ligand in human pancreatic adenocarcinoma. Int. J. Cancer 2003, 106, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, G.; te Kronnie, G.; Basso, G. The effects of siRNA-mediated inhibition of E2A-PBX1 on EB-1 and Wnt16b expression in the 697 pre-B leukemia cell line. Haematologica 2006, 91, 765–771. [Google Scholar] [PubMed]
- Abiatari, I.; Gillen, S.; DeOliveira, T.; Klose, T.; Bo, K.; Giese, N.A.; Friess, H.; Kleeff, J. The microtubule-associated protein MAPRE2 is involved in perineural invasion of pancreatic cancer cells. Int. J. Oncol. 2009, 35, 1111–1116. [Google Scholar] [PubMed][Green Version]
- Kimura, Y.; Morita, T.; Hayashi, K.; Miki, T.; Sobue, K. Myocardin functions as an effective inducer of growth arrest and differentiation in human uterine leiomyosarcoma cells. Cancer Res. 2010, 70, 501–511. [Google Scholar] [CrossRef]
- de Oliveira, S.I.; Andrade, L.N.; Onuchic, A.C.; Nonogaki, S.; Fernandes, P.D.; Pinheiro, M.C.; Rohde, C.B.; Chammas, R.; Jancar, S. Platelet-activating factor receptor (PAF-R)-dependent pathways control tumour growth and tumour response to chemotherapy. BMC Cancer 2010, 10, 200. [Google Scholar] [CrossRef]
- Legoffic, A.; Calvo, E.; Cano, C.; Folch-Puy, E.; Barthet, M.; Delpero, J.R.; Ferres-Maso, M.; Dagorn, J.C.; Closa, D.; Iovanna, J. The reg4 gene, amplified in the early stages of pancreatic cancer development, is a promising therapeutic target. PLoS ONE 2009, 4, e7495. [Google Scholar] [CrossRef]
- Violette, S.; Festor, E.; Pandrea-Vasile, I.; Mitchell, V.; Adida, C.; Dussaulx, E.; Lacorte, J.M.; Chambaz, J.; Lacasa, M.; Lesuffleur, T. Reg IV, a new member of the regenerating gene family, is overexpressed in colorectal carcinomas. Int. J. Cancer 2003, 103, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Bishnupuri, K.S.; Luo, Q.; Murmu, N.; Houchen, C.W.; Anant, S.; Dieckgraefe, B.K. Reg IV activates the epidermal growth factor receptor/Akt/AP-1 signaling pathway in colon adenocarcinomas. Gastroenterology 2006, 130, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Oue, N.; Sasahira, T.; Yi, L.; Moriwaka, Y.; Shimomoto, T.; Fujii, K.; Ohmori, H.; Yasui, W. Reg IV enhances peritoneal metastasis in gastric carcinomas. Cell Prolif. 2009, 42, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Oue, N.; Hamai, Y.; Mitani, Y.; Matsumura, S.; Oshimo, Y.; Aung, P.P.; Kuraoka, K.; Nakayama, H.; Yasui, W. Gene expression profile of gastric carcinoma: Identification of genes and tags potentially involved in invasion, metastasis, and carcinogenesis by serial analysis of gene expression. Cancer Res. 2004, 64, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lai, M.; Lv, B.; Gu, X.; Wang, H.; Zhu, Y.; Zhu, Y.; Shao, L.; Wang, G. Overexpression of Reg IV in colorectal adenoma. Cancer Lett. 2003, 200, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Takehara, A.; Eguchi, H.; Ohigashi, H.; Ishikawa, O.; Kasugai, T.; Hosokawa, M.; Katagiri, T.; Nakamura, Y.; Nakagawa, H. Novel tumor marker REG4 detected in serum of patients with resectable pancreatic cancer and feasibility for antibody therapy targeting REG4. Cancer Sci. 2006, 97, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- South, S.T.; Brothman, A.R. Clinical laboratory implementation of cytogenomic microarrays. Cytogenet. Genome Res. 2011, 135, 203–211. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, N.; Dijkhuizen, T.; Hehir-Kwa, J.Y.; Carter, N.P.; Feuk, L.; Firth, H.V.; Kuhn, R.M.; Ledbetter, D.H.; Martin, C.L.; van Ravenswaaij-Arts, C.M.; et al. Diagnostic interpretation of array data using public databases and internet sources. Hum. Mutat. 2012, 33, 930–940. [Google Scholar] [CrossRef]
- Haraksingh, R.R.; Abyzov, A.; Gerstein, M.; Urban, A.E.; Snyder, M. Genome-wide mapping of copy number variation in humans: Comparative analysis of high resolution array platforms. PLoS ONE 2011, 6, e27859. [Google Scholar] [CrossRef]
- Perry, G.H.; Ben-Dor, A.; Tsalenko, A.; Sampas, N.; Rodriguez-Revenga, L.; Tran, C.W.; Scheffer, A.; Steinfeld, I.; Tsang, P.; Yamada, N.A.; et al. The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet. 2008, 82, 685–695. [Google Scholar] [CrossRef]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T.; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Vermeesch, J.R.; Brady, P.D.; Sanlaville, D.; Kok, K.; Hastings, R.J. Genome-wide arrays: Quality criteria and platforms to be used in routine diagnostics. Hum. Mutat. 2012, 33, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Chen, S.; Hu, Y.; Huang, W. Single-cell RNA transcriptome reveals the intra-tumoral heterogeneity and regulators underlying tumor progression in metastatic pancreatic ductal adenocarcinoma. Cell Death Discov. 2021, 7, 331. [Google Scholar] [CrossRef]
- Mahdipour-Shirayeh, A.; Erdmann, N.; Leung-Hagesteijn, C.; Tiedemann, R.E. sciCNV: High-throughput paired profiling of transcriptomes and DNA copy number variations at single-cell resolution. Brief. Bioinform. 2022, 23, bbab413. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Bai, S.; Henderson, Y.C.; Lin, Y.; Schalck, A.; Yan, Y.; Kumar, T.; Hu, M.; Sei, E.; Davis, A.; et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 2021, 39, 599–608. [Google Scholar] [CrossRef]
- Fu, X.; Patel, H.P.; Coppola, S.; Xu, L.; Cao, Z.; Lenstra, T.L.; Grima, R. Quantifying how post-transcriptional noise and gene copy number variation bias transcriptional parameter inference from mRNA distributions. eLife 2022, 11, e82493. [Google Scholar] [CrossRef]
- Erickson, A.; He, M.; Berglund, E.; Marklund, M.; Mirzazadeh, R.; Schultz, N.; Kvastad, L.; Andersson, A.; Bergenstrahle, L.; Bergenstrahle, J.; et al. Spatially resolved clonal copy number alterations in benign and malignant tissue. Nature 2022, 608, 360–367. [Google Scholar] [CrossRef]
- Mu, Q.; Wang, J. CNAPE: A Machine Learning Method for Copy Number Alteration Prediction from Gene Expression. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 306–311. [Google Scholar] [CrossRef]
- Canto, M.I.; Goggins, M.; Yeo, C.J.; Griffin, C.; Axilbund, J.E.; Brune, K.; Ali, S.Z.; Jagannath, S.; Petersen, G.M.; Fishman, E.K.; et al. Screening for pancreatic neoplasia in high-risk individuals: An EUS-based approach. Clin. Gastroenterol. Hepatol. 2004, 2, 606–621. [Google Scholar] [CrossRef]
- Fanale, D.; Iovanna, J.L.; Calvo, E.L.; Berthezene, P.; Belleau, P.; Dagorn, J.C.; Bronte, G.; Cicero, G.; Bazan, V.; Rolfo, C.; et al. Germline copy number variation in the YTHDC2 gene: Does it have a role in finding a novel potential molecular target involved in pancreatic adenocarcinoma susceptibility? Expert Opin. Ther. Targets 2014, 18, 841–850. [Google Scholar] [CrossRef]
- Lin, B.; Pan, Y.; Yu, D.; Dai, S.; Sun, H.; Chen, S.; Zhang, J.; Xiang, Y.; Huang, C. Screening and Identifying m6A Regulators as an Independent Prognostic Biomarker in Pancreatic Cancer Based on The Cancer Genome Atlas Database. Biomed. Res. Int. 2021, 2021, 5573628. [Google Scholar] [CrossRef] [PubMed]
- Laurila, E.; Savinainen, K.; Kuuselo, R.; Karhu, R.; Kallioniemi, A. Characterization of the 7q21-q22 amplicon identifies ARPC1A, a subunit of the Arp2/3 complex, as a regulator of cell migration and invasion in pancreatic cancer. Genes Chromosomes Cancer. 2009, 48, 330–339. [Google Scholar] [CrossRef]
- Lin, Z.; Liu, J.; Xiao, X.; Meng, J. Expression and prognostic significance of epithelial cell transforming sequence 2 in invasive breast cancer. Chin. J. Phys. Train. 2023, 46, 780–785. [Google Scholar]
- Rausch, V.; Krieg, A.; Camps, J.; Behrens, B.; Beier, M.; Wangsa, D.; Heselmeyer-Haddad, K.; Baldus, S.E.; Knoefel, W.T.; Ried, T.; et al. Array comparative genomic hybridization of 18 pancreatic ductal adenocarcinomas and their autologous metastases. BMC Res. Notes. 2017, 10, 560. [Google Scholar] [CrossRef] [PubMed]
- Qi, B.; Liu, H.; Dong, Y.; Shi, X.; Zhou, Q.; Zeng, F.; Bao, N.; Li, Q.; Yuan, Y.; Yao, L.; et al. The nine ADAMs family members serve as potential biomarkers for immune infiltration in pancreatic adenocarcinoma. PeerJ. 2020, 8, e9736. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yu, Y.; Zong, K.; Lv, P.; Gu, Y. Up-regulation of IGF2BP2 by multiple mechanisms in pancreatic cancer promotes cancer proliferation by activating the PI3K/Akt signaling pathway. J. Exp. Clin. Cancer. Res. 2019, 38, 497. [Google Scholar] [CrossRef]
- Zhong, H.; Shi, Q.; Wen, Q.; Chen, J.; Li, X.; Ruan, R.; Zeng, S.; Dai, X.; Xiong, J.; Li, L.; et al. Pan-cancer analysis reveals potential of FAM110A as a prognostic and immunological biomarker in human cancer. Front. Immunol. 2023, 14, 1058627. [Google Scholar] [CrossRef]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef]
- McCarroll, S.A. Extending genome-wide association studies to copy-number variation. Hum. Mol. Genet. 2008, 17, R135–R142. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oketch, D.J.A.; Giulietti, M.; Piva, F. Copy Number Variations in Pancreatic Cancer: From Biological Significance to Clinical Utility. Int. J. Mol. Sci. 2024, 25, 391. https://doi.org/10.3390/ijms25010391
Oketch DJA, Giulietti M, Piva F. Copy Number Variations in Pancreatic Cancer: From Biological Significance to Clinical Utility. International Journal of Molecular Sciences. 2024; 25(1):391. https://doi.org/10.3390/ijms25010391
Chicago/Turabian StyleOketch, Daisy J. A., Matteo Giulietti, and Francesco Piva. 2024. "Copy Number Variations in Pancreatic Cancer: From Biological Significance to Clinical Utility" International Journal of Molecular Sciences 25, no. 1: 391. https://doi.org/10.3390/ijms25010391
APA StyleOketch, D. J. A., Giulietti, M., & Piva, F. (2024). Copy Number Variations in Pancreatic Cancer: From Biological Significance to Clinical Utility. International Journal of Molecular Sciences, 25(1), 391. https://doi.org/10.3390/ijms25010391