

Nrf2 Pathway and Oxidative Stress as a Common Target for Treatment of Diabetes and Its Comorbidities
 , , , and
, , , and
Abstract
1. Introduction
2. How Diabetes Induces Oxidative Stress
3. Diabetes and Cardiovascular Disease
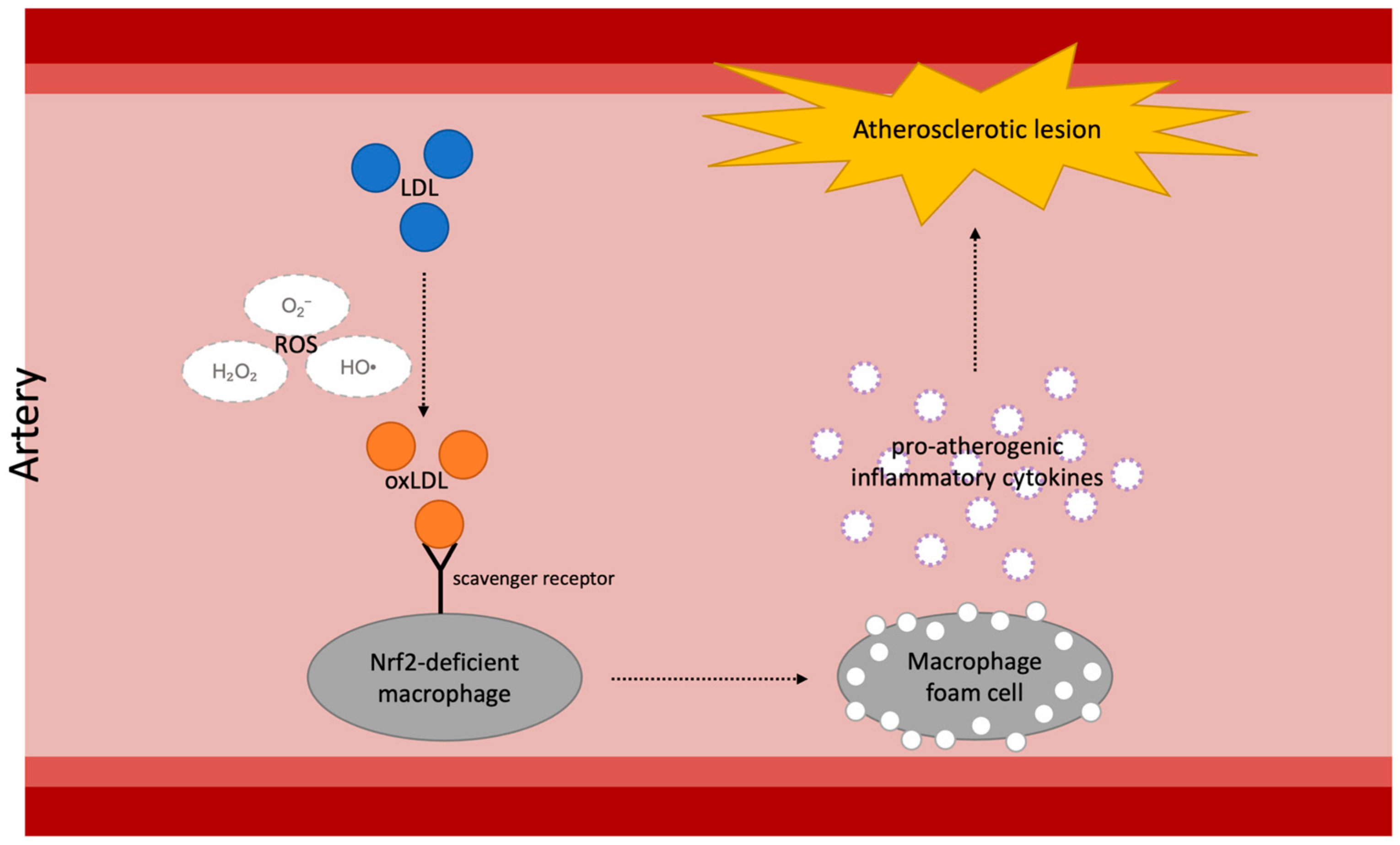
3.1. Diabetes and Atherosclerosis
3.2. Nrf2-Targeting Treatment in Diabetic Atherosclerosis
3.3. Diabetes and Cardiomyopathy
3.4. Nrf2-Targeting Treatment in Diabetic Cardiomyopathy
4. Diabetes and Kidney Disease
5. Diabetes and Liver Diseases
5.1. Diabetes and Progression of Non-Alcoholic Liver Disease
5.2. Diabetes and Alcoholic Liver Disease
5.3. Effects of Diabetes Drugs on Liver Responsse to Toxins
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centers for Disease Control and Prevention. 2022 National Diabetes Statistics Report; US Department of Health and Human Services: Atlanta, GA, USA, 2022.
- Chawla, A.; Chawla, R.; Jaggi, S. Microvasular and macrovascular complications in diabetes mellitus: Distinct or continuum? Indian J. Endocrinol. Metab. 2016, 20, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Hohn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Orasanu, G.; Plutzky, J. The pathologic continuum of diabetic vascular disease. J. Am. Coll. Cardiol. 2009, 53, S35–S42. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, R.I.; Rema, M.; Pradeepa, R.; Deepa, M.; Shanthirani, C.S.; Deepa, R.; Mohan, V. Prevalence and risk factors of diabetic nephropathy in an urban south indian population: The chennai urban rural epidemiology study (cures 45). Diabetes Care 2007, 30, 2019–2024. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.I.; Stratton, I.M.; Neil, H.A.; Yudkin, J.S.; Matthews, D.R.; Cull, C.A.; Wright, A.D.; Turner, R.C.; Holman, R.R. Association of systolic blood pressure with macrovascular and microvascular complications of type 2 diabetes (ukpds 36): Prospective observational study. BMJ 2000, 321, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Chen, L.; Hatch, G.M. Berberine as a therapy for type 2 diabetes and its complications: From mechanism of action to clinical studies. Biochem. Cell Biol. 2015, 93, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Filippatos, G.; Butler, J.; Farmakis, D.; Zannad, F.; Ofstad, A.P.; Ferreira, J.P.; Green, J.B.; Rosenstock, J.; Schnaidt, S.; Brueckmann, M.; et al. Empagliflozin for heart failure with preserved left ventricular ejection fraction with and without diabetes. Circulation 2022, 146, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Kamboj, P.; Talukdar, N.C.; Banerjee, S.K. Therapeutic benefit of dillenia indica in diabetes and its associated complications. J. Diabetes Res. 2019, 2019, 4632491. [Google Scholar] [CrossRef]
- Kawanami, D.; Takashi, Y.; Tanabe, M. Significance of metformin use in diabetic kidney disease. Int. J. Mol. Sci. 2020, 21, 4239. [Google Scholar] [CrossRef]
- Serina, J.J.C.; Castilho, P. Using polyphenols as a relevant therapy to diabetes and its complications, a review. Crit. Rev. Food Sci. Nutr. 2022, 62, 8355–8387. [Google Scholar] [CrossRef]
- Araki, E.; Nishikawa, T. Oxidative stress: A cause and therapeutic target of diabetic complications. J. Diabetes Investig. 2010, 1, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Sheetz, M.J.; King, G.L. Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. JAMA 2002, 288, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Tesfamariam, B. Free radicals in diabetic endothelial cell dysfunction. Free Radic. Biol. Med. 1994, 16, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Vikramadithyan, R.K.; Hu, Y.; Noh, H.L.; Liang, C.P.; Hallam, K.; Tall, A.R.; Ramasamy, R.; Goldberg, I.J. Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. J. Clin. Investig. 2005, 115, 2434–2443. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Ho, E.C.; Lam, K.S.; Chung, S.K. Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol. 2003, 14, S233–S236. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Chung, S.S. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. 1999, 13, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Chung, S.K.; Chung, S.S. Demonstration that polyol accumulation is responsible for diabetic cataract by the use of transgenic mice expressing the aldose reductase gene in the lens. Proc. Natl. Acad. Sci. USA 1995, 92, 2780–2784. [Google Scholar] [CrossRef]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase c--dependent activation of nad(p)h oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef]
- Donnelly, R.; Idris, I.; Forrester, J.V. Protein kinase c inhibition and diabetic retinopathy: A shot in the dark at translational research. Br. J. Ophthalmol. 2004, 88, 145–151. [Google Scholar] [CrossRef]
- Aiello, L.P.; Avery, R.L.; Arrigg, P.G.; Keyt, B.A.; Jampel, H.D.; Shah, S.T.; Pasquale, L.R.; Thieme, H.; Iwamoto, M.A.; Park, J.E.; et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994, 331, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Mansoor, S.; Sharma, A.; Sapkal, A.; Sheth, J.; Falatoonzadeh, P.; Kuppermann, B.; Kenney, M. Diabetic retinopathy and vegf. Open Ophthalmol. J. 2013, 7, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.L.; Schmidt, A.M. Protein glycation: A firm link to endothelial cell dysfunction. Circ. Res. 2004, 95, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Candido, R.; Forbes, J.M.; Thomas, M.C.; Thallas, V.; Dean, R.G.; Burns, W.C.; Tikellis, C.; Ritchie, R.H.; Twigg, S.M.; Cooper, M.E.; et al. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ. Res. 2003, 92, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Du, Y.; Miller, C.M.; Kern, T.S. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radic. Biol. Med. 2003, 35, 1491–1499. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- David, J.A.; Rifkin, W.J.; Rabbani, P.S.; Ceradini, D.J. The nrf2/keap1/are pathway and oxidative stress as a therapeutic target in type ii diabetes mellitus. J. Diabetes Res. 2017, 2017, 4826724. [Google Scholar] [CrossRef]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The anti-inflammatory and anti-oxidant mechanisms of the keap1/nrf2/are signaling pathway in chronic diseases. Aging Dis. 2019, 10, 637–651. [Google Scholar] [CrossRef]
- Stefanson, A.L.; Bakovic, M. Dietary regulation of keap1/nrf2/are pathway: Focus on plant-derived compounds and trace minerals. Nutrients 2014, 6, 3777–3801. [Google Scholar] [CrossRef]
- Jian, Z.; Li, K.; Liu, L.; Zhang, Y.; Zhou, Z.; Li, C.; Gao, T. Heme oxygenase-1 protects human melanocytes from h2o2-induced oxidative stress via the nrf2-are pathway. J. Investig. Dermatol. 2011, 131, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Albert-Garay, J.S.; Riesgo-Escovar, J.R.; Salceda, R. High glucose concentrations induce oxidative stress by inhibiting nrf2 expression in rat muller retinal cells in vitro. Sci. Rep. 2022, 12, 1261. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Small maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the keap1-nrf2 regulatory pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 6379–6384. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.Y.; Ma, Z.; Segar, L. Spontaneously diabetic ins2(+/akita):Apoe-deficient mice exhibit exaggerated hypercholesterolemia and atherosclerosis. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E145–E154. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Liu, J.; Wang, W.; Wang, M.; Zhao, F.; Sun, J.; Liu, J.; Deng, Q.; Zhao, D. High sdldl cholesterol can be used to reclassify individuals with low cardiovascular risk for early intervention: Findings from the chinese multi-provincial cohort study. J. Atheroscler. Thromb. 2020, 27, 695–710. [Google Scholar] [CrossRef]
- Tribble, D.L.; Rizzo, M.; Chait, A.; Lewis, D.M.; Blanche, P.J.; Krauss, R.M. Enhanced oxidative susceptibility and reduced antioxidant content of metabolic precursors of small, dense low-density lipoproteins. Am. J. Med. 2001, 110, 103–110. [Google Scholar] [CrossRef]
- Jin, J.L.; Zhang, H.W.; Cao, Y.X.; Liu, H.H.; Hua, Q.; Li, Y.F.; Zhang, Y.; Wu, N.Q.; Zhu, C.G.; Xu, R.X.; et al. Association of small dense low-density lipoprotein with cardiovascular outcome in patients with coronary artery disease and diabetes: A prospective, observational cohort study. Cardiovasc. Diabetol. 2020, 19, 45. [Google Scholar] [CrossRef]
- Miller, M. Low-density lipoprotein triglycerides: Widening the atherogenic landscape in cvd risk assessment. J. Am. Coll. Cardiol. 2018, 72, 170–172. [Google Scholar] [CrossRef]
- Yen, F.T.; Roitel, O.; Bonnard, L.; Notet, V.; Pratte, D.; Stenger, C.; Magueur, E.; Bihain, B.E. Lipolysis stimulated lipoprotein receptor: A novel molecular link between hyperlipidemia, weight gain, and atherosclerosis in mice. J. Biol. Chem. 2008, 283, 25650–25659. [Google Scholar] [CrossRef]
- Narvekar, P.; Berriel Diaz, M.; Krones-Herzig, A.; Hardeland, U.; Strzoda, D.; Stohr, S.; Frohme, M.; Herzig, S. Liver-specific loss of lipolysis-stimulated lipoprotein receptor triggers systemic hyperlipidemia in mice. Diabetes 2009, 58, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Petrie, J.R.; Chaturvedi, N.; Ford, I.; Brouwers, M.; Greenlaw, N.; Tillin, T.; Hramiak, I.; Hughes, A.D.; Jenkins, A.J.; Klein, B.E.K.; et al. Cardiovascular and metabolic effects of metformin in patients with type 1 diabetes (removal): A double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Li, W.B.; Liu, J.B.; Lu, J.W.; Feng, J.F. Autophagy: Novel applications of nonsteroidal anti-inflammatory drugs for primary cancer. Cancer Med. 2018, 7, 471–484. [Google Scholar] [CrossRef] [PubMed]
- You, G.; Long, X.; Song, F.; Huang, J.; Tian, M.; Xiao, Y.; Deng, S.; Wu, Q. Metformin activates the ampk-mtor pathway by modulating lncrna tug1 to induce autophagy and inhibit atherosclerosis. Drug Des. Devel. Ther. 2020, 14, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Osonoi, Y.; Mita, T.; Azuma, K.; Nakajima, K.; Masuyama, A.; Goto, H.; Nishida, Y.; Miyatsuka, T.; Fujitani, Y.; Koike, M.; et al. Defective autophagy in vascular smooth muscle cells enhances cell death and atherosclerosis. Autophagy 2018, 14, 1991–2006. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Fan, G.; Li, Y.; Zhou, Q. Tug1 represses apoptosis, autophagy, and inflammatory response by regulating mir-27a-3p/slit2 in lipopolysaccharide-treated vascular endothelial cells. J. Surg. Res. 2020, 256, 345–354. [Google Scholar] [CrossRef]
- Shi, Z.; Zhu, Q.; Fan, J. Lncrna tug1 promotes atherosclerosis progression by targeting mir-382-5p. Int. J. Clin. Exp. Pathol. 2021, 14, 972–979. [Google Scholar] [PubMed]
- Forouzandeh, F.; Salazar, G.; Patrushev, N.; Xiong, S.; Hilenski, L.; Fei, B.; Alexander, R.W. Metformin beyond diabetes: Pleiotropic benefits of metformin in attenuation of atherosclerosis. J. Am. Heart Assoc. 2014, 3, e001202. [Google Scholar] [CrossRef]
- Ichiki, T.; Miyazaki, R.; Kamiharaguchi, A.; Hashimoto, T.; Matsuura, H.; Kitamoto, S.; Tokunou, T.; Sunagawa, K. Resveratrol attenuates angiotensin ii-induced senescence of vascular smooth muscle cells. Regul. Pept. 2012, 177, 35–39. [Google Scholar] [CrossRef]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef]
- Du, J.; Zhu, M.; Li, H.; Liang, G.; Li, Y.; Feng, S. Metformin attenuates cardiac remodeling in mice through the nrf2/keap1 signaling pathway. Exp. Ther. Med. 2020, 20, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, Y.; Sato, K.; Watanabe, T.; Nohtomi, K.; Terasaki, M.; Nagashima, M.; Hirano, T. A glucagon-like peptide-1 analog liraglutide suppresses macrophage foam cell formation and atherosclerosis. Peptides 2014, 54, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, M.; Watanabe, T.; Terasaki, M.; Tomoyasu, M.; Nohtomi, K.; Kim-Kaneyama, J.; Miyazaki, A.; Hirano, T. Native incretins prevent the development of atherosclerotic lesions in apolipoprotein e knockout mice. Diabetologia 2011, 54, 2649–2659. [Google Scholar] [CrossRef]
- Deng, C.; Cao, J.; Han, J.; Li, J.; Li, Z.; Shi, N.; He, J. Liraglutide activates the nrf2/ho-1 antioxidant pathway and protects brain nerve cells against cerebral ischemia in diabetic rats. Comput. Intell. Neurosci. 2018, 2018, 3094504. [Google Scholar] [CrossRef] [PubMed]
- Felez-Nobrega, M.; Werneck, A.O.; Bauman, A.; Haro, J.M.; Koyanagi, A. Active school commuting in adolescents from 28 countries across africa, the americas, and asia: A temporal trends study. Int. J. Behav. Nutr. Phys. Act. 2023, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Park, S.H.; Yun, J.M.; Nam, T.G.; Kim, Y.E.; Kim, D.O.; Kim, Y.J. Effect of cinnamon water extract on monocyte-to-macrophage differentiation and scavenger receptor activity. BMC Complement. Altern. Med. 2014, 14, 90. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.C.; Chung, Y.L.; Wu, M.L.; Chuang, S.M. Cinnamaldehyde enhances nrf2 nuclear translocation to upregulate phase ii detoxifying enzyme expression in hepg2 cells. J. Agric. Food Chem. 2011, 59, 5164–5171. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.M.; Pereira, A.; Seica, R.M. Cinnamaldehyde supplementation reverts endothelial dysfunction in rat models of diet-induced obesity: Role of nf-e2-related factor-2. Antioxidants 2022, 12, 82. [Google Scholar] [CrossRef]
- Wondrak, G.T.; Villeneuve, N.F.; Lamore, S.D.; Bause, A.S.; Jiang, T.; Zhang, D.D. The cinnamon-derived dietary factor cinnamic aldehyde activates the nrf2-dependent antioxidant response in human epithelial colon cells. Molecules 2010, 15, 3338–3355. [Google Scholar] [CrossRef]
- Wang, J.; Huang, X.; Liu, H.; Chen, Y.; Li, P.; Liu, L.; Li, J.; Ren, Y.; Huang, J.; Xiong, E.; et al. Empagliflozin ameliorates diabetic cardiomyopathy via attenuating oxidative stress and improving mitochondrial function. Oxidative Med. Cell. Longev. 2022, 2022, 1122494. [Google Scholar] [CrossRef]
- Gu, Y.; Ma, C.T.; Gu, H.L.; Shi, L.; Tian, X.T.; Xu, W.Q. Sitagliptin improves cardiac function after myocardial infarction through activation of autophagy in streptozotocin-induced diabetic mice. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8973–8983. [Google Scholar] [PubMed]
- Kong, L.; Deng, J.; Zhou, X.; Cai, B.; Zhang, B.; Chen, X.; Chen, Z.; Wang, W. Sitagliptin activates the p62-keap1-nrf2 signalling pathway to alleviate oxidative stress and excessive autophagy in severe acute pancreatitis-related acute lung injury. Cell Death Dis. 2021, 12, 928. [Google Scholar] [CrossRef]
- Huang, S.; Huang, Y.; Lin, W.; Wang, L.; Yang, Y.; Li, P.; Xiao, L.; Chen, Y.; Chu, Q.; Yuan, X. Sitagliptin alleviates radiation-induced intestinal injury by activating nrf2-antioxidant axis, mitigating nlrp3 inf-lammasome activation, and reversing gut microbiota disorder. Oxidative Med. Cell. Longev. 2022, 2022, 2586305. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, T.; Welungoda, I.; Widdop, R.E.; Simpson, R.W.; Dear, A.E. The glp-1 receptor agonist liraglutide inhibits progression of vascular disease via effects on atherogenesis, plaque stability and endothelial function in an apoe(−/−) mouse model. Diabetes Vasc. Dis. Res. 2013, 10, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Rakipovski, G.; Rolin, B.; Nohr, J.; Klewe, I.; Frederiksen, K.S.; Augustin, R.; Hecksher-Sorensen, J.; Ingvorsen, C.; Polex-Wolf, J.; Knudsen, L.B. The glp-1 analogs liraglutide and semaglutide reduce atherosclerosis in apoe(−/−) and ldlr(−/−) mice by a mechanism that includes inflammatory pathways. JACC Basic Transl. Sci. 2018, 3, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yuan, Y.; Li, Y.; Rao, X. Effect of liraglutide on atherosclerosis in patients with impaired glucose tolerance: A double-blind, randomized controlled clinical trial. Exp. Ther. Med. 2023, 25, 249. [Google Scholar] [CrossRef] [PubMed]
- Neto, J.; Damasceno, M.M.C.; Ciol, M.A.; de Freitas, R.; de Araujo, M.F.M.; de Souza Teixeira, C.R.; Carvalho, G.C.N.; de Siqueira Coelho Lisboa, K.W.; de Souza, D.F.; de Menezes Nogueira, J.; et al. Analysis of the effectiveness of cinnamon (cinnamomum verum) in the reduction of glycemic and lipidic levels of adults with type 2 diabetes: A study protocol. Medicine 2020, 99, e18553. [Google Scholar] [CrossRef] [PubMed]
- Deyno, S.; Eneyew, K.; Seyfe, S.; Tuyiringire, N.; Peter, E.L.; Muluye, R.A.; Tolo, C.U.; Ogwang, P.E. Efficacy and safety of cinnamon in type 2 diabetes mellitus and pre-diabetes patients: A meta-analysis and meta-regression. Diabetes Res. Clin. Pract. 2019, 156, 107815. [Google Scholar] [CrossRef]
- Nayak, I.N.; Chinta, R.; Jetti, R. Anti-atherosclerotic potential of aqueous extract of cinnamomum zeylanicum bark against glucocorticoid induced atherosclerosis in wistar rats. J. Clin. Diagn. Res. 2017, 11, FC19–FC23. [Google Scholar] [CrossRef]
- Jin, S.; Cho, K.H. Water extracts of cinnamon and clove exhibits potent inhibition of protein glycation and anti-atherosclerotic activity in vitro and in vivo hypolipidemic activity in zebrafish. Food Chem. Toxicol. 2011, 49, 1521–1529. [Google Scholar] [CrossRef]
- Ruotsalainen, A.K.; Inkala, M.; Partanen, M.E.; Lappalainen, J.P.; Kansanen, E.; Makinen, P.I.; Heinonen, S.E.; Laitinen, H.M.; Heikkila, J.; Vatanen, T.; et al. The absence of macrophage nrf2 promotes early atherogenesis. Cardiovasc. Res. 2013, 98, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [PubMed]
- von Bibra, H.; St John Sutton, M. Diastolic dysfunction in diabetes and the metabolic syndrome: Promising potential for diagnosis and prognosis. Diabetologia 2010, 53, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M. Diabetes and myocardial infarction. Baillieres Best Pract. Res. Clin. Endocrinol. Metab. 1999, 13, 331–343. [Google Scholar] [CrossRef]
- Andersen, M.J.; Ersboll, M.; Axelsson, A.; Gustafsson, F.; Hassager, C.; Kober, L.; Borlaug, B.A.; Boesgaard, S.; Skovgaard, L.T.; Moller, J.E. Sildenafil and diastolic dysfunction after acute myocardial infarction in patients with preserved ejection fraction: The sildenafil and diastolic dysfunction after acute myocardial infarction (sidami) trial. Circulation 2013, 127, 1200–1208. [Google Scholar] [CrossRef]
- Clark, R.J.; McDonough, P.M.; Swanson, E.; Trost, S.U.; Suzuki, M.; Fukuda, M.; Dillmann, W.H. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear o-glcnacylation. J. Biol. Chem. 2003, 278, 44230–44237. [Google Scholar] [CrossRef]
- Mueller, T.; Ouyang, X.; Johnson, M.S.; Qian, W.J.; Chatham, J.C.; Darley-Usmar, V.; Zhang, J. New insights into the biology of protein o-glcnacylation: Approaches and observations. Front. Aging 2020, 1, 620382. [Google Scholar] [CrossRef]
- Baudoin, L.; Issad, T. O-glcnacylation and inflammation: A vast territory to explore. Front. Endocrinol. 2014, 5, 235. [Google Scholar] [CrossRef]
- Kawase, Y.; Hajjar, R.J. The cardiac sarcoplasmic/endoplasmic reticulum calcium atpase: A potent target for cardiovascular diseases. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 554–565. [Google Scholar] [CrossRef]
- Gao, Y.; Wells, L.; Comer, F.I.; Parker, G.J.; Hart, G.W. Dynamic o-glycosylation of nuclear and cytosolic proteins: Cloning and characterization of a neutral, cytosolic beta-n-acetylglucosaminidase from human brain. J. Biol. Chem. 2001, 276, 9838–9845. [Google Scholar] [CrossRef]
- Haltiwanger, R.S.; Blomberg, M.A.; Hart, G.W. Glycosylation of nuclear and cytoplasmic proteins. Purification and characterization of a uridine diphospho-n-acetylglucosamine:Polypeptide beta-n-acetylglucosaminyltransferase. J. Biol. Chem. 1992, 267, 9005–9013. [Google Scholar] [CrossRef] [PubMed]
- Hammoudi, N.; Jeong, D.; Singh, R.; Farhat, A.; Komajda, M.; Mayoux, E.; Hajjar, R.; Lebeche, D. Empagliflozin improves left ventricular diastolic dysfunction in a genetic model of type 2 diabetes. Cardiovasc. Drugs Ther. 2017, 31, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Pabel, S.; Wagner, S.; Bollenberg, H.; Bengel, P.; Kovacs, A.; Schach, C.; Tirilomis, P.; Mustroph, J.; Renner, A.; Gummert, J.; et al. Empagliflozin directly improves diastolic function in human heart failure. Eur. J. Heart Fail. 2018, 20, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Chiasson, J.L.; Josse, R.G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M.; Group, S.-N.T.R. Acarbose treatment and the risk of cardiovascular disease and hypertension in patients with impaired glucose tolerance: The stop-niddm trial. JAMA 2003, 290, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Picatoste, B.; Ramirez, E.; Caro-Vadillo, A.; Iborra, C.; Ares-Carrasco, S.; Egido, J.; Tunon, J.; Lorenzo, O. Sitagliptin reduces cardiac apoptosis, hypertrophy and fibrosis primarily by insulin-dependent mechanisms in experimental type-ii diabetes. Potential roles of glp-1 isoforms. PLoS ONE 2013, 8, e78330. [Google Scholar] [CrossRef]
- Yang, M.; Xi, N.; Gao, M.; Yu, Y. Sitagliptin mitigates hypoxia/reoxygenation (h/r)-induced injury in cardiomyocytes by mediating sirtuin 3 (sirt3) and autophagy. Bioengineered 2022, 13, 13162–13173. [Google Scholar] [CrossRef]
- De Geest, B.; Mishra, M. Role of oxidative stress in diabetic cardiomyopathy. Antioxidants 2022, 11, 784. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.Y.; Wen, T.J.; Cheng, Y.H.; Tsai, Y.T.; Chiang, C.Y.; Chien, C.T. Diabetes upregulates oxidative stress and downregulates cardiac protection to exacerbate myocardial ischemia/reperfusion injury in rats. Antioxidants 2020, 9, 679. [Google Scholar] [CrossRef]
- Dinic, S.; Arambasic, J.; Mihailovic, M.; Uskokovic, A.; Grdovic, N.; Markovic, J.; Karadzic, B.; Poznanovic, G.; Vidakovic, M. Decreased o-glcnacylation of the key proteins in kinase and redox signalling pathways is a novel mechanism of the beneficial effect of alpha-lipoic acid in diabetic liver. Br. J. Nutr. 2013, 110, 401–412. [Google Scholar] [CrossRef]
- Arambasic, J.; Mihailovic, M.; Uskokovic, A.; Dinic, S.; Grdovic, N.; Markovic, J.; Poznanovic, G.; Bajec, D.; Vidakovic, M. Alpha-lipoic acid upregulates antioxidant enzyme gene expression and enzymatic activity in diabetic rat kidneys through an o-glcnac-dependent mechanism. Eur. J. Nutr. 2013, 52, 1461–1473. [Google Scholar] [CrossRef]
- Chen, P.H.; Chi, J.T.; Boyce, M. Keap1 has a sweet spot: A new connection between intracellular glycosylation and redox stress signaling in cancer cells. Mol. Cell. Oncol. 2017, 4, e1361501. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Smith, T.J.; Wu, J.; Siesser, P.F.; Bisnett, B.J.; Khan, F.; Hogue, M.; Soderblom, E.; Tang, F.; Marks, J.R.; et al. Glycosylation of keap1 links nutrient sensing to redox stress signaling. EMBO J. 2017, 36, 2233–2250. [Google Scholar] [CrossRef] [PubMed]
- Burlew, B.S.; Weber, K.T. Cardiac fibrosis as a cause of diastolic dysfunction. Herz 2002, 27, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Hovind, P.; Tarnow, L.; Rossing, P.; Jensen, B.R.; Graae, M.; Torp, I.; Binder, C.; Parving, H.H. Predictors for the development of microalbuminuria and macroalbuminuria in patients with type 1 diabetes: Inception cohort study. BMJ 2004, 328, 1105. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, C.E.; Christensen, C.K. Predicting diabetic nephropathy in insulin-dependent patients. N. Engl. J. Med. 1984, 311, 89–93. [Google Scholar] [CrossRef] [PubMed]
- de Boer, I.H.; Rue, T.C.; Cleary, P.A.; Lachin, J.M.; Molitch, M.E.; Steffes, M.W.; Sun, W.; Zinman, B.; Brunzell, J.D.; Diabetes, C.; et al. Long-term renal outcomes of patients with type 1 diabetes mellitus and microalbuminuria: An analysis of the diabetes control and complications trial/epidemiology of diabetes interventions and complications cohort. Arch. Intern. Med. 2011, 171, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Gerasimova, M.; Rose, M.A.; Masuda, T.; Satriano, J.; Mayoux, E.; Koepsell, H.; Thomson, S.C.; Rieg, T. Sglt2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic akita mice. Am. J. Physiol.-Renal. Physiol. 2014, 306, F194–F204. [Google Scholar] [CrossRef] [PubMed]
- Halimi, S.; Verges, B. Adverse effects and safety of sglt-2 inhibitors. Diabetes Metab. 2014, 40, S28–S34. [Google Scholar] [CrossRef]
- Lim, J.-H.; Kwon, S.; Noh, H.W.; Jeon, S.; Jung, H.-Y.; Choi, J.-Y.; Park, S.-H.; Kim, C.-D.; Kim, Y.-L.; Lee, J.P.; et al. Sodium-Glucose Cotransporter 2 Inhibitors in Kidney Transplant Recipients. Available online: https://www.asn-online.org/education/kidneyweek/2021/program-abstract.aspx?controlId=3606944 (accessed on 21 December 2023).
- Arora, M.K.; Sarup, Y.; Tomar, R.; Singh, M.; Kumar, P. Amelioration of diabetes-induced diabetic nephropathy by aloe vera: Implication of oxidative stress and hyperlipidemia. J. Diet. Suppl. 2019, 16, 227–244. [Google Scholar] [CrossRef]
- Chatterjee, P.; Mukherjee, A.; Nandy, S. Protective effects of the aqueous leaf extract of aloe barbadensis on gentamicin and cisplatin–induced nephrotoxic rats. Asian Pac. J. Trop. Biomed. 2012, 2, S1754–S1763. [Google Scholar] [CrossRef]
- Chang, T.T.; Chen, Y.A.; Li, S.Y.; Chen, J.W. Nrf-2 mediated heme oxygenase-1 activation contributes to the anti-inflammatory and renal protective effects of ginkgo biloba extract in diabetic nephropathy. J. Ethnopharmacol. 2021, 266, 113474. [Google Scholar] [CrossRef]
- Cheng, D.; Liang, B.; Li, Y. Antihyperglycemic effect of ginkgo biloba extract in streptozotocin-induced diabetes in rats. Biomed. Res. Int. 2013, 2013, 162724. [Google Scholar] [CrossRef]
- Dare, A.; Channa, M.L.; Nadar, A. L-ergothioneine and its combination with metformin attenuates renal dysfunction in type-2 diabetic rat model by activating nrf2 antioxidant pathway. Biomed. Pharmacother. 2021, 141, 111921. [Google Scholar] [CrossRef]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomlinson, J.W. Non-alcoholic fatty liver disease and diabetes. Metabolism 2016, 65, 1096–1108. [Google Scholar] [CrossRef]
- Ezhilarasan, D.; Lakshmi, T. A molecular insight into the role of antioxidants in nonalcoholic fatty liver diseases. Oxidative Med. Cell. Longev. 2022, 2022, 9233650. [Google Scholar] [CrossRef]
- Pais, R.; Charlotte, F.; Fedchuk, L.; Bedossa, P.; Lebray, P.; Poynard, T.; Ratziu, V.; Group, L.S. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J. Hepatol. 2013, 59, 550–556. [Google Scholar] [CrossRef]
- Kotronen, A.; Juurinen, L.; Hakkarainen, A.; Westerbacka, J.; Corner, A.; Bergholm, R.; Yki-Jarvinen, H. Liver fat is increased in type 2 diabetic patients and underestimated by serum alanine aminotransferase compared with equally obese nondiabetic subjects. Diabetes Care 2008, 31, 165–169. [Google Scholar] [CrossRef]
- Bae, J.C.; Cho, Y.K.; Lee, W.Y.; Seo, H.I.; Rhee, E.J.; Park, S.E.; Park, C.Y.; Oh, K.W.; Sung, K.C.; Kim, B.I. Impact of nonalcoholic fatty liver disease on insulin resistance in relation to hba1c levels in nondiabetic subjects. Am. J. Gastroenterol. 2010, 105, 2389–2395. [Google Scholar] [CrossRef]
- Dharmalingam, M.; Yamasandhi, P.G. Nonalcoholic fatty liver disease and type 2 diabetes mellitus. Indian J. Endocrinol. Metab. 2018, 22, 421–428. [Google Scholar] [CrossRef]
- Ford, R.J.; Fullerton, M.D.; Pinkosky, S.L.; Day, E.A.; Scott, J.W.; Oakhill, J.S.; Bujak, A.L.; Smith, B.K.; Crane, J.D.; Blumer, R.M.; et al. Metformin and salicylate synergistically activate liver ampk, inhibit lipogenesis and improve insulin sensitivity. Biochem. J. 2015, 468, 125–132. [Google Scholar] [CrossRef]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: A randomized trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; De Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011, 31, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Min-DeBartolo, J.; Schlerman, F.; Akare, S.; Wang, J.; McMahon, J.; Zhan, Y.; Syed, J.; He, W.; Zhang, B.; Martinez, R.V. Thrombospondin-i is a critical modulator in non-alcoholic steatohepatitis (nash). PLoS ONE 2019, 14, e0226854. [Google Scholar] [CrossRef]
- Yao, M.; Rogers, N.M.; Csanyi, G.; Rodriguez, A.I.; Ross, M.A.; St Croix, C.; Knupp, H.; Novelli, E.M.; Thomson, A.W.; Pagano, P.J.; et al. Thrombospondin-1 activation of signal-regulatory protein-alpha stimulates reactive oxygen species production and promotes renal ischemia reperfusion injury. J. Am. Soc. Nephrol. 2014, 25, 1171–1186. [Google Scholar] [CrossRef] [PubMed]
- Hosseini-Fard, S.R.; Dehpour, A.R.; Emamgholipour, S.; Golestani, A. Naltrexone protects against bdl-induced cirrhosis in wistar rats by attenuating thrombospondin-1 and enhancing antioxidant defense system via nrf-2. Life Sci. 2022, 300, 120576. [Google Scholar] [CrossRef] [PubMed]
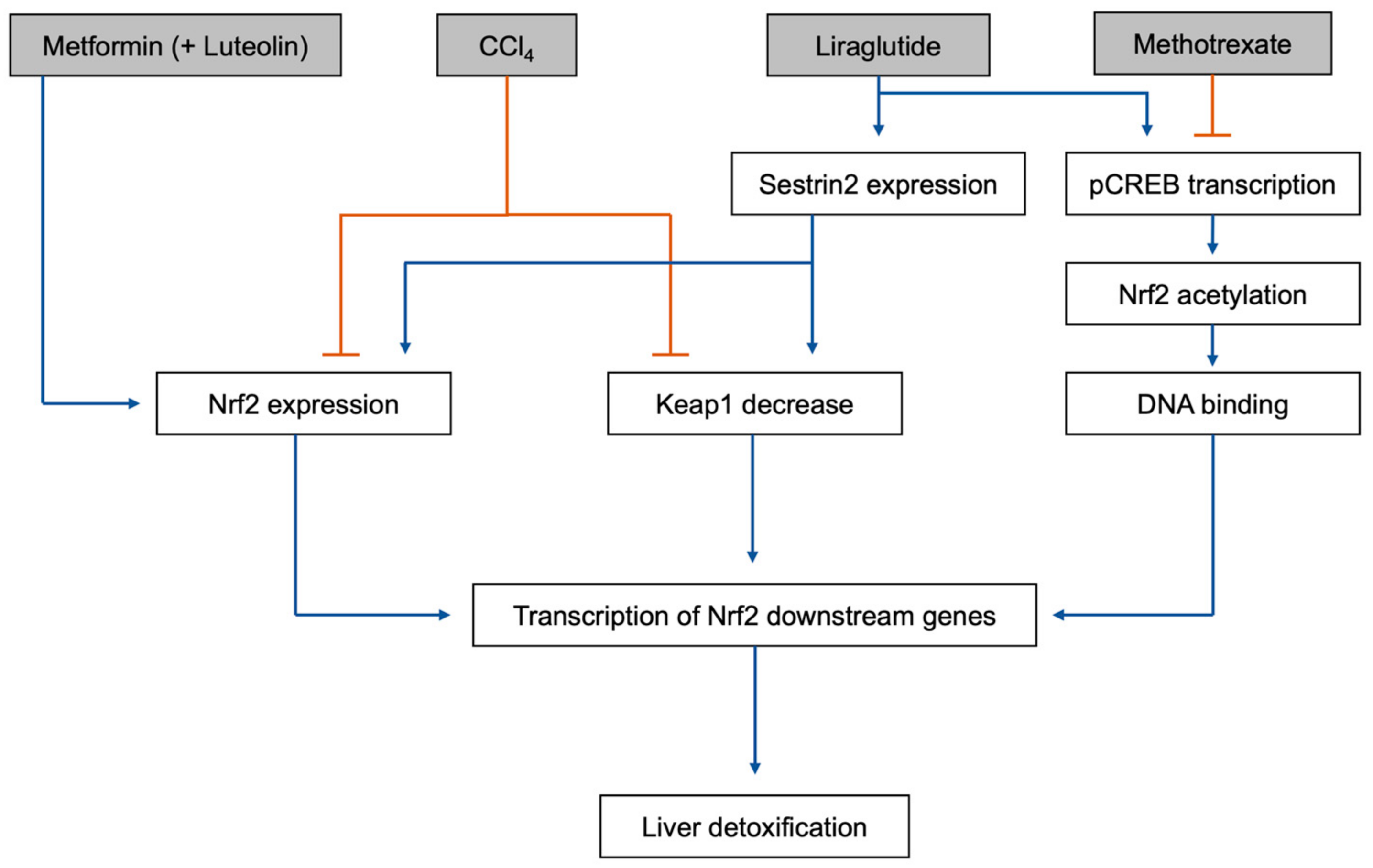
- Yan, Y.; Jun, C.; Lu, Y.; Jiangmei, S. Combination of metformin and luteolin synergistically protects carbon tetrachloride-induced hepatotoxicity: Mechanism involves antioxidant, anti-inflammatory, antiapoptotic, and nrf2/ho-1 signaling pathway. Biofactors 2019, 45, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, Y.; Liu, J.; Ye, J.; Yuan, W.; Jiang, H.; Wang, Z.; Jiang, H.; Wan, J. Recent insights into the biological functions of sestrins in health and disease. Cell. Physiol. Biochem. 2017, 43, 1731–1741. [Google Scholar] [CrossRef] [PubMed]
- Huttl, M.; Markova, I.; Miklankova, D.; Zapletalova, I.; Poruba, M.; Haluzik, M.; Vaneckova, I.; Malinska, H. In a prediabetic model, empagliflozin improves hepatic lipid metabolism independently of obesity and before onset of hyperglycemia. Int. J. Mol. Sci. 2021, 22, 11513. [Google Scholar] [CrossRef]
- Guo, M.; Gu, L.; Hui, H.; Li, X.; Jin, L. Extracts of dracocephalum tanguticum maxim ameliorate acute alcoholic liver disease via regulating transcription factors in mice. Front. Pharmacol. 2022, 13, 830532. [Google Scholar] [CrossRef]
- Qiu, P.; Dong, Y.; Li, B.; Kang, X.J.; Gu, C.; Zhu, T.; Luo, Y.Y.; Pang, M.X.; Du, W.F.; Ge, W.H. Dihydromyricetin modulates p62 and autophagy crosstalk with the keap-1/nrf2 pathway to alleviate ethanol-induced hepatic injury. Toxicol. Lett. 2017, 274, 31–41. [Google Scholar] [CrossRef]
- Khalaf, M.M.; Hassanein, E.H.M.; Shalkami, A.S.; Hemeida, R.A.M.; Mohamed, W.R. Diallyl disulfide attenuates methotrexate-induced hepatic oxidative injury, inflammation and apoptosis and enhances its anti-tumor activity. Curr. Mol. Pharmacol. 2022, 15, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Wang, N.; Xiao, Y.; Deng, Y.; Zha, A.; Tan, B.; Wang, J.; Yin, Y.; Liao, P. Ellagic acid ameliorates paraquat-induced liver injury associated with improved gut microbial profile. Environ. Pollut. 2022, 293, 118572. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.Q.; Ding, J.; Zhang, L.; Liu, C.M. Protective effects of ursolic acid in an experimental model of liver fibrosis through nrf2/are pathway. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Bi, H.; Li, X.; Cui, L.; He, W.; Tian, Y.; Liu, F.; Gao, G.; Wang, Z.; Chen, N.; et al. Up-regulation of nrf2/p62/keap1 involves in the anti-fibrotic effect of combination of monoammonium glycyrrhizinate and cysteine hydrochloride induced by ccl4. Eur. J. Pharmacol. 2021, 913, 174628. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; Leon, R.; Lopez, M.G.; Oliva, B.; et al. Transcription factor nrf2 as a therapeutic target for chronic diseases: A systems medicine approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed]
- Abdelaziz, A.I.; Mantawy, E.M.; Gad, A.M.; Fawzy, H.M.; Azab, S.S. Activation of pcreb/nrf-2 signaling mediates re-positioning of liraglutide as hepato-protective for methotrexate -induced liver injury (mili). Food Chem. Toxicol. 2019, 132, 110719. [Google Scholar] [CrossRef] [PubMed]
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L. Susceptibility of nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell. Biol. 2014, 34, 3305–3320. [Google Scholar]
- Okamura, T.; Hashimoto, Y.; Hamaguchi, M.; Obora, A.; Kojima, T.; Fukui, M. Effect of alcohol consumption and the presence of fatty liver on the risk for incident type 2 diabetes: A population-based longitudinal study. BMJ Open Diabetes Res. Care 2020, 8, e001629. [Google Scholar] [CrossRef]
- Aberg, F.; Helenius-Hietala, J.; Puukka, P.; Farkkila, M.; Jula, A. Interaction between alcohol consumption and metabolic syndrome in predicting severe liver disease in the general population. Hepatology 2018, 67, 2141–2149. [Google Scholar] [CrossRef]
- Kiechl, S.; Willeit, J.; Rungger, G.; Egger, G.; Oberhollenzer, F.; Bonora, E. Alcohol consumption and atherosclerosis: What is the relation? Prospective results from the bruneck study. Stroke 1998, 29, 900–907. [Google Scholar] [CrossRef]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The role of oxidative stress and antioxidants in liver diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, C.K.; Lavoie, H.A.; Dipette, D.J.; Singh, U.S. Resveratrol restores nrf2 level and prevents ethanol-induced toxic effects in the cerebellum of a rodent model of fetal alcohol spectrum disorders. Mol. Pharmacol. 2011, 80, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liang, X.C.; Zhang, H.; Wu, Q.L.; Qu, L.; Sun, Q. Quercetin protects rat dorsal root ganglion neurons against high glucose-induced injury in vitro through nrf-2/ho-1 activation and nf-kappab inhibition. Acta Pharmacol. Sin. 2013, 34, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Movahed, A.; Nabipour, I.; Lieben Louis, X.; Thandapilly, S.J.; Yu, L.; Kalantarhormozi, M.; Rekabpour, S.J.; Netticadan, T. Antihyperglycemic effects of short term resveratrol supplementation in type 2 diabetic patients. Evid.-Based Complement. Alternat. Med. 2013, 2013, 851267. [Google Scholar] [CrossRef]
- Golbidi, S.; Badran, M.; Laher, I. Antioxidant and anti-inflammatory effects of exercise in diabetic patients. Exp. Diabetes Res. 2012, 2012, 941868. [Google Scholar] [CrossRef] [PubMed]
- Fathi, R.; Nasiri, K.; Akbari, A.; Ahmadi-KaniGolzar, F.; Farajtabar, Z. Exercise protects against ethanol-induced damage in rat heart and liver through the inhibition of apoptosis and activation of nrf2/keap-1/ho-1 pathway. Life Sci. 2020, 256, 117958. [Google Scholar] [CrossRef]
- Freigang, S.; Ampenberger, F.; Spohn, G.; Heer, S.; Shamshiev, A.T.; Kisielow, J.; Hersberger, M.; Yamamoto, M.; Bachmann, M.F.; Kopf, M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 2011, 41, 2040–2051. [Google Scholar] [CrossRef]
- Grant, D.M. Detoxification pathways in the liver. J. Inherit. Metab. Dis. 1991, 14, 421–430. [Google Scholar] [CrossRef]
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and inhibitors of nrf2: A review of their potential for clinical development. Oxidative Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Chian, S.; Li, Y.Y.; Wang, X.J.; Tang, X.W. Luteolin sensitizes two oxaliplatin-resistant colorectal cancer cell lines to chemotherapeutic drugs via inhibition of the nrf2 pathway. Asian Pac. J. Cancer Prev. 2014, 15, 2911–2916. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, H.; Fan, L.; Wu, X.; Xin, A.; Ren, H.; Wang, X.J. Luteolin inhibits nrf2 leading to negative regulation of the nrf2/are pathway and sensitization of human lung carcinoma a549 cells to therapeutic drugs. Free Radic. Biol. Med. 2011, 50, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.M.; Ke, Z.P.; Wang, J.N.; Yang, J.Y.; Chen, S.Y.; Chen, H. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma bel-7402/adm cells to doxorubicin via inhibiting pi3k/akt/nrf2 pathway. Carcinogenesis 2013, 34, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, Y.; Li, W.; Miao, H.; Zhang, H.; Zhou, Y.; Li, Z.; You, Q.; Zhao, L.; Guo, Q. Wogonin reverses multi-drug resistance of human myelogenous leukemia k562/a02 cells via downregulation of mrp1 expression by inhibiting nrf2/are signaling pathway. Biochem. Pharmacol. 2014, 92, 220–234. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Sun, K.; Wang, X.; Pan, H.; Zhu, J.; Ji, X.; Li, X. Chrysin suppresses proliferation, migration, and invasion in glioblastoma cell lines via mediating the erk/nrf2 signaling pathway. Drug Des. Devel. Ther. 2018, 12, 721–733. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, H.X.; Zhu, J.Q.; Dou, Y.X.; Xian, Y.F.; Lin, Z.X. Natural nrf2 inhibitors: A review of their potential for cancer treatment. Int. J. Biol. Sci. 2023, 19, 3029–3041. [Google Scholar] [CrossRef]
- Milani, L.; Galindo, C.M.; Turin de Oliveira, N.M.; Corso, C.R.; Adami, E.R.; Stipp, M.C.; Beltrame, O.C.; Acco, A. The glp-1 analog liraglutide attenuates acute liver injury in mice. Ann. Hepatol. 2019, 18, 918–928. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Disease | Drug/Substance | Action in Nrf2 and Other Antioxidative Pathways | References |
|---|---|---|---|
| Atherosclerosis | Metformin | SOD1 upregulation | [49,51] |
| ROS inhibition | |||
| Nrf2 upregulation | [52] | ||
| Keap1 downregulation | |||
| HO-1 upregulation | |||
| Upregulation of Nrf2 translocation | |||
| Liraglutide | Inhibition of oxLDL uptake | [53] | |
| CD36 downregulation | [54] | ||
| Nrf2/HO-1 pathway activation | [55] | ||
| Promotion of SOD and MPO activities | |||
| Promotion of Nrf2 activities | [56] | ||
| C. verum/C. cassia | CD36 downregulation | [57] | |
| SRA downregulation | |||
| Upregulation of Nrf2 translocation | [58] | ||
| Nrf2 upregulation | [59] | ||
| Keap1 regulation | [60] | ||
| Cardiac fibrosis | Empagliflozin | Nrf2 upregulation | [61] |
| SOD2 upregulation | |||
| Upregulation of Nrf2 translocation | |||
| Sitagliptin | Downregulation of inflammatory cytokines | [62] | |
| Upregulation of Nrf2 translocation | [63] | ||
| Nrf2 upregulation | [64] | ||
| HO-1 upregulation |
| Disease | Drug/Substance | Action in Nrf2 and Other Antioxidative Pathways | References |
|---|---|---|---|
| Diabetic nephropathy | A. vera | Reduction of oxidative stress | [102] |
| GBE | Nrf2 upregulation | [103,104] | |
| HO-1 upregulation | |||
| Metformin with L-egt | Nrf2 upregulation | [105] | |
| Upregulation of cytoprotective genes and antioxidative enzymes | |||
| Downregulation of TGF-β1 gene | |||
| Inhibition of the NF-kB inflammatory gene |
| Disease | Drug/Substance | Action in Nrf2 and Other Antioxidative Pathways | References |
|---|---|---|---|
| Nonalcoholic fatty liver disease | Naltrexone | Nrf2 upregulation | [115] |
| THBS-1 downregulation | |||
| NOX-1 downregulation | |||
| Metformin | Nrf2 upregulation | [118] | |
| HO-1 upregulation | |||
| Liraglutide | Nrf2 upregulation | [119] | |
| HO-1 upregulation | |||
| Sestrin2 upregulation | |||
| Keap1 degradation | |||
| Empagliflozin | Nrf2 upregulation | [120] | |
| Normalization of cytochrome p450 expression | |||
| Alcoholic liver disease | D. tanguticum | Nrf2 upregulation | [121] |
| Keap1 downregulation | |||
| Dihydromyricetin | Nrf2 upregulation | [122] | |
| Keap1 downregulation | |||
| HO-1 downregulation | |||
| Alleviation of disordered NF-κB and Nrf2 localization | |||
| Toxin-induced liver damage | Diallyl disulfide | Nrf2 upregulation and Keap1 downregulation | [123] |
| Ellagic acid | [124] | ||
| Ursolic acid | [125] | ||
| Monoammonium glycyrrhizinate | [126] | ||
| Cysteine hydrochloride | |||
| Metformin with luteolin | Nrf2 upregulation | [118,127] | |
| HO-1 upregulation | |||
| Liraglutide | pCREB upregulation | [128] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, M.; Cruz Cisneros, L.; Cho, E.J.; Alexander, M.; Kimelman, F.A.; Swentek, L.; Ferrey, A.; Tantisattamo, E.; Ichii, H. Nrf2 Pathway and Oxidative Stress as a Common Target for Treatment of Diabetes and Its Comorbidities. Int. J. Mol. Sci. 2024, 25, 821. https://doi.org/10.3390/ijms25020821
Yi M, Cruz Cisneros L, Cho EJ, Alexander M, Kimelman FA, Swentek L, Ferrey A, Tantisattamo E, Ichii H. Nrf2 Pathway and Oxidative Stress as a Common Target for Treatment of Diabetes and Its Comorbidities. International Journal of Molecular Sciences. 2024; 25(2):821. https://doi.org/10.3390/ijms25020821
Chicago/Turabian StyleYi, Michelle, Leslie Cruz Cisneros, Eric J. Cho, Michael Alexander, Francesca A. Kimelman, Lourdes Swentek, Antoney Ferrey, Ekamol Tantisattamo, and Hirohito Ichii. 2024. "Nrf2 Pathway and Oxidative Stress as a Common Target for Treatment of Diabetes and Its Comorbidities" International Journal of Molecular Sciences 25, no. 2: 821. https://doi.org/10.3390/ijms25020821
APA StyleYi, M., Cruz Cisneros, L., Cho, E. J., Alexander, M., Kimelman, F. A., Swentek, L., Ferrey, A., Tantisattamo, E., & Ichii, H. (2024). Nrf2 Pathway and Oxidative Stress as a Common Target for Treatment of Diabetes and Its Comorbidities. International Journal of Molecular Sciences, 25(2), 821. https://doi.org/10.3390/ijms25020821