
HTRA2/OMI-Mediated Mitochondrial Quality Control Alters Macrophage Polarization Affecting Systemic Chronic Inflammation

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Dysregulated Macrophage Polarization Promotes SCI Progression
3. HtrA2 Is a Novel Inflammatory Mediator and a Potential Target for Anti-Inflammatory Therapies
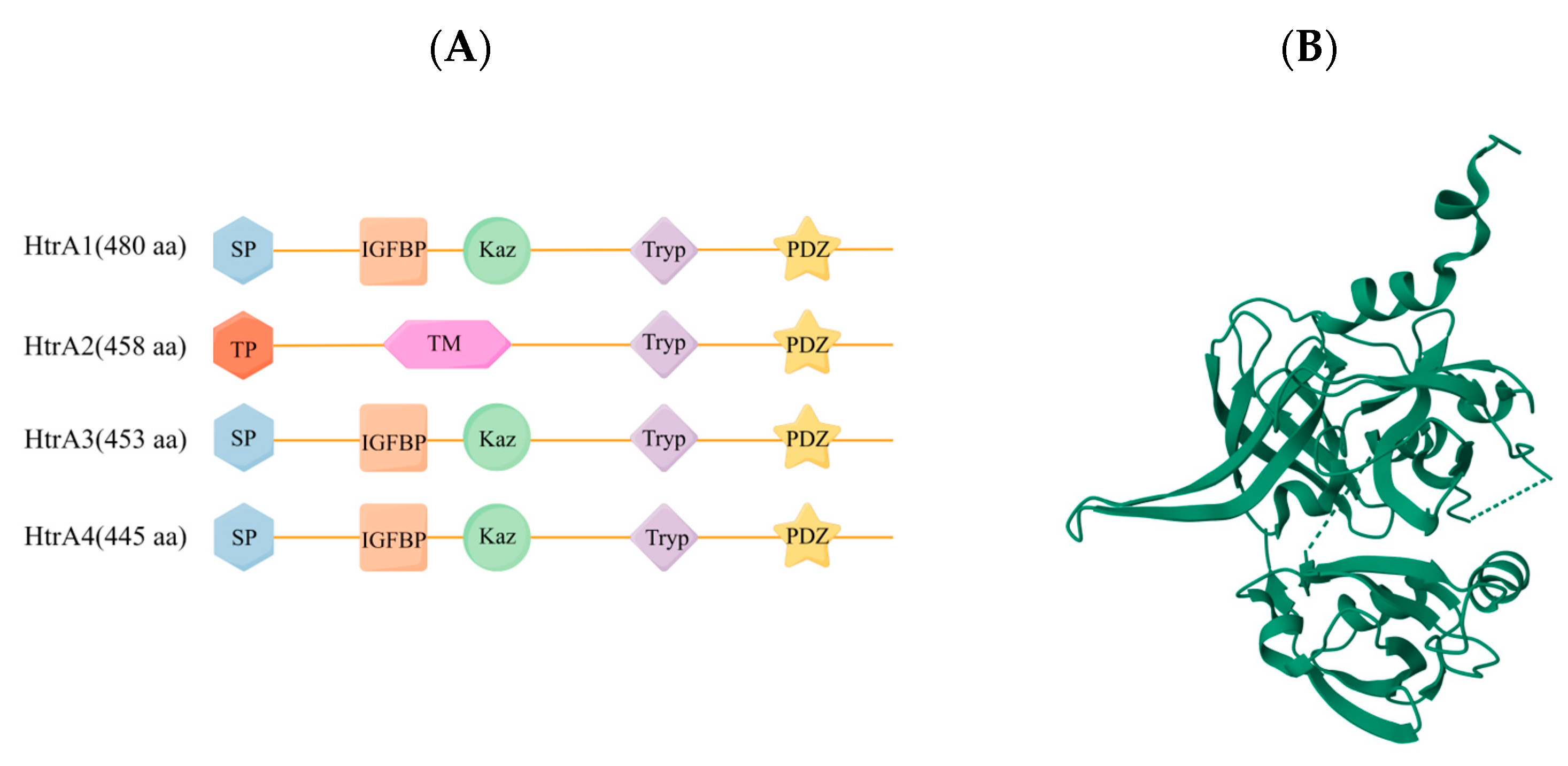
3.1. Structural Features of HtrA2
3.2. Functions of HtrA2 in MQC
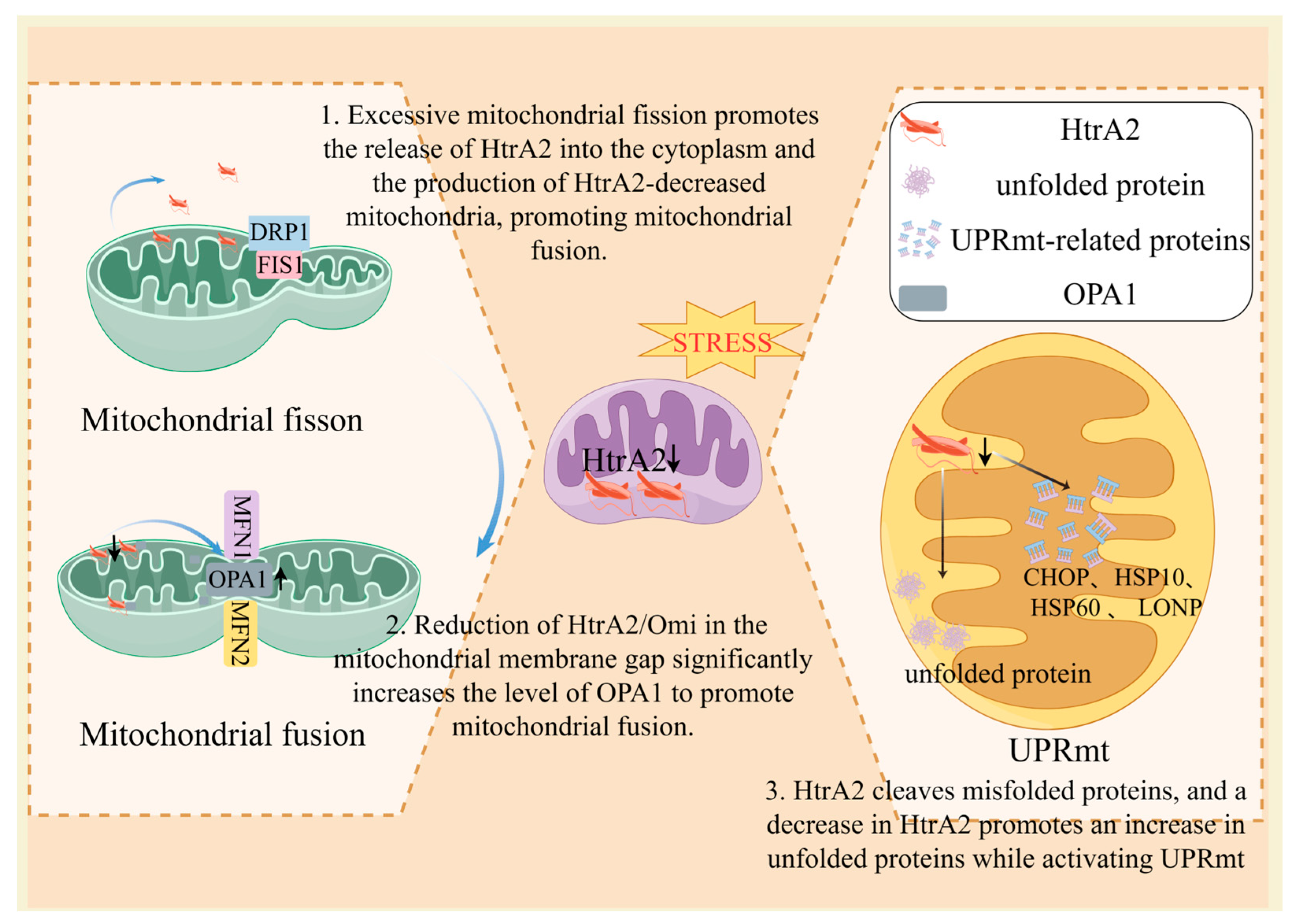
3.2.1. HtrA2 Dysregulation Triggers UPRmt
3.2.2. HtrA2 Dysregulation Induces an Imbalance in Mitochondrial Dynamics
4. HtrA2-Mediated Modulation of Mitochondrial Quality Control Signaling Is Involved in the Regulation of Macrophage Polarization Phenotype in SCI
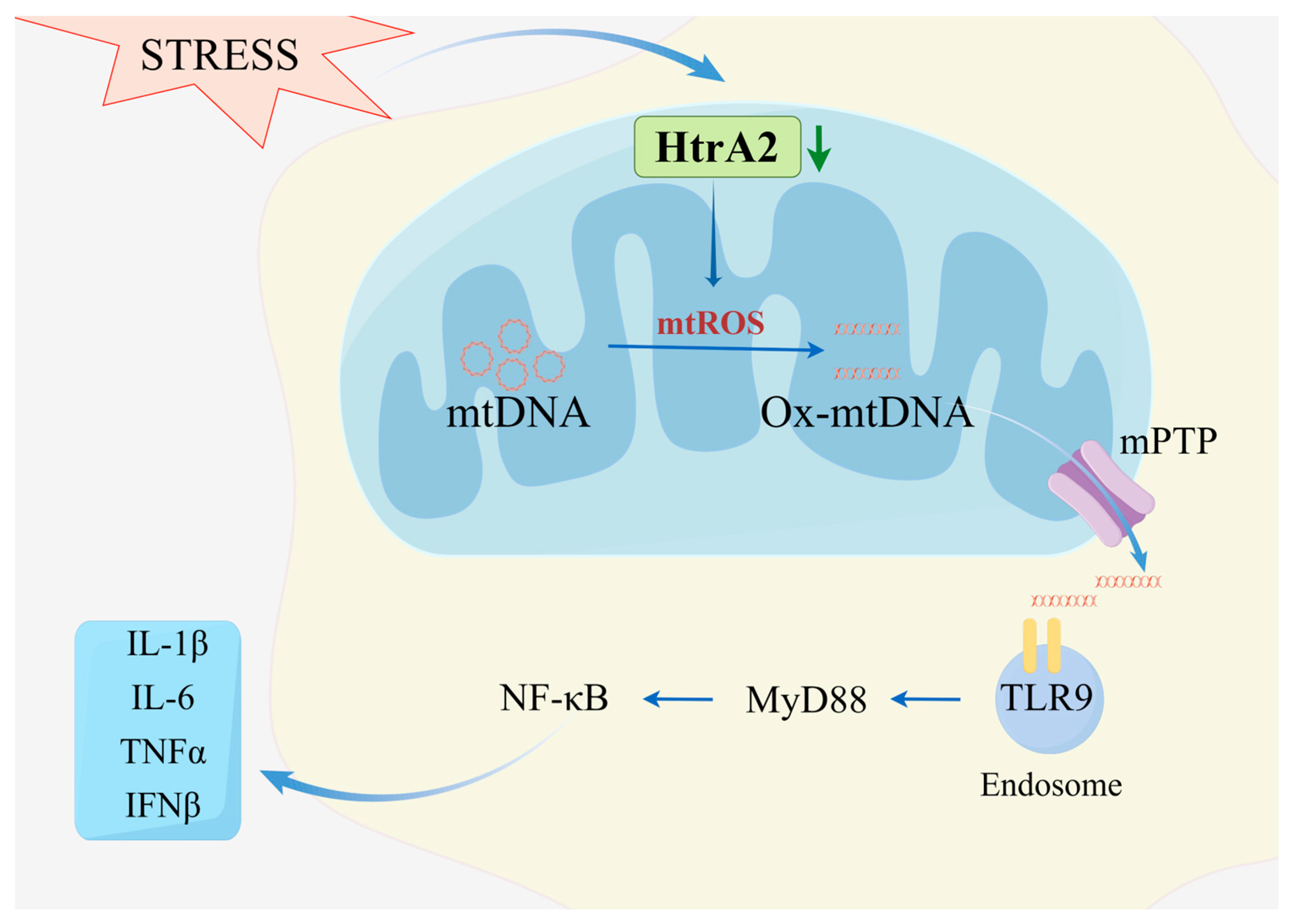
4.1. Aberrant HtrA2 Function Promotes ROS Generation Inducing Ox-mtDNA-Catalyzed Macrophage Immune Activation
4.2. OXPHOS Complex Abnormalities Promote Pro-Inflammatory Macrophages Polarization
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, W.B.S.; Meng, J.; Zhang, J. Does Low Grade Systemic Inflammation Have a Role in Chronic Pain? Front. Mol. Neurosci. 2021, 14, 785214. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-Aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef]
- Liston, A.; Masters, S.L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 2017, 17, 208–214. [Google Scholar] [CrossRef]
- Frank, D.; Vince, J.E. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef]
- Alivernini, S.; MacDonald, L.; Elmesmari, A.; Finlay, S.; Tolusso, B.; Gigante, M.R.; Petricca, L.; Di Mario, C.; Bui, L.; Perniola, S.; et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat. Med. 2020, 26, 1295–1306. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Shao, L.W.; Peng, Q.; Dong, M.; Gao, K.; Li, Y.; Li, Y.; Li, C.Y.; Liu, Y. Histone deacetylase HDA-1 modulates mitochondrial stress response and longevity. Nat. Commun. 2020, 11, 4639. [Google Scholar] [CrossRef]
- Yao, B.F.; Luo, X.J.; Peng, J. A review for the correlation between optic atrophy 1-dependent mitochondrial fusion and cardiovascular disorders. Int. J. Biol. Macromol. 2023, 254 Pt 2, 127910. [Google Scholar] [CrossRef]
- Ng, M.Y.W.; Wai, T.; Simonsen, A. Quality control of the mitochondrion. Dev. Cell 2021, 56, 881–905. [Google Scholar] [CrossRef]
- Kawano, I.; Bazila, B.; Ježek, P.; Dlasková, A. Mitochondrial Dynamics and Cristae Shape Changes during Metabolic Reprogramming. Antioxid. Redox Signal. 2023, 39, 684–707. [Google Scholar] [CrossRef]
- Faccio, L.; Fusco, C.; Chen, A.; Martinotti, S.; Bonventre, J.V.; Zervos, A.S. Characterization of a novel human serine protease that has extensive homology to bacterial heat shock endoprotease HtrA and is regulated by kidney ischemia. J. Biol. Chem. 2000, 275, 2581–2588. [Google Scholar] [CrossRef]
- Goo, H.G.; Rhim, H.; Kang, S. Pathogenic Role of Serine Protease HtrA2/Omi in Neurodegenerative Diseases. Curr. Protein Pept. Sci. 2017, 18, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Shen, L.; Zhang, P.; Lin, F.; Ma, J.; Wu, Y.; Yu, H.; Sun, L. Inhibition of High-Temperature Requirement Protein A2 Protease Activity Represses Myogenic Differentiation via UPRmt. Int. J. Mol. Sci. 2022, 23, 11761. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, F.; Hao, F.; Ju, R. The expression of HtrA2 and its diagnostic value in patients with hepatocellular carcinoma. Medicine 2018, 97, e0128. [Google Scholar] [CrossRef]
- Dagda, R.K.; Chu, C.T. Mitochondrial quality control: Insights on how Parkinson’s disease related genes PINK1, parkin, and Omi/HtrA2 interact to maintain mitochondrial homeostasis. J. Bioenerg. Biomembr. 2009, 41, 473–479. [Google Scholar] [CrossRef]
- Xu, Z.; Lin, J.; Xie, Y.; Tang, H.; Xie, J.; Zeng, R. HtrA2 is required for inflammatory responses in BMDMs via controlling TRAF2 stability in collagen-induced arthritis. Mol. Immunol. 2021, 129, 78–85. [Google Scholar] [CrossRef]
- Wang, X. Inhibition of HtrA2 alleviates inflammatory response and cell apoptosis in lipopolysaccharide-induced acute pneumonia in rats. Mol. Med. Rep. 2020, 22, 3127–3134. [Google Scholar] [CrossRef]
- Wang, Y.; Li, N.; Zhang, X.; Horng, T. Mitochondrial metabolism regulates macrophage biology. J. Biol. Chem. 2021, 297, 100904. [Google Scholar] [CrossRef]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Lai, R.C.; Sim, W.K.; Lim, S.K. Therapeutic Efficacy of Mesenchymal Stem/Stromal Cell Small Extracellular Vesicles in Alleviating Arthritic Progression by Restoring Macrophage Balance. Biomolecules 2023, 13, 1501. [Google Scholar] [CrossRef]
- Tardito, S.; Martinelli, G.; Soldano, S.; Paolino, S.; Pacini, G.; Patane, M.; Alessandri, E.; Smith, V.; Cutolo, M. Macrophage M1/M2 polarization and rheumatoid arthritis: A systematic review. Autoimmun. Rev. 2019, 18, 102397. [Google Scholar] [CrossRef]
- Yu, H.; Chang, Q.; Sun, T.; He, X.; Wen, L.; An, J.; Feng, J.; Zhao, Y. Metabolic reprogramming and polarization of microglia in Parkinson’s disease: Role of inflammasome and iron. Ageing Res. Rev. 2023, 90, 102032. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; Di Cera, E. Evolution of peptidase diversity. J. Biol. Chem. 2008, 283, 30010–30014. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Nero, T.L.; Wang, Y.; Parker, M.W.; Nie, G. Activity-modulating monoclonal antibodies to the human serine protease HtrA3 provide novel insights into regulating HtrA proteolytic activities. PLoS ONE 2014, 9, e108235. [Google Scholar] [CrossRef] [PubMed]
- Nie, G.Y.; Hampton, A.; Li, Y.; Findlay, J.K.; Salamonsen, L.A. Identification and cloning of two isoforms of human high-temperature requirement factor A3 (HtrA3), characterization of its genomic structure and comparison of its tissue distribution with HtrA1 and HtrA2. Biochem. J. 2003, 371 Pt 1, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Vande Walle, L.; Lamkanfi, M.; Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi: An overview. Cell Death Differ. 2008, 15, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Jeong, G.H.; Nam, M.K.; Hur, W.; Heo, S.; Lee, S.; Choi, E.; Park, J.H.; Park, Y.; Kim, W.U.; Rhim, H.; et al. Role of high-temperature requirement serine protease A 2 in rheumatoid inflammation. Arthritis Res. Ther. 2023, 25, 96. [Google Scholar] [CrossRef]
- Koch, A.E. Review: Angiogenesis: Implications for rheumatoid arthritis. Arthritis Rheum. 1998, 41, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.M.; Datta, P.; Srinivasula, S.M.; Ji, W.; Gupta, S.; Zhang, Z.; Davies, E.; Hajnóczky, G.; Saunders, T.L.; Van Keuren, M.L.; et al. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature 2003, 425, 721–727. [Google Scholar] [CrossRef]
- Lee, S.H.; Moon, Y.M.; Seo, H.B.; Kim, S.Y.; Kim, E.K.; Yi, J.; Nam, M.K.; Min, J.K.; Park, S.H.; Rhim, H.; et al. HtrA2 suppresses autoimmune arthritis and regulates activation of STAT3. Sci. Rep. 2016, 6, 39393. [Google Scholar] [CrossRef]
- Hu, Q.; Li, B.; Xu, R.; Chen, D.; Mu, C.; Fei, E.; Wang, G. The protease Omi cleaves the mitogen-activated protein kinase kinase MEK1 to inhibit microglial activation. Sci. Signal. 2012, 5, ra61. [Google Scholar] [CrossRef]
- Feng, L.; Li, Z.; Xiong, Y.; Yan, T.; Fu, C.; Zeng, Q.; Wang, H. HtrA2 Independently Predicts Poor Prognosis and Correlates with Immune Cell Infiltration in Hepatocellular Carcinoma. J. Oncol. 2023, 2023, 4067418. [Google Scholar] [CrossRef]
- Singh, S.; Datta, G.; Jain, S.; Thakur, V.; Arora, P.; Muneer, A.; Asad, M.; Ali, S.; Mohmmed, A. Dual role of an essential HtrA2/Omi protease in the human malaria parasite: Maintenance of mitochondrial homeostasis and induction of apoptosis-like cell death under cellular stress. PLoS Pathog. 2022, 18, e1010932. [Google Scholar] [CrossRef] [PubMed]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Chaganti, L.K.; Kuppili, R.R.; Bose, K. Intricate structural coordination and domain plasticity regulate activity of serine protease HtrA2. FASEB J. 2013, 27, 3054–3066. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.M.; Morrison, A.; Klupsch, K.; Fedele, V.; Moisoi, N.; Teismann, P.; Abuin, A.; Grau, E.; Geppert, M.; Livi, G.P.; et al. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol. Cell. Biol. 2004, 24, 9848–9862. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Shi, S.; Ju, W.; Faul, C.; Tsai, S.Y.; Mundel, P.; Böttinger, E.P. Mpv17l protects against mitochondrial oxidative stress and apoptosis by activation of Omi/HtrA2 protease. Proc. Natl. Acad. Sci. USA 2008, 105, 14106–14111. [Google Scholar] [CrossRef] [PubMed]
- Toyama, Y.; Harkness, R.W.; Kay, L.E. Structural basis of protein substrate processing by human mitochondrial high-temperature requirement A2 protease. Proc. Natl. Acad. Sci. USA 2022, 119, e2203172119. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Imai, Y.; Nakayama, H.; Takahashi, K.; Takio, K.; Takahashi, R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell 2001, 8, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Radke, S.; Chander, H.; Schäfer, P.; Meiss, G.; Krüger, R.; Schulz, J.B.; Germain, D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef] [PubMed]
- Moisoi, N.; Klupsch, K.; Fedele, V.; East, P.; Sharma, S.; Renton, A.; Plun-Favreau, H.; Edwards, R.E.; Teismann, P.; Esposti, M.D.; et al. Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ. 2009, 16, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Desideri, E.; Martins, L.M. Mitochondrial Stress Signalling: HTRA2 and Parkinson’s Disease. Int. J. Cell Biol. 2012, 2012, 607929. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.; Kaplitt, M.G. Novel mitochondrial substrates of omi indicate a new regulatory role in neurodegenerative disorders. PLoS ONE 2009, 4, e7100. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Germain, D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 2011, 124 Pt 9, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Rainbolt, T.K.; Saunders, J.M.; Wiseman, R.L. YME1L degradation reduces mitochondrial proteolytic capacity during oxidative stress. EMBO Rep. 2015, 16, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef]
- Skorko-Glonek, J.; Zurawa-Janicka, D.; Koper, T.; Jarzab, M.; Figaj, D.; Glaza, P.; Lipinska, B. HtrA protease family as therapeutic targets. Curr. Pharm. Des. 2013, 19, 977–1009. [Google Scholar] [CrossRef]
- Zhang, C.; He, A.; Liu, S.; He, Q.; Luo, Y.; He, Z.; Chen, Y.; Tao, A.; Yan, J. Inhibition of HtrA2 alleviated dextran sulfate sodium (DSS)-induced colitis by preventing necroptosis of intestinal epithelial cells. Cell Death Dis. 2019, 10, 344. [Google Scholar] [CrossRef]
- Klupsch, K.; Downward, J. The protease inhibitor Ucf-101 induces cellular responses independently of its known target, HtrA2/Omi. Cell Death Differ. 2006, 13, 2157–2159. [Google Scholar] [CrossRef]
- Hur, W.; Kang, B.Y.; Kim, S.M.; Lee, G.W.; Kim, J.H.; Nam, M.K.; Rhim, H.; Yoon, S.K. Serine Protease HtrA2/Omi Deficiency Impairs Mitochondrial Homeostasis and Promotes Hepatic Fibrogenesis via Activation of Hepatic Stellate Cells. Cells 2019, 8, 1119. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Sun, L.K.; Su, J.; Yan, W.Y.; Jin, Y.; Luo, X.; Jiang, X.R.; Wang, H.L. Serine protease HtrA2/Omi regulates adaptive mitochondrial reprogramming in the brain cortex after ischemia/reperfusion injury via UCP2-SIRT3-PGC1 axis. Hum. Cell 2022, 35, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Rath, E.; Berger, E.; Messlik, A.; Nunes, T.; Liu, B.; Kim, S.C.; Hoogenraad, N.; Sans, M.; Sartor, R.B.; Haller, D. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 2012, 61, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Begum, M.E.; Sen, D. DOR agonist (SNC-80) exhibits anti-parkinsonian effect via downregulating UPR/oxidative stress signals and inflammatory response in vivo. Neurosci. Lett. 2018, 678, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Cipolat, S.; de Brito, O.M.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117 Pt 26, 6535–6546. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: Regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Kieper, N.; Holmström, K.M.; Ciceri, D.; Fiesel, F.C.; Wolburg, H.; Ziviani, E.; Whitworth, A.J.; Martins, L.M.; Kahle, P.J.; Krüger, R. Modulation of mitochondrial function and morphology by interaction of Omi/HtrA2 with the mitochondrial fusion factor OPA1. Exp. Cell Res. 2010, 316, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; He, F.; Zhao, X.; Zhang, Y.; Chu, X.; Hua, C.; Qu, Y.; Duan, Y.; Ming, L. YAP Inhibits the Apoptosis and Migration of Human Rectal Cancer Cells via Suppression of JNK-Drp1-Mitochondrial Fission-HtrA2/Omi Pathways. Cell. Physiol. Biochem. 2017, 44, 2073–2089. [Google Scholar] [CrossRef] [PubMed]
- Moehlman, A.T.; Youle, R.J. Mitochondrial Quality Control and Restraining Innate Immunity. Annu. Rev. Cell Dev. Biol. 2020, 36, 265–289. [Google Scholar] [CrossRef] [PubMed]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling Inflamm-Aging through Mitochondrial Dysfunction: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef]
- Hernández-Aguilera, A.; Rull, A.; Rodríguez-Gallego, E.; Riera-Borrull, M.; Luciano-Mateo, F.; Camps, J.; Menéndez, J.A.; Joven, J. Mitochondrial dysfunction: A basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediat. Inflamm. 2013, 2013, 135698. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Martins, L.M.; Iaccarino, I.; Tenev, T.; Gschmeissner, S.; Totty, N.F.; Lemoine, N.R.; Savopoulos, J.; Gray, C.W.; Creasy, C.L.; Dingwall, C.; et al. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J. Biol. Chem. 2002, 277, 439–444. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.M.; Tarkowski, A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Nam, M.K.; Seong, Y.; Jeong, G.H.; Yoo, S.A.; Rhim, H. HtrA2 regulates α-Synuclein-mediated mitochondrial reactive oxygen species production in the mitochondria of microglia. Biochem. Biophys. Res. Commun. 2023, 638, 84–93. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.J.; Boshuizen, M.C.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Padgett, L.E.; Burg, A.R.; Lei, W.; Tse, H.M. Loss of NADPH oxidase-derived superoxide skews macrophage phenotypes to delay type 1 diabetes. Diabetes 2015, 64, 937–946. [Google Scholar] [CrossRef]
- Han, C.; Sheng, Y.; Wang, J.; Zhou, X.; Li, W.; Zhang, C.; Guo, L.; Yang, Y. NOX4 promotes mucosal barrier injury in inflammatory bowel disease by mediating macrophages M1 polarization through ROS. Int. Immunopharmacol. 2022, 104, 108361. [Google Scholar] [CrossRef] [PubMed]
- Torres-Castro, I.; Arroyo-Camarena, Ú.D.; Martínez-Reyes, C.P.; Gómez-Arauz, A.Y.; Dueñas-Andrade, Y.; Hernández-Ruiz, J.; Béjar, Y.L.; Zaga-Clavellina, V.; Morales-Montor, J.; Terrazas, L.I.; et al. Human monocytes and macrophages undergo M1-type inflammatory polarization in response to high levels of glucose. Immunol. Lett. 2016, 176, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Swain, M.M.; Pal, A. Hyperglycemia-induced inflammation caused down-regulation of 8-oxoG-DNA glycosylase levels in murine macrophages is mediated by oxidative-nitrosative stress-dependent pathways. Int. J. Biochem. Cell Biol. 2016, 73, 82–98. [Google Scholar] [CrossRef]
- Jin, Z.; Wei, W.; Yang, M.; Du, Y.; Wan, Y. Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization. Cell Metab. 2014, 20, 483–498. [Google Scholar] [CrossRef]
- Goo, H.G.; Jung, M.K.; Han, S.S.; Rhim, H.; Kang, S. HtrA2/Omi deficiency causes damage and mutation of mitochondrial DNA. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2013, 1833, 1866–1875. [Google Scholar] [CrossRef]
- Xiaopeng, C.; Jin, T. Perfluorooctane sulfonate (PFOS) causes aging damage in the liver through the mt-DNA-mediated NLRP3 signaling pathway. Ecotoxicol. Environ. Saf. 2023, 262, 115121. [Google Scholar] [CrossRef]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2023, 23, 159–173. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Lin, M.M.; Liu, N.; Qin, Z.H.; Wang, Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol. Sin. 2022, 43, 2439–2447. [Google Scholar] [CrossRef]
- Huang, H.; Sun, Z.; Xu, J.; Wang, L.; Zhao, J.; Li, J.; Zhang, S.; Yuan, F.; Liu, M.; Fang, Z. Yang-Xin-Shu-Mai granule alleviates atherosclerosis by regulating macrophage polarization via the TLR9/MyD88/NF-κB signaling pathway. J. Ethnopharmacol. 2024, 318, 116868. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, S.; Liu, W.; Lv, X.; Wang, B.; Hu, B.; Shao, Z. Bardoxolone methyl breaks the vicious cycle between M1 macrophages and senescent nucleus pulposus cells through the Nrf2/STING/NF-κB pathway. Int. Immunopharmacol. 2023, 127, 111262. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Q.; Wang, Y.; Zhang, C.; Xu, S.; Luo, M.; Yang, S. Caragana sinica (Buc’hoz) Rehd. (jin ji er) polysaccharide regulates the immune function and intestinal microbiota of cyclophosphamide (CTX) induced immunosuppressed mice. J. Ethnopharmacol. 2024, 322, 117551. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS−) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, J.; Milojevic, J.; Melacini, G.; Ortega, J. A new function of human HtrA2 as an amyloid-beta oligomerization inhibitor. J. Alzheimer. Dis. 2009, 17, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.J.; Jamal, M.; Hong, S.T. The function of bacterial HtrA is evolutionally conserved in mammalian HtrA2/Omi. Sci. Rep. 2020, 10, 5284. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X. The role of HtrA2/Omi in regulating mitochondrial unfolded proteinresponse and its role in cerebral ischemia reperfusion injury. Master’s Thesis, Jilin University, Changchun, China, 2017. [Google Scholar]
- Wu, Y.; Zhou, K.; Liu, B.; Xu, J.; Lei, L.; Hu, J.; Cheng, X.; Zhong, F.; Wang, S. Glial Activation, Mitochondrial Imbalance, and Akt/mTOR Signaling May Be Potential Mechanisms of Cognitive Impairment in Heart Failure Mice. Neurotox. Res. 2023, 41, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Alvarez, R.; De Francesco, E.M.; Fiorillo, M.; Sotgia, F.; Lisanti, M.P. Mitochondrial Fission Factor (MFF) Inhibits Mitochondrial Metabolism and Reduces Breast Cancer Stem Cell (CSC) Activity. Front. Oncol. 2020, 10, 1776. [Google Scholar] [CrossRef] [PubMed]
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a Cellular Hub in Infection and Inflammation. Int. J. Mol. Sci. 2021, 22, 11338. [Google Scholar] [CrossRef] [PubMed]
- Temelie, M.; Talpur, R.; Dominguez-Prieto, M.; Dantas Silva, A.; Cenusa, C.; Craciun, L.; Savu, D.I.; Moisoi, N. Impaired Integrated Stress Response and Mitochondrial Integrity Modulate Genotoxic Stress Impact and Lower the Threshold for Immune Signalling. Int. J. Mol. Sci. 2023, 24, 5891. [Google Scholar] [CrossRef]
- Sharma, S.; Kaufmann, T.; Biswas, S. Impact of inhibitor of apoptosis proteins on immune modulation and inflammation. Immunol. Cell Biol. 2017, 95, 236–243. [Google Scholar] [CrossRef]
- Nadella, V.; Mohanty, A.; Sharma, L.; Yellaboina, S.; Mollenkopf, H.J.; Mazumdar, V.B.; Palaparthi, R.; Mylavarapu, M.B.; Maurya, R.; Kurukuti, S.; et al. Inhibitors of Apoptosis Protein Antagonists (Smac Mimetic Compounds) Control Polarization of Macrophages during Microbial Challenge and Sterile Inflammatory Responses. Front. Immunol. 2017, 8, 1792. [Google Scholar] [CrossRef]
- Lecis, D.; De Cesare, M.; Perego, P.; Conti, A.; Corna, E.; Drago, C.; Seneci, P.; Walczak, H.; Colombo, M.P.; Delia, D.; et al. Smac mimetics induce inflammation and necrotic tumour cell death by modulating macrophage activity. Cell Death Dis. 2013, 4, e920. [Google Scholar] [CrossRef]
- Kavanagh, E.; Rodhe, J.; Burguillos, M.A.; Venero, J.L.; Joseph, B. Regulation of caspase-3 processing by cIAP2 controls the switch between pro-inflammatory activation and cell death in microglia. Cell Death Dis. 2014, 5, e1565. [Google Scholar] [CrossRef]
- Yang, Q.H.; Church-Hajduk, R.; Ren, J.; Newton, M.L.; Du, C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003, 17, 1487–1496. [Google Scholar] [CrossRef]
- Xu, R.; Hu, Q.; Ma, Q.; Liu, C.; Wang, G. The protease Omi regulates mitochondrial biogenesis through the GSK3β/PGC-1α pathway. Cell Death Dis. 2014, 5, e1373. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Fernandes-Alnemri, T.; Alnemri, E.S. A novel role for the mitochondrial HTRA2/OMI protease in aging. Autophagy 2013, 9, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Cilenti, L.; Ambivero, C.T.; Ward, N.; Alnemri, E.S.; Germain, D.; Zervos, A.S. Inactivation of Omi/HtrA2 protease leads to the deregulation of mitochondrial Mulan E3 ubiquitin ligase and increased mitophagy. Biochim Biophys Acta BBA—Mol. Cell Res. 2014, 1843, 1295–1307. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Yan, X.; Yuan, Y.; Li, R.; Zhao, Y.; Fu, J.; Wang, J.; Su, J. HTRA2/OMI-Mediated Mitochondrial Quality Control Alters Macrophage Polarization Affecting Systemic Chronic Inflammation. Int. J. Mol. Sci. 2024, 25, 1577. https://doi.org/10.3390/ijms25031577
Liu Q, Yan X, Yuan Y, Li R, Zhao Y, Fu J, Wang J, Su J. HTRA2/OMI-Mediated Mitochondrial Quality Control Alters Macrophage Polarization Affecting Systemic Chronic Inflammation. International Journal of Molecular Sciences. 2024; 25(3):1577. https://doi.org/10.3390/ijms25031577
Chicago/Turabian StyleLiu, Qingqing, Xiaoyu Yan, Yuan Yuan, Runyuan Li, Yuanxin Zhao, Jiaying Fu, Jian Wang, and Jing Su. 2024. "HTRA2/OMI-Mediated Mitochondrial Quality Control Alters Macrophage Polarization Affecting Systemic Chronic Inflammation" International Journal of Molecular Sciences 25, no. 3: 1577. https://doi.org/10.3390/ijms25031577
APA StyleLiu, Q., Yan, X., Yuan, Y., Li, R., Zhao, Y., Fu, J., Wang, J., & Su, J. (2024). HTRA2/OMI-Mediated Mitochondrial Quality Control Alters Macrophage Polarization Affecting Systemic Chronic Inflammation. International Journal of Molecular Sciences, 25(3), 1577. https://doi.org/10.3390/ijms25031577