
Organelle Genomes of Epipogium roseum Provide Insight into the Evolution of Mycoheterotrophic Orchids



Abstract
1. Introduction
2. Results
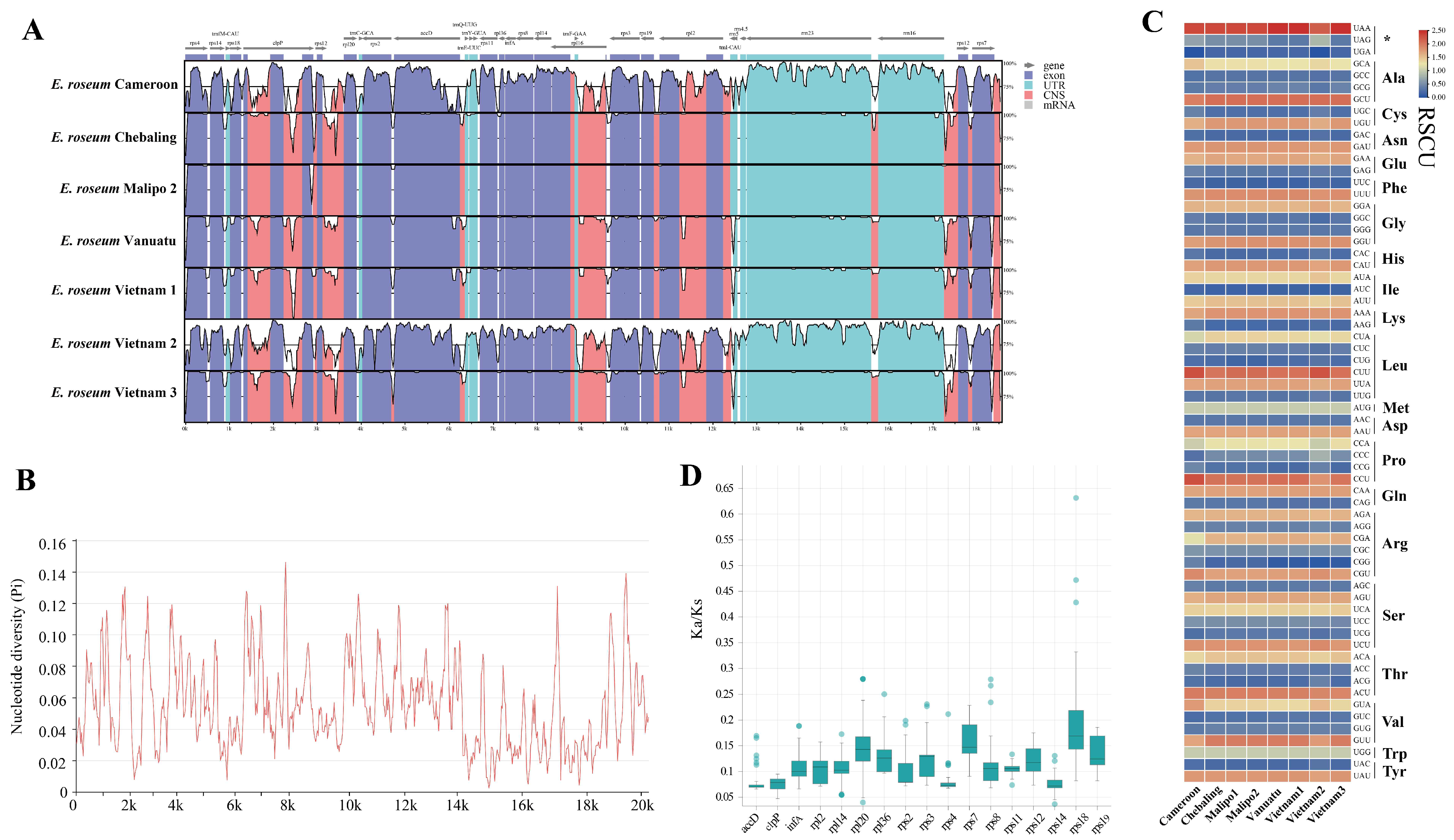
2.1. Characterization and Comparative Analysis of Plastomes
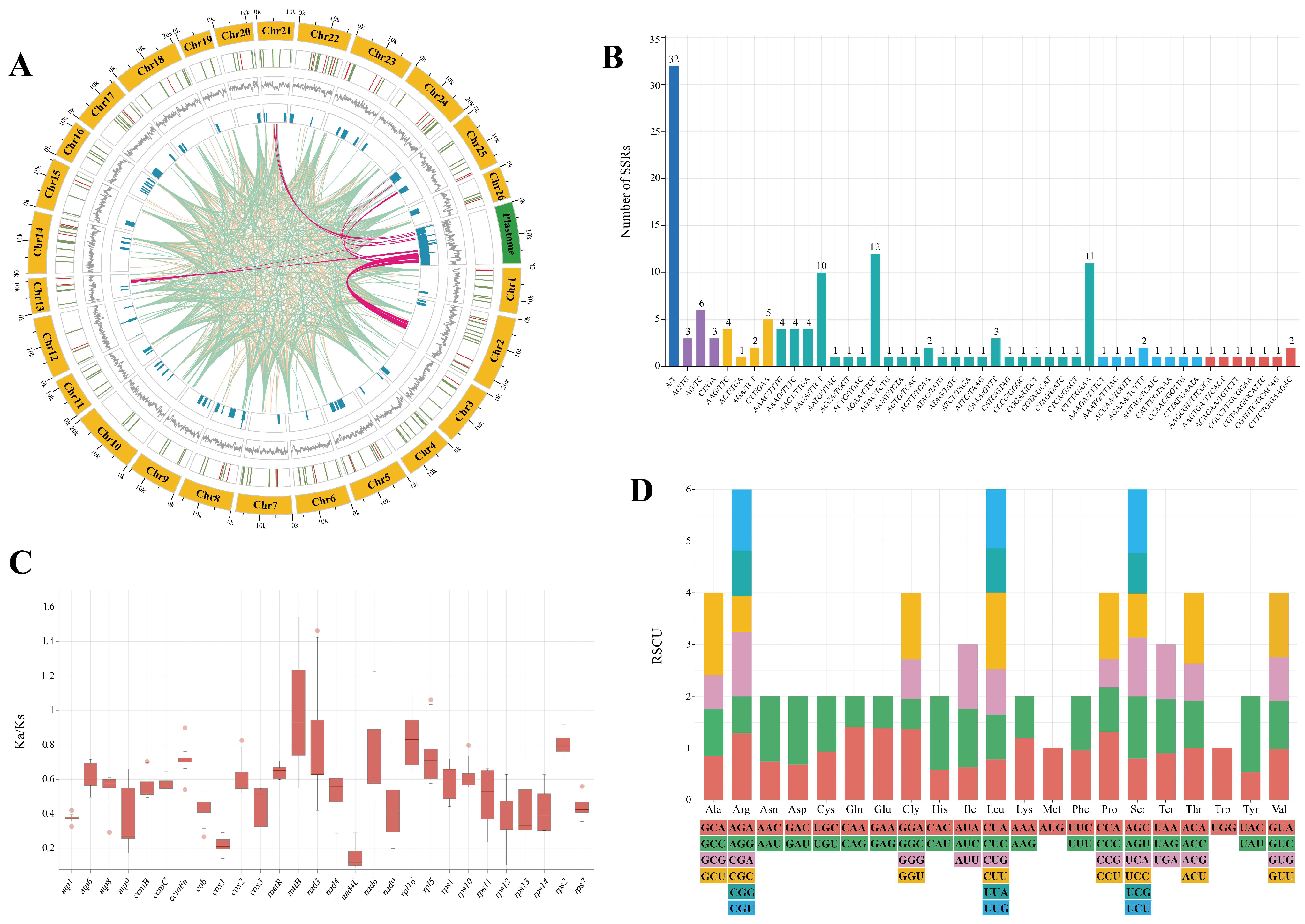
2.2. Comprehensive Analysis of Mitogenome Characteristics
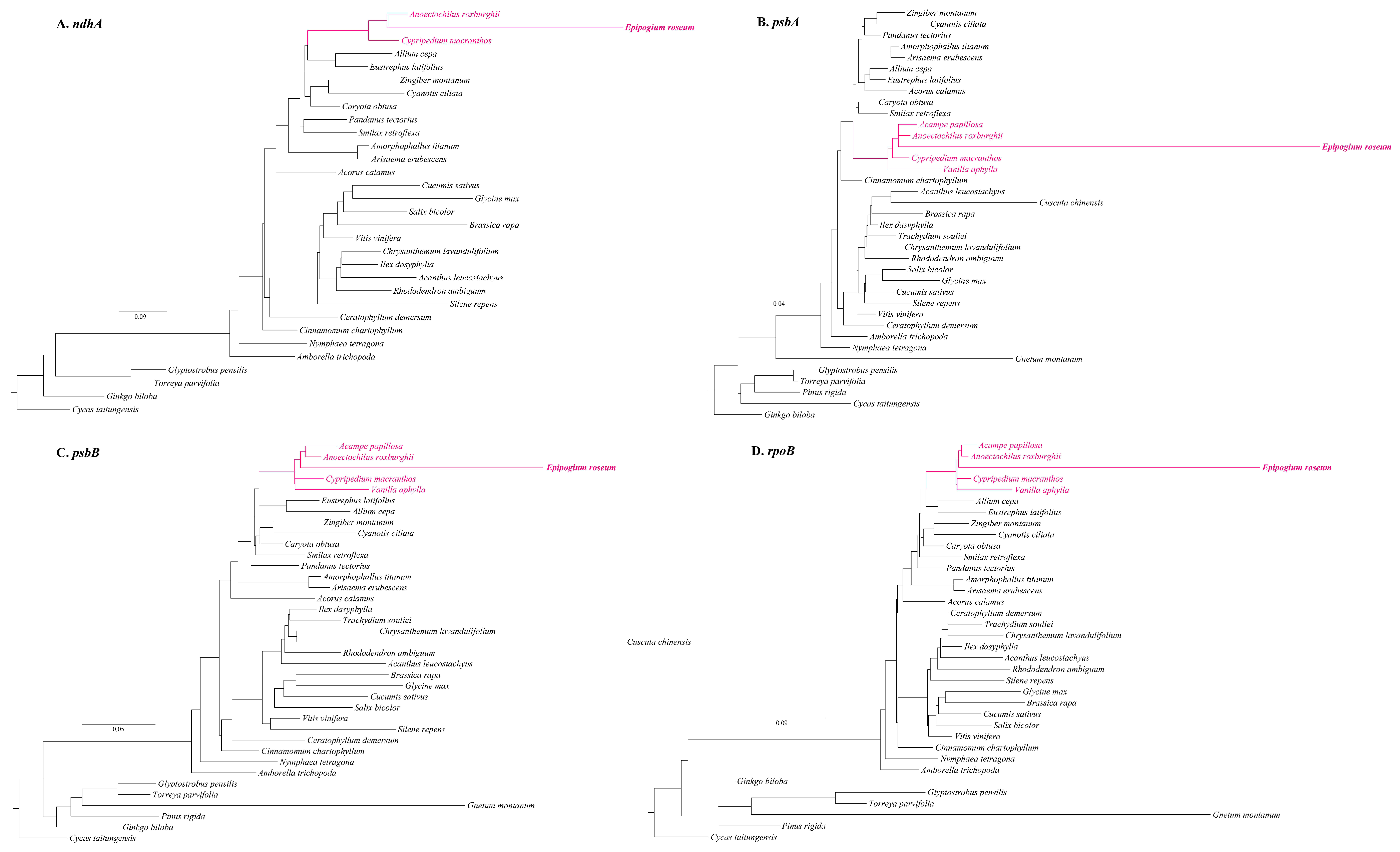
2.3. Migration of Plastid Regions in Mitogenome
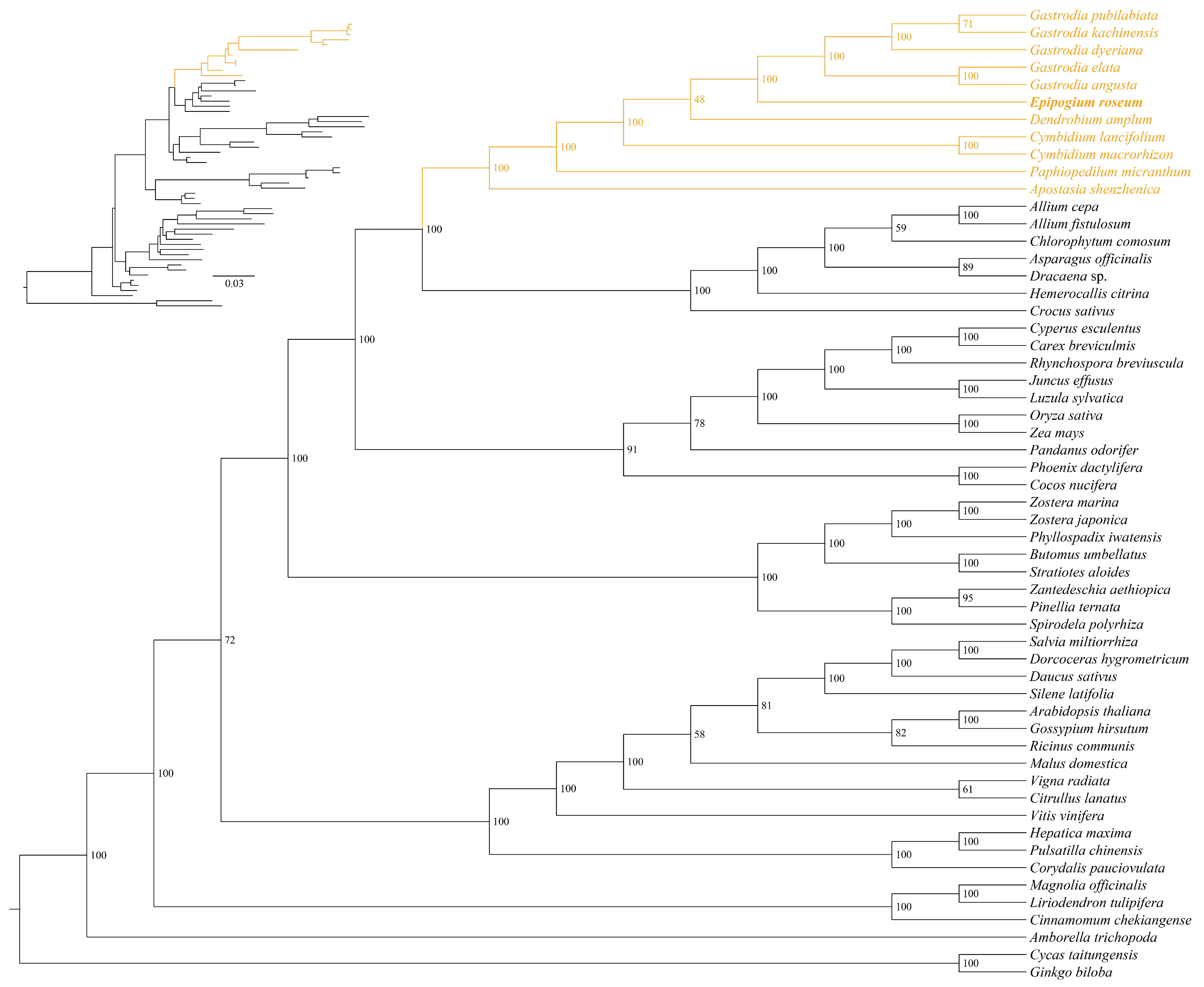
2.4. Phylogenetic Position of E. roseum
3. Discussion
4. Materials and Methods
4.1. Sample Sampling, DNA Extraction and Sequencing, Genome Assembly and Annotation
4.2. Genome Characters and Comparative Genomics
4.3. Gene Transfer and Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leake, J.R. The biology of myco-heterotrophic (‘saprophytic’) plants. New Phytol. 1994, 127, 171–216. [Google Scholar] [CrossRef]
- Bidartondo, M.I. The evolutionary ecology of myco-heterotrophy. New Phytol. 2005, 167, 335–352. [Google Scholar] [CrossRef]
- Li, M.-H.; Liu, K.-W.; Li, Z.; Lu, H.-C.; Ye, Q.-L.; Zhang, D.; Wang, J.-Y.; Li, Y.-F.; Zhong, Z.-M.; Liu, X.; et al. Genomes of leafy and leafless Platanthera orchids illuminate the evolution of mycoheterotrophy. Nat. Plants 2022, 8, 373–388. [Google Scholar] [CrossRef]
- Suetsugu, K.; Matsubayashi, J. Evidence for mycorrhizal cheating in Apostasia nipponica, an early-diverging member of the Orchidaceae. New Phytol. 2021, 229, 2302–2310. [Google Scholar] [CrossRef]
- Motomura, H.; Marc-André, S.; Martos, F.; Kagawa, A. Mycoheterotrophy evolved from mixotrophic ancestors: Evidence in Cymbidium (orchidaceae). Ann. Bot. 2010, 106, 573. [Google Scholar] [CrossRef]
- Yagame, T.; Orihara, T.; Selosse, M.A.; Yamato, M.; Iwase, K. Mixotrophy of Platanthera minor, an orchid associated with ectomycorrhiza-forming Ceratobasidiaceae fungi. New Phytol. 2012, 193, 178–187. [Google Scholar] [CrossRef]
- Wang, J.; Kan, S.; Liao, X.; Zhou, J.; Tembrock, L.R.; Daniell, H.; Jin, S.; Wu, Z. Plant organellar genomes: Much done, much more to do. Trends Plant Sci. 2024. [Google Scholar] [CrossRef]
- Wu, Z.; Sloan, D.B. Recombination and intraspecific polymorphism for the presence and absence of entire chromosomes in mitochondrial genomes. Heredity 2019, 122, 647–659. [Google Scholar] [CrossRef]
- Lin, Y.; Li, P.; Zhang, Y.; Akhter, D.; Pan, R.; Fu, Z.; Huang, M.; Li, X.; Feng, Y. Unprecedented organelle genomic variations in morning glories reveal independent evolutionary scenarios of parasitic plants and the diversification of plant mitochondrial complexes. BMC Biol. 2022, 20, 49. [Google Scholar] [CrossRef]
- Organelle Genome Resources Database. Available online: https://www.ncbi.nlm.nih.gov/genome/organelle/ (accessed on 22 November 2023).
- Chase, M.W.; Cameron, K.M.; Freudenstein, J.V.; Pridgeon, A.M.; Salazar, G.; Van den Berg, C.; Schuiteman, A. An updated classification of Orchidaceae. Bot. J. Linn. Soc. 2015, 177, 151–174. [Google Scholar] [CrossRef]
- Ke, S.J.; Liu, D.K.; Tu, X.D.; He, X.; Zhang, M.M.; Zhu, M.J.; Zhang, D.Y.; Zhang, C.L.; Lan, S.R.; Liu, Z.J. Apostasia Mitochondrial Genome Analysis and Monocot Mitochondria Phylogenomics. Int. J. Mol. Sci. 2023, 24, 7837. [Google Scholar] [CrossRef]
- Li, X.; Zhe, M.; Huang, Y.; Fan, W.; Yang, J.; Zhu, A. The Evolution of Mitochondrial Genomes between Two Cymbidium Sister Species: Dozens of Circular Chromosomes and the Maintenance and Deterioration of Genome Synteny. Genes 2023, 14, 864. [Google Scholar] [CrossRef]
- Yuan, Y.; Jin, X.; Liu, J.; Zhao, X.; Zhou, J.; Wang, X.; Wang, D.; Lai, C.; Xu, W.; Huang, J.; et al. The Gastrodia elata genome provides insights into plant adaptation to heterotrophy. Nat. Commun 2018, 9, 1615. [Google Scholar] [CrossRef]
- Yang, J.X.; Dierckxsens, N.; Bai, M.Z.; Guo, Y.Y. Multichromosomal mitochondrial genome of Paphiopedilum micranthum: Compact and fragmented genome, and rampant intracellular gene transfer. Int. J. Mol. Sci. 2023, 24, 3976. [Google Scholar] [CrossRef]
- Kim, Y.K.; Jo, S.; Cheon, S.H.; Hong, J.R.; Kim, K.J. Ancient Horizontal Gene Transfers from Plastome to Mitogenome of a Nonphotosynthetic Orchid, Gastrodia pubilabiata (Epidendroideae, Orchidaceae). Int. J. Mol. Sci. 2023, 24, 11448. [Google Scholar] [CrossRef]
- Woodson, J.D.; Chory, J. Coordination of gene expression between organellar and nuclear genomes. Nat. Rev. Genet. 2008, 9, 383–395. [Google Scholar] [CrossRef]
- Keeling, P.J.; Palmer, J.D. Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 2008, 9, 605–618. [Google Scholar] [CrossRef]
- You, C.; Cui, T.; Zhang, C.; Zang, S.; Su, Y.; Que, Y. Assembly of the Complete Mitochondrial Genome of Gelsemium elegans Revealed the Existence of Homologous Conformations Generated by a Repeat Mediated Recombination. Int. J. Mol. Sci. 2022, 24, 527. [Google Scholar] [CrossRef]
- Park, S.; Grewe, F.; Zhu, A.; Ruhlman, T.A.; Sabir, J.; Mower, J.P.; Jansen, R.K. Dynamic evolution of Geranium mitochondrial genomes through multiple horizontal and intracellular gene transfers. New Phytol. 2015, 208, 570–583. [Google Scholar] [CrossRef]
- Zhou, S.; Zhi, X.; Yu, R.; Liu, Y.; Zhou, R. Factors contributing to mitogenome size variation and a recurrent intracellular DNA transfer in Melastoma. BMC Genom. 2023, 24, 370. [Google Scholar] [CrossRef]
- Zhou, X.; Lin, H.; Fan, X.L.; Gao, J.Y. Autonomous self-pollination and insect visitation in a saprophytic orchid, Epipogium roseum (D. Don) Lindl. Aust. J. Bot. 2012, 60, 154–159. [Google Scholar] [CrossRef]
- Schelkunov, M.I.; Shtratnikova, V.Y.; Nuraliev, M.S.; Selosse, M.A.; Penin, A.A.; Logacheva, M.D. Exploring the limits for reduction of plastid genomes: A case study of the mycoheterotrophic orchids Epipogium aphyllum and Epipogium roseum. Genome Biol. Evol. 2015, 7, 1179–1191. [Google Scholar] [CrossRef]
- Kim, Y.K.; Jo, S.; Cheon, S.H.; Joo, M.J.; Hong, J.R.; Kwak, M.; Kim, K.J. Plastome evolution and phylogeny of Orchidaceae, with 24 new sequences. Front. Plant Sci. 2020, 11, 22. [Google Scholar] [CrossRef]
- Li, L.; Wu, Q.; Fang, L.; Wu, K.; Li, M.; Zeng, S. Comparative chloroplast genomics and phylogenetic analysis of Thuniopsis and closely related genera within Coelogyninae (Orchidaceae). Front. Genet. 2022, 13, 850201. [Google Scholar] [CrossRef]
- Zhao, Z.; Zeng, M.Y.; Wu, Y.W.; Li, J.W.; Zhou, Z.; Liu, Z.J.; Li, M.H. Characterization and Comparative Analysis of the Complete Plastomes of Five Epidendrum (Epidendreae, Orchidaceae) Species. Int. J. Mol. Sci. 2023, 24, 14437. [Google Scholar] [CrossRef]
- Zhou, C.Y.; Zeng, M.Y.; Gao, X.; Zhao, Z.; Li, R.; Wu, Y.; Liu, Z.J.; Zhang, D.; Li, M.H. Characteristics and Comparative Analysis of Seven Complete Plastomes of Trichoglottis s.l. (Aeridinae, Orchidaceae). Int. J. Mol. Sci. 2023, 24, 14544. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Liao, X.Z.; Zhang, X.N.; Tembrock, L.R.; Broz, A. Genomic architectural variation of plant mitochondria—A review of multichromosomal structuring. J. Syst. Evol. 2022, 60, 160–168. [Google Scholar] [CrossRef]
- Yang, Z.; Ni, Y.; Lin, Z.; Yang, L.; Chen, G.; Nijiati, N.; Hu, Y.; Chen, X. De novo assembly of the complete mitochondrial genome of sweet potato (Ipomoea batatas [L.] Lam) revealed the existence of homologous conformations generated by the repeat-mediated recombination. BMC Plant Biol. 2022, 22, 285. [Google Scholar] [CrossRef]
- Goremykin, V.V.; Salamini, F.; Velasco, R.; Viola, R. Mitochondrial DNA of Vitis vinifera and the issue of rampant horizontal gene transfer. Mol. Biol. Evol. 2009, 26, 99–110. [Google Scholar] [CrossRef]
- Wang, S.; Li, D.; Yao, X.; Song, Q.; Wang, Z.; Zhang, Q.; Zhong, C.; Liu, Y.; Huang, H. Evolution and diversification of kiwifruit mitogenomes through extensive whole-genome rearrangement and mosaic loss of intergenic sequences in a highly variable region. Genome Biol. Evol. 2019, 11, 1192–1206. [Google Scholar] [CrossRef]
- Niu, Y.; Gao, C.; Liu, J. Complete mitochondrial genomes of three Mangifera species, their genomic structure and gene transfer from chloroplast genomes. BMC Genom. 2022, 23, 147. [Google Scholar] [CrossRef]
- Rice, D.W.; Alverson, A.J.; Richardson, A.O.; Young, G.J.; Sanchez-Puerta, M.V.; Munzinger, J.; Barry, K.; Boore, J.L.; Zhang, Y.; DePamphilis, C.W.; et al. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 2013, 342, 1468–1473. [Google Scholar] [CrossRef]
- Gandini, C.L.; Sanchez-Puerta, M.V. Foreign plastid sequences in plant mitochondria are frequently acquired via mitochondrion-to-mitochondrion horizontal transfer. Sci. Rep. 2017, 7, 43402. [Google Scholar] [CrossRef]
- Li, M.H.; Zhang, G.Q.; Lan, S.R.; Liu, Z.J.; China Phylogeny Consortium. A molecular phylogeny of Chinese orchids. J. Syst. Evol. 2016, 54, 349–362. [Google Scholar] [CrossRef]
- Molvray, M.; Kores, P.J.; Chase, M.W. Polyphyly of mycoheterotrophic orchids and functional influences on floral and molecular characteristics. In Monocots: Systematics and Evolution; Wilson, K.L., Mossison, D.A., Eds.; CSIRO: Melbourne, Australia, 2000; pp. 441–448. [Google Scholar]
- Zhang, G.; Hu, Y.; Huang, M.Z.; Huang, W.C.; Liu, D.K.; Zhang, D.; Hu, H.; Downing, J.L.; Liu, Z.J.; Ma, H. Comprehensive phylogenetic analyses of Orchidaceae using nuclear genes and evolutionary insights into epiphytism. J. Integr. Plant. Biol. 2023, 65, 1204–1225. [Google Scholar] [CrossRef]
- Arseneau, J.R.; Steeves, R.; Laflamme, M. Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol. Ecol. Resour. 2017, 17, 686–693. [Google Scholar] [CrossRef]
- Liu, D.K.; Tu, X.D.; Zhao, Z.; Zeng, M.Y.; Zhang, S.; Ma, L.; Zhang, G.Q.; Wang, M.M.; Liu, Z.J.; Lan, S.R.; et al. Plastid phylogenomic data yield new and robust insights into the phylogeny of Cleisostoma–Gastrochilus clades (Orchidaceae, Aeridinae). Mol. Phyl. Evol. 2020, 145, 106729. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; De Pamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.; Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinform. 2012, 13, 238. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. Organellar Genome DRAW (OGDRAW) version 1.3. 1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Brudno, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Baĵoglou, S. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19, i54–i62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Xia, X. DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Gao, H.; Kong, J. Distribution characteristics and biological function of tandem repeat sequences in the genomes of different organisms. Zool. Res. 2005, 26, 555–564. [Google Scholar]
- Kurĵ, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- ChiPlot. Available online: https://www.chiplot.online/ (accessed on 20 November 2023).
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated Mega BLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Length (bp) | GC Content (%) | Genes | PacBio Reads Coverage |
|---|---|---|---|---|
| Chr 1 | 14,127 | 42.5 | rps7 | 22.27 |
| Chr 2 | 23,000 | 45.8 | ψ atp4, atp8, nad4L | 22.24 |
| Chr 3 | 17,940 | 44.3 | nad2 * | 15.70 |
| Chr 4 | 14,746 | 44.8 | psbE, trnQ-TTC | 10.66 |
| Chr 5 | 17,464 | 46.2 | atp1, ccmFn, ψ nad5 | 10.49 |
| Chr 6 | 16,756 | 44.7 | atp9 | 17.81 |
| Chr 7 | 17,441 | 44.8 | rps2 | 20.79 |
| Chr 8 | 16,696 | 44.7 | atp6, cox2 **, ψ nad3, rps11, trnM-CAT | 16.41 |
| Chr 9 | 15,231 | 45.4 | cob, ψ nad1, rpl5, rps14, trnC-GCA | 12.31 |
| Chr 10 | 20,234 | 46.6 | nad4 *** | 20.48 |
| Chr 11 | 12,241 | 45.2 | ψ nad5 | 10.43 |
| Chr 12 | 18,964 | 45.6 | nad3, rps12, trnE-TTC, trnH-GTG | 19.73 |
| Chr 13 | 10,976 | 44.3 | trnD-GTC, trn-GTT | 14.64 |
| Chr 14 | 19,264 | 46.4 | atp9, rps1, trnY-GTA | 22.61 |
| Chr 15 | 15,510 | 45.8 | matR | 12.59 |
| Chr 16 | 11,633 | 48.0 | ψ nad7, trnE-TTC | 6.56 |
| Chr 17 | 15,703 | 46.0 | ψ rpl2, rpl16, rps19 | 12.40 |
| Chr 18 | 20,493 | 45.7 | mttB, rps13 | 30.44 |
| Chr 19 | 9973 | 44.7 | ccmB | 6.38 |
| Chr 20 | 11,816 | 45.4 | ψ nad5 | 5.82 |
| Chr 21 | 11,460 | 41.6 | nad9, trnF-GAA, trnW-CCA | 15.88 |
| Chr 22 | 16,388 | 44.8 | cox1, trnL-CAA | 15.47 |
| Chr 23 | 19,053 | 45.0 | ccmC, nad6, rps10 * | 21.72 |
| Chr 24 | 20,245 | 45.3 | cox3 | 21.19 |
| Chr 25 | 17,333 | 45.1 | rrn5, rrn18, rrn26, trnM-CAT | 32.24 |
| Chr 26 | 9865 | 45.4 | ψ ccmFc | 6.56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Z.; Li, Y.; Zhai, J.-W.; Liu, Z.-J.; Li, M.-H. Organelle Genomes of Epipogium roseum Provide Insight into the Evolution of Mycoheterotrophic Orchids. Int. J. Mol. Sci. 2024, 25, 1578. https://doi.org/10.3390/ijms25031578
Zhao Z, Li Y, Zhai J-W, Liu Z-J, Li M-H. Organelle Genomes of Epipogium roseum Provide Insight into the Evolution of Mycoheterotrophic Orchids. International Journal of Molecular Sciences. 2024; 25(3):1578. https://doi.org/10.3390/ijms25031578
Chicago/Turabian StyleZhao, Zhuang, Yuanyuan Li, Jun-Wen Zhai, Zhong-Jian Liu, and Ming-He Li. 2024. "Organelle Genomes of Epipogium roseum Provide Insight into the Evolution of Mycoheterotrophic Orchids" International Journal of Molecular Sciences 25, no. 3: 1578. https://doi.org/10.3390/ijms25031578
APA StyleZhao, Z., Li, Y., Zhai, J.-W., Liu, Z.-J., & Li, M.-H. (2024). Organelle Genomes of Epipogium roseum Provide Insight into the Evolution of Mycoheterotrophic Orchids. International Journal of Molecular Sciences, 25(3), 1578. https://doi.org/10.3390/ijms25031578