

Eternal Youth: A Comprehensive Exploration of Gene, Cellular, and Pharmacological Anti-Aging Strategies


Abstract
1. Introduction
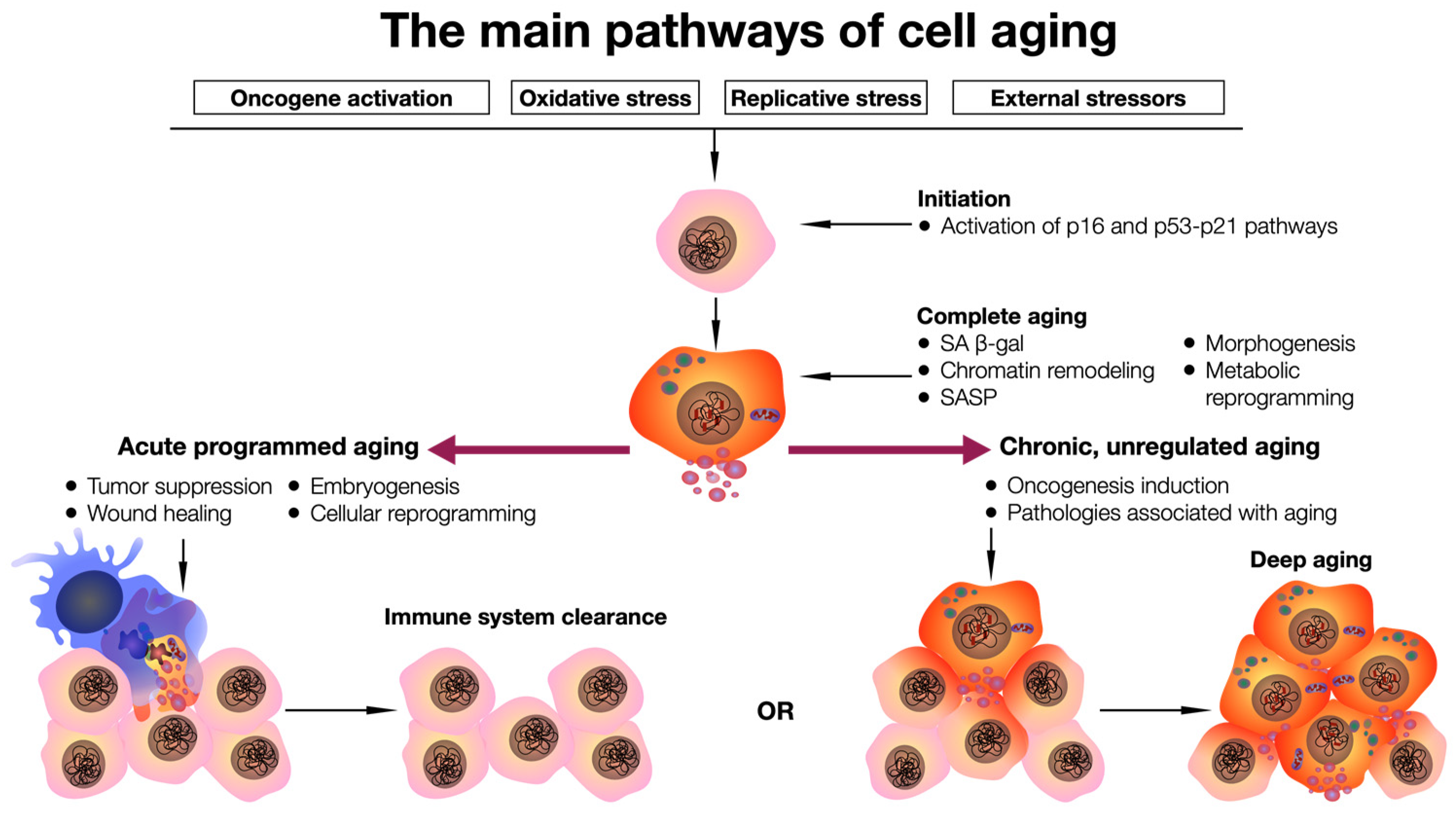
2. Cellular and Organ-Specific Aging
3. The Use of Senolytic Agents

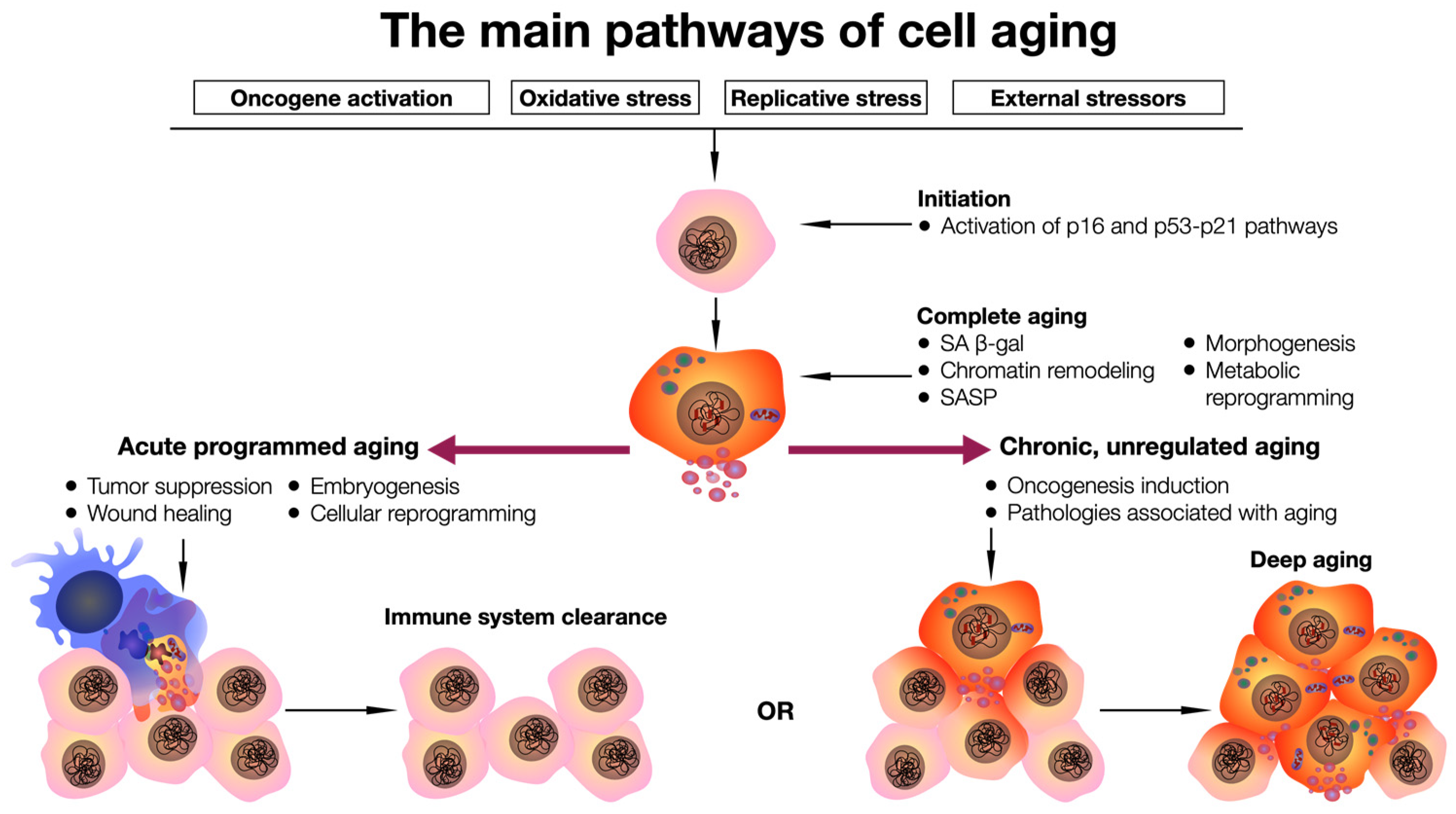{kind=link}
| Name | Mechanism of Action | Model Used | Effect | Reference |
|---|---|---|---|---|
| A-1155463 | Selective inhibitor of BCL-XL | SCID-Beige mice with xenograft of BCL-XL-dependent lung carcinoma cells of the NCI-H146 lung carcinoma cell line | Causes reversible thrombocytopenia in mice and inhibits small-cell lung cancer xenograft growth in vivo after repeated administration | [67] |
| IMR-90 cell line, primary human preadipocytes, HUVECs | Induces apoptosis of senescent HUVEC and IMR-90 cells, but not preadipocytes | [66] | ||
| Gastric cancer cell lines (23132/87, SNU216, NCI-N87, MKN1, AGS, HGC27, SNU719). Human multiple myeloma cell lines (MM1S, KMS12PE, KMS12BM) | Has a cytostatic effect on tumor cells | [68] | ||
| A-1331852 | Selective inhibitor of BCL-XL | Mouse model of glioblastoma multiforme, U251 and SNB-19 cells | Promoted the destruction of U251 cells | [69] |
| Cardiac glycosides | Inducers of apoptosis through the Na+/K+-ATPase pump | PDX-immunodeficient NMRInu/nu mice with xenografts of A549 (lung adenocarcinoma) and IMR-90 (normal human lung cells) cells | In vivo inhibition of xenografted tumors in mice after treatment | [70] |
| Curcumin | Decreased expression of the opioid nociceptin receptor gene (OPRL1) | Human neuroglia cell line T98G | Reduces OPRL1 gene expression associated with pain syndromes | [71] |
| Inhibition of the mitogen-activated protein kinase (MAPK)/transcriptional nuclear factor κB (NF-κB) signaling pathway | C57BL/6 mice and primary hepatocytes isolated from the livers of C57BL/6 mice | Inhibits the MAPK signaling pathway in the liver in old mice and p38 in old mice with diet-induced obesity. Improves insulin homeostasis and reduces body weight in old mice | [72] | |
| Dasatinib + quercetin | Suppression of SRC family kinase inhibitors | DU-145 and LNCaP prostate cancer cells | Inhibits the cell adhesion, migration, and invasion of prostate cancer cells at low nanomolar concentrations | [73] |
| Decreased mRNA levels of genes encoding proinflammatory cytokines: IL-1β, tumor necrosis factor α (TNF-α) and chemokine (C-X-C motif) ligand 8b.1 (CXCL-8b.1) | Danio rerio fish larvae deficient in the serine protease inhibitor protein Spint1a | Has an anti-inflammatory effect and reduces chronic inflammation | [7] | |
| Fisetin | Blocks the signaling pathway PI3K/AKT/mTOR/p16INK4a | Ercc1−/∆ mice (human progeroid syndrome model) and aged wild-type mice, human fibroblasts (IMR-90) | Tissue-specifically reduces cellular senescence in mouse adipose tissue and human cells | [74] |
| FOXO4-related peptide | Blocks the interaction of transcription factor FOXO4 with p53, leading to apoptosis | Human chondrocytes of early and late passages | Removes (eliminates) senescent cells in the late passage chondrocyte population in vitro | [75] |
| Geldanamycin | HSP90 inhibitor | Primary embryonic fibroblasts from Ercc1−/−/mesenchymal stem cells (MSCs) from the bone marrow of Ercc1−/Δ and Ercc1−/∆ mice | Prolongs the lifespan of mice with a progeria model, delays the onset of several age-related symptoms, and reduces the expression of p16INK4a | [76] |
| Luteolin | Inhibitor of mTOR signaling pathway | Human bladder cancer cell lines T24, 5637 (with p53 mutation), and RT-4 and rat bladder cancer cell line BC31 (with p53 mutation) in vitro/rats with bladder cancer model | Inhibits cell survival and induces cell cycle arrest in the G2/M phase; p21 activation in bladder cancer cells | [77] |
| Navitoclax (previously ABT263) | BCL-2 inhibitor | Human skin xenograft in immunodeficient mice | Causes selective elimination of senescent dermal fibroblasts | [78] |
| Nutlin-3a | E3 ubiquitin ligase inhibitor MDM2/p53 | A chemically induced aging mouse model, an Alu-induced geographic retinal atrophy model, and aged mice | Has a senolytic effect; reduces levels of aging markers, SASP components, and ocular pigment deposits | [79] |
| Piperlongumin | Inhibitor of extracellular signal-regulated kinase (ERK) 1/2 | Senescent human fibroblasts of WI-38 lineage | Shows moderate selectivity in reducing the viability of ionizing-radiation-induced senescent fibroblasts of the WI-38 lineage | [80] |
| Rapamycin | Inhibitor of mTOR signaling pathway | Nrf2-KO fibroblasts (nuclear factor Nrf2 knockout) in vivo, Nrf2-KO mice in vitro | Increases Nrf2 levels, which activates autophagy, and reduces induction of cellular senescence in vitro. In mice, Nrf2-KO reduces the concentration of proinflammatory cytokines in serum and adipose tissue in vivo | [81] |
| Resorcin | HSP90 inhibitor | Primary embryonic fibroblasts from Ercc1−/− and Ercc1−/∆ mice | Reduces the number of senescent mouse embryonic cells | [76] |
| Tanespimycin (17-AAG) | HSP90 inhibitor | An isogenic model of BAX knockout in human colon carcinoma cell line HCT116 in vitro and in tumor xenografts in vivo. | Causes a cytostatic antiproliferative effect on tumor cells through inhibition of oncogenes | [82] |
| Alvespimycin (17-DMAG) | HSP90 inhibitor | Primary embryonic fibroblasts from Ercc1−/− and Ercc1−/∆ mice | Prolongs the lifespan of Ercc1−/∆ mice, delays the onset of several age-related symptoms, and reduces p16INK4a expression | [76] |
4. Cell and Genetically Modified Cell-Based Therapy
5. Possibilities for Gene Therapy Based on Recombinant Adeno-Associated Viruses
5.1. Sirtuins
5.2. Telomerase
5.3. The Aging Suppressor Gene Klotho
5.4. Fibroblast Growth Factor 21
5.5. Using Other Genes to Treat Age-Related Diseases
5.5.1. Treatment of Age-Related Macular Degeneration
5.5.2. Cognitive Dysfunction/Neurodegenerative Diseases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Ros, M.; Carrascosa, J.M. Current nutritional and pharmacological anti-aging interventions. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165612. [Google Scholar] [CrossRef]
- Austad, S.N.; Hoffman, J.M. Is antagonistic pleiotropy ubiquitous in aging biology? Evol. Med. Public Health 2018, 2018, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, A.; Reddy, P.; Belmonte, J.C.I. Anti-Aging Strategies Based on Cellular Reprogramming. Trends Mol. Med. 2016, 22, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Pagliai, G.; Sofi, F.; Dinu, M.; Sticchi, E.; Vannetti, F.; Molino Lova, R.; Ordovas, J.M.; Gori, A.M.; Marcucci, R.; Giusti, B.; et al. CLOCK gene polymorphisms and quality of aging in a cohort of nonagenarians—The MUGELLO Study. Sci. Rep. 2019, 9, 1472. [Google Scholar] [CrossRef] [PubMed]
- Crouch, J.; Shvedova, M.; Thanapaul, R.; Botchkarev, V.; Roh, D. Epigenetic Regulation of Cellular Senescence. Cells 2022, 11, 672. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Silva, D.; Canton-Sandoval, J.; Martinez-Navarro, F.J.; Perez-Sanchez, H.; de Oliveira, S.; Mulero, V.; Alcaraz-Perez, F.; Cayuela, M.L. Senescence-Independent Anti-Inflammatory Activity of the Senolytic Drugs Dasatinib, Navitoclax, and Venetoclax in Zebrafish Models of Chronic Inflammation. Int. J. Mol. Sci. 2022, 23, 10468. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Noguchi, M.; Kodama, T.; Shimosato, Y.; Koide, T.; Naruke, T.; Singh, G.; Katyal, S.L. Papillary adenoma of type 2 pneumocytes. Am. J. Surg. Pathol. 1986, 10, 134–139. [Google Scholar] [CrossRef]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory cytokines, aging, and age-related diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, M.A.; Jaw, J.; Shochet, L.R.; Robson, K.J.; Ooi, J.D.; Brouwer, E.; Heeringa, P.; Holdsworth, S.R.; Kitching, A.R. Ageing enhances cellular immunity to myeloperoxidase and experimental anti-myeloperoxidase glomerulonephritis. Rheumatology 2022, 61, 2132–2143. [Google Scholar] [CrossRef]
- Rustenhoven, J.; Pavlou, G.; Storck, S.E.; Dykstra, T.; Du, S.; Wan, Z.; Quintero, D.; Scallan, J.P.; Smirnov, I.; Kamm, R.D.; et al. Age-related alterations in meningeal immunity drive impaired CNS lymphatic drainage. J. Exp. Med. 2023, 220, e20221929. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.M.; Pesic, M.; Engel, E.; Ziegler, P.K.; Diefenhardt, M.; Kennel, K.B.; Buettner, F.; Conche, C.; Petrocelli, V.; Elwakeel, E.; et al. Inflammatory fibroblasts mediate resistance to neoadjuvant therapy in rectal cancer. Cancer Cell 2022, 40, 168–184.e13. [Google Scholar] [CrossRef] [PubMed]
- Faust, H.J.; Zhang, H.; Han, J.; Wolf, M.T.; Jeon, O.H.; Sadtler, K.; Pena, A.N.; Chung, L.; Maestas, D.R., Jr.; Tam, A.J.; et al. IL-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J. Clin. Investig. 2020, 130, 5493–5507. [Google Scholar] [CrossRef]
- Amarya, S.; Singh, K.; Sabharwal, M. Ageing Process and Physiological Changes. In Gerontology; InTech: London, UK, 2018. [Google Scholar]
- Burton, D.G.; Krizhanovsky, V. Physiological and pathological consequences of cellular senescence. Cell Mol. Life Sci. 2014, 71, 4373–4386. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Park, S.S.; Choi, Y.W.; Kim, J.H.; Kim, H.S.; Park, T.J. Senescent tumor cells: An overlooked adversary in the battle against cancer. Exp. Mol. Med. 2021, 53, 1834–1841. [Google Scholar] [CrossRef]
- Missiaen, R.; Anderson, N.M.; Kim, L.C.; Nance, B.; Burrows, M.; Skuli, N.; Carens, M.; Riscal, R.; Steensels, A.; Li, F.; et al. GCN2 inhibition sensitizes arginine-deprived hepatocellular carcinoma cells to senolytic treatment. Cell Metab. 2022, 34, 1151–1167.e1157. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Saretzki, G. Telomeres, Telomerase and Ageing. Subcell. Biochem. 2018, 90, 221–308. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Kim, S.H.; Lim, C.S.; Rubio, M. Cellular senescence, cancer and aging: The telomere connection. Exp. Gerontol. 2001, 36, 1619–1637. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, X.; Ding, X.; Wang, F.; Geng, X. Telomere and its role in the aging pathways: Telomere shortening, cell senescence and mitochondria dysfunction. Biogerontology 2019, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Pettersson, L.; Hoglund, M.; Heidenblad, M.; Gorunova, L.; Wiegant, J.; Mertens, F.; Dal Cin, P.; Mitelman, F.; Mandahl, N. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc. Natl. Acad. Sci. USA 2000, 97, 5357–5362. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Yan, G.; Song, X.; Xie, L.; Zhou, Y.; Li, J.; Hu, X.; Li, Z.; Hu, J.; Zhang, Y.; et al. Deubiquitylation and stabilization of p21 by USP11 is critical for cell-cycle progression and DNA damage responses. Proc. Natl. Acad. Sci. USA 2018, 115, 4678–4683. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Hwang, H.J.; Kang, D.; Park, H.A.; Lee, H.C.; Jeong, D.; Lee, K.; Park, H.J.; Ko, Y.G.; Lee, J.S. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene 2019, 38, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Chandeck, C.; Mooi, W.J. Oncogene-induced cellular senescence. Adv. Anat. Pathol. 2010, 17, 42–48. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Takasugi, M.; Yoshida, Y.; Hara, E.; Ohtani, N. The role of cellular senescence and SASP in tumour microenvironment. FEBS J. 2023, 290, 1348–1361. [Google Scholar] [CrossRef]
- Safwan-Zaiter, H.; Wagner, N.; Wagner, K.D. P16INK4A-More Than a Senescence Marker. Life 2022, 12, 1332. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- McConnell, B.B.; Starborg, M.; Brookes, S.; Peters, G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr. Biol. 1998, 8, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Horsley, G.C. Baggage from the past. Am. J. Nurs. 1988, 88, 60–63. [Google Scholar] [PubMed]
- Ortiz-Montero, P.; Londono-Vallejo, A.; Vernot, J.P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal 2017, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Olan, I.; Handa, T.; Narita, M. Beyond SAHF: An integrative view of chromatin compartmentalization during senescence. Curr. Opin. Cell Biol. 2023, 83, 102206. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Jaul, E.; Barron, J. Age-Related Diseases and Clinical and Public Health Implications for the 85 Years Old and Over Population. Front. Public Health 2017, 5, 335. [Google Scholar] [CrossRef]
- Belur Nagaraj, S.; Kieneker, L.M.; Pena, M.J. Kidney Age Index (KAI): A novel age-related biomarker to estimate kidney function in patients with diabetic kidney disease using machine learning. Comput. Methods Programs Biomed. 2021, 211, 106434. [Google Scholar] [CrossRef]
- Morris, J.F.; Temple, W. Spirometric “lung age” estimation for motivating smoking cessation. Prev. Med. 1985, 14, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.E.; Cropley, V.; Maier, A.B.; Lautenschlager, N.T.; Breakspear, M.; Zalesky, A. Heterogeneous aging across multiple organ systems and prediction of chronic disease and mortality. Nat. Med. 2023, 29, 1221–1231. [Google Scholar] [CrossRef]
- Oh, H.S.; Rutledge, J.; Nachun, D.; Palovics, R.; Abiose, O.; Moran-Losada, P.; Channappa, D.; Urey, D.Y.; Kim, K.; Sung, Y.J.; et al. Organ aging signatures in the plasma proteome track health and disease. Nature 2023, 624, 164–172. [Google Scholar] [CrossRef]
- Schaum, N.; Lehallier, B.; Hahn, O.; Palovics, R.; Hosseinzadeh, S.; Lee, S.E.; Sit, R.; Lee, D.P.; Losada, P.M.; Zardeneta, M.E.; et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 2020, 583, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Alavi, P.; Yousef Abdualla, R.; Brown, D.; Mojiri, A.; Nagendran, J.; Lewis, J.; Bourque, S.L.; Jahroudi, N. Aging Is Associated With Organ-Specific Alterations in the Level and Expression Pattern of von Willebrand Factor. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 2183–2196. [Google Scholar] [CrossRef]
- Schadel, P.; Troisi, F.; Czapka, A.; Gebert, N.; Pace, S.; Ori, A.; Werz, O. Aging drives organ-specific alterations of the inflammatory microenvironment guided by immunomodulatory mediators in mice. FASEB J. 2021, 35, e21558. [Google Scholar] [CrossRef] [PubMed]
- Safwan-Zaiter, H.; Wagner, N.; Michiels, J.F.; Wagner, K.D. Dynamic Spatiotemporal Expression Pattern of the Senescence-Associated Factor p16Ink4a in Development and Aging. Cells 2022, 11, 541. [Google Scholar] [CrossRef] [PubMed]
- Strzyz, P. Senescent fibroblasts support lung repair. Nat. Rev. Mol. Cell Biol. 2022, 23, 775. [Google Scholar] [CrossRef]
- Yun, J.; Hansen, S.; Morris, O.; Madden, D.T.; Libeu, C.P.; Kumar, A.J.; Wehrfritz, C.; Nile, A.H.; Zhang, Y.; Zhou, L.; et al. Senescent cells perturb intestinal stem cell differentiation through Ptk7 induced noncanonical Wnt and YAP signaling. Nat. Commun. 2023, 14, 156. [Google Scholar] [CrossRef]
- Guo, Y.; Ayers, J.L.; Carter, K.T.; Wang, T.; Maden, S.K.; Edmond, D.; Newcomb, P.P.; Li, C.; Ulrich, C.; Yu, M.; et al. Senescence-associated tissue microenvironment promotes colon cancer formation through the secretory factor GDF15. Aging Cell 2019, 18, e13013. [Google Scholar] [CrossRef]
- Slieker, R.C.; Relton, C.L.; Gaunt, T.R.; Slagboom, P.E.; Heijmans, B.T. Age-related DNA methylation changes are tissue-specific with ELOVL2 promoter methylation as exception. Epigenet. Chromatin 2018, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B., Jr.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of beta Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142.e4. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, B.; Gasek, N.S.; Zhou, Y.; Cohn, R.L.; Martin, D.E.; Zuo, W.; Flynn, W.F.; Guo, C.; Jellison, E.R.; et al. Targeting p21(Cip1) highly expressing cells in adipose tissue alleviates insulin resistance in obesity. Cell Metab. 2022, 34, 75–89.e8. [Google Scholar] [CrossRef] [PubMed]
- Saccon, T.D.; Nagpal, R.; Yadav, H.; Cavalcante, M.B.; Nunes, A.D.C.; Schneider, A.; Gesing, A.; Hughes, B.; Yousefzadeh, M.; Tchkonia, T.; et al. Senolytic Combination of Dasatinib and Quercetin Alleviates Intestinal Senescence and Inflammation and Modulates the Gut Microbiome in Aged Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Hense, J.D.; Garcia, D.N.; Isola, J.V.; Alvarado-Rincon, J.A.; Zanini, B.M.; Prosczek, J.B.; Stout, M.B.; Mason, J.B.; Walsh, P.T.; Brieno-Enriquez, M.A.; et al. Senolytic treatment reverses obesity-mediated senescent cell accumulation in the ovary. Geroscience 2022, 44, 1747–1759. [Google Scholar] [CrossRef]
- Alharbi, K.S.; Afzal, O.; Altamimi, A.S.A.; Almalki, W.H.; Kazmi, I.; Al-Abbasi, F.A.; Alzarea, S.I.; Makeen, H.A.; Albratty, M. A study of the molecular mechanism of quercetin and dasatinib combination as senolytic in alleviating age-related and kidney diseases. J. Food Biochem. 2022, 46, e14471. [Google Scholar] [CrossRef]
- Murakami, T.; Inagaki, N.; Kondoh, H. Cellular Senescence in Diabetes Mellitus: Distinct Senotherapeutic Strategies for Adipose Tissue and Pancreatic beta Cells. Front. Endocrinol. 2022, 13, 869414. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Selective anti-cancer agents as anti-aging drugs. Cancer Biol. Ther. 2013, 14, 1092–1097. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Bourgeois, B.; Madl, T. Regulation of cellular senescence via the FOXO4-p53 axis. FEBS Lett. 2018, 592, 2083–2097. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Yoon, N.G.; Lee, J.E.; Hu, S.; Yoon, S.; Kim, S.Y.; Hong, J.H.; Nam, D.; Chae, Y.C.; Park, J.B.; et al. Unleashing the full potential of Hsp90 inhibitors as cancer therapeutics through simultaneous inactivation of Hsp90, Grp94, and TRAP1. Exp. Mol. Med. 2020, 52, 79–91. [Google Scholar] [CrossRef]
- Chen, D.D.; Peng, X.; Wang, Y.; Jiang, M.; Xue, M.; Shang, G.; Liu, X.; Jia, X.; Liu, B.; Lu, Y.; et al. HSP90 acts as a senomorphic target in senescent retinal pigmental epithelial cells. Aging 2021, 13, 21547–21570. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-X(L) inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.F.; Hasvold, L.; Wang, L.; Wang, X.; Petros, A.M.; Park, C.H.; Boghaert, E.R.; Catron, N.D.; Chen, J.; Colman, P.M.; et al. Discovery of a Potent and Selective BCL-XL Inhibitor with in Vivo Activity. ACS Med. Chem. Lett. 2014, 5, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhang, L.; Wang, C.; Li, Z.; Luo, M.; Xie, G.; Yang, X.; Li, M.; Ren, S.; Zhao, D.; et al. Anti-apoptotic protein BCL-XL as a therapeutic vulnerability in gastric cancer. Anim. Models Exp. Med. 2023, 6, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.; Southon, A.G.; Saleh, E.; Brinkmann, K.; Ke, F.; Iliopoulos, M.; Cross, R.S.; Jenkins, M.R.; Nhu, D.; Wang, Z.; et al. BH3 mimetic drugs cooperate with Temozolomide, JQ1 and inducers of ferroptosis in killing glioblastoma multiforme cells. Cell Death Differ. 2022, 29, 1335–1348. [Google Scholar] [CrossRef]
- Triana-Martinez, F.; Picallos-Rabina, P.; Da Silva-Alvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreiros, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef]
- Seo, E.J.; Efferth, T.; Panossian, A. Curcumin downregulates expression of opioid-related nociceptin receptor gene (OPRL1) in isolated neuroglia cells. Phytomedicine 2018, 50, 285–299. [Google Scholar] [CrossRef]
- Lee, D.Y.; Lee, S.J.; Chandrasekaran, P.; Lamichhane, G.; O’Connell, J.F.; Egan, J.M.; Kim, Y. Dietary Curcumin Attenuates Hepatic Cellular Senescence by Suppressing the MAPK/NF-kappaB Signaling Pathway in Aged Mice. Antioxidants 2023, 12, 1165. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Kim, D.; Cheng, J.Q.; Zhang, S.; Lee, J.H.; Buettner, R.; Mirosevich, J.; Lee, F.Y.; Jove, R. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005, 65, 9185–9189. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; He, Y.; Makarcyzk, M.J.; Lin, H. Senolytic Peptide FOXO4-DRI Selectively Removes Senescent Cells From in vitro Expanded Human Chondrocytes. Front. Bioeng. Biotechnol. 2021, 9, 677576. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Naiki, T.; Naiki-Ito, A.; Suzuki, S.; Kato, H.; Nozaki, S.; Nagai, T.; Etani, T.; Nagayasu, Y.; Ando, R.; et al. Luteolin suppresses bladder cancer growth via regulation of mechanistic target of rapamycin pathway. Cancer Sci. 2020, 111, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Takaya, K.; Ishii, T.; Asou, T.; Kishi, K. Navitoclax (ABT-263) Rejuvenates Human Skin by Eliminating Senescent Dermal Fibroblasts in a Mouse/Human Chimeric Model. Rejuvenation Res. 2023, 26, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Kim, C. Nutlin-3a for age-related macular degeneration. Aging 2022, 14, 5614–5616. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, J.; Liu, X.; Zhang, X.; Zhang, S.; Zhang, X.; Zhou, D.; Zheng, G. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging 2016, 8, 2915–2926. [Google Scholar] [CrossRef]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef]
- Powers, M.V.; Valenti, M.; Miranda, S.; Maloney, A.; Eccles, S.A.; Thomas, G.; Clarke, P.A.; Workman, P. Mode of cell death induced by the HSP90 inhibitor 17-AAG (tanespimycin) is dependent on the expression of pro-apoptotic BAX. Oncotarget 2013, 4, 1963–1975. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.T.; Hilgaertner, J.; Simonson, C.; Ablin, R.J.; Badowski, M. Cell-based therapy for epithelial wounds. Cytotherapy 2012, 14, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Pippias, M.; Jager, K.J.; Asberg, A.; Berger, S.P.; Finne, P.; Heaf, J.G.; Kerschbaum, J.; Lempinen, M.; Magaz, A.; Massy, Z.A.; et al. Young deceased donor kidneys show a survival benefit over older donor kidneys in transplant recipients aged 20-50 years: A study by the ERA-EDTA Registry. Nephrol. Dial. Transplant. 2020, 35, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Iske, J.; Matsunaga, T.; Zhou, H.; Tullius, S.G. Donor and Recipient Age-Mismatches: The Potential of Transferring Senescence. Front. Immunol. 2021, 12, 671479. [Google Scholar] [CrossRef] [PubMed]
- Keren, A.; Bertolini, M.; Keren, Y.; Ullmann, Y.; Paus, R.; Gilhar, A. Human organ rejuvenation by VEGF-A: Lessons from the skin. Sci. Adv. 2022, 8, eabm6756. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wei, J.; Da Fonseca Ferreira, A.; Wang, H.; Zhang, L.; Zhang, Q.; Bellio, M.A.; Chu, X.M.; Khan, A.; Jayaweera, D.; et al. Rejuvenation of Senescent Endothelial Progenitor Cells by Extracellular Vesicles Derived From Mesenchymal Stromal Cells. JACC Basic Transl. Sci. 2020, 5, 1127–1141. [Google Scholar] [CrossRef]
- Grigorian-Shamagian, L.; Liu, W.; Fereydooni, S.; Middleton, R.C.; Valle, J.; Cho, J.H.; Marban, E. Cardiac and systemic rejuvenation after cardiosphere-derived cell therapy in senescent rats. Eur. Heart J. 2017, 38, 2957–2967. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Goodell, M.A.; Rando, T.A. Ageing and rejuvenation of tissue stem cells and their niches. Nat. Rev. Mol. Cell Biol. 2023, 24, 45–62. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Zhang, C.F.; Fu, B.C.; Sun, L.; Wang, X.Q.; Chen, W.; Liu, W.; Liu, K.Y.; Du, G.Q.; Ma, C.Y.; et al. Sirtuin3 protects aged human mesenchymal stem cells against oxidative stress and enhances efficacy of cell therapy for ischaemic heart diseases. J. Cell Mol. Med. 2018, 22, 5504–5517. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Tazetdinova, L.G.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Application of Mesenchymal Stem Cells for Therapeutic Agent Delivery in Anti-tumor Treatment. Front. Pharmacol. 2018, 9, 259. [Google Scholar] [CrossRef]
- Awad, M.E.; Hussein, K.A.; Helwa, I.; Abdelsamid, M.F.; Aguilar-Perez, A.; Mohsen, I.; Hunter, M.; Hamrick, M.W.; Isales, C.M.; Elsalanty, M.; et al. Meta-Analysis and Evidence Base for the Efficacy of Autologous Bone Marrow Mesenchymal Stem Cells in Knee Cartilage Repair: Methodological Guidelines and Quality Assessment. Stem Cells Int. 2019, 2019, 3826054. [Google Scholar] [CrossRef] [PubMed]
- Gasanova, S.Y. Cell therapy for destructive pancreatitis. Khirurgiia 2022, 9, 50–55. [Google Scholar] [CrossRef]
- Gjerde, C.; Mustafa, K.; Hellem, S.; Rojewski, M.; Gjengedal, H.; Yassin, M.A.; Feng, X.; Skaale, S.; Berge, T.; Rosen, A.; et al. Cell therapy induced regeneration of severely atrophied mandibular bone in a clinical trial. Stem Cell Res. Ther. 2018, 9, 213. [Google Scholar] [CrossRef] [PubMed]
- Zarei, F.; Abbaszadeh, A. Application of Cell Therapy for Anti-Aging Facial Skin. Curr. Stem Cell Res. Ther. 2019, 14, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, T.; Hou, X.; Zhou, Z.; Zhang, F.; Ma, H.; Wu, X.; Jiang, J. Exosomes secreted by mesenchymal stem cells delay brain aging by upregulating SIRT1 expression. Sci. Rep. 2023, 13, 13213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Y.; Gao, T.; Xu, R.J.; Sun, L.; Zhang, C.F.; Bai, L.; Chen, W.; Liu, K.Y.; Zhou, Y.; Jiao, X.; et al. SIRT3 Transfection of Aged Human Bone Marrow-Derived Mesenchymal Stem Cells Improves Cell Therapy-Mediated Myocardial Repair. Rejuvenation Res. 2020, 23, 453–464. [Google Scholar] [CrossRef]
- Primorac, D.; Molnar, V.; Rod, E.; Jelec, Z.; Cukelj, F.; Matisic, V.; Vrdoljak, T.; Hudetz, D.; Hajsok, H.; Boric, I. Knee Osteoarthritis: A Review of Pathogenesis and State-of-the-Art Non-Operative Therapeutic Considerations. Genes 2020, 11, 854. [Google Scholar] [CrossRef]
- Andia, I.; Maffulli, N.; Burgos-Alonso, N. Stromal vascular fraction technologies and clinical applications. Expert. Opin. Biol. Ther. 2019, 19, 1289–1305. [Google Scholar] [CrossRef]
- Rowe, G.; Tracy, E.; Beare, J.E.; LeBlanc, A.J. Cell therapy rescues aging-induced beta-1 adrenergic receptor and GRK2 dysfunction in the coronary microcirculation. Geroscience 2022, 44, 329–348. [Google Scholar] [CrossRef]
- Mautner, K.; Bowers, R.; Easley, K.; Fausel, Z.; Robinson, R. Functional Outcomes Following Microfragmented Adipose Tissue Versus Bone Marrow Aspirate Concentrate Injections for Symptomatic Knee Osteoarthritis. Stem Cells Transl. Med. 2019, 8, 1149–1156. [Google Scholar] [CrossRef]
- Weng, H.P.; Cheng, Y.Y.; Lee, H.L.; Hsu, T.Y.; Chang, Y.T.; Shen, Y.A. Enhanced Platelet-Rich Plasma (ePRP) Stimulates Wound Healing through Effects on Metabolic Reprogramming in Fibroblasts. Int. J. Mol. Sci. 2021, 22, 12623. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Luo, X.; Xiong, Y.; Liu, G.; Wang, J.; Chen, X.; Mi, B. Platelet-rich plasma versus hyaluronic acid in knee osteoarthritis: A meta-analysis with the consistent ratio of injection. J. Orthop. Surg. 2020, 28, 2309499019887660. [Google Scholar] [CrossRef] [PubMed]
- Grether-Beck, S.; Marini, A.; Jaenicke, T.; Goessens-Ruck, P.; McElwee, K.J.; Hoffmann, R.; Krutmann, J. Autologous Cell Therapy for Aged Human Skin: A Randomized, Placebo-Controlled, Phase-I Study. Skin Pharmacol. Physiol. 2020, 33, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Iwasaki, Y.; Makabe, K.; Kamao, H.; Mandai, M.; Shiina, T.; Ogasawara, K.; Hirami, Y.; Kurimoto, Y.; Takahashi, M. Successful Transplantation of Retinal Pigment Epithelial Cells from MHC Homozygote iPSCs in MHC-Matched Models. Stem Cell Rep. 2016, 7, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Chung, S.; Yun, K.; Kim, B.; So, S.; Kang, S.; Kang, E.; Lee, J.Y. Long-term effects of human induced pluripotent stem cell-derived retinal cell transplantation in Pde6b knockout rats. Exp. Mol. Med. 2021, 53, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Futatsugi, Y.; Ishida, M.; Edo, A.; Takahashi, M. Retinal Pigment Epithelial Cells Derived from Induced Pluripotent Stem (iPS) Cells Suppress or Activate T Cells via Costimulatory Signals. Int. J. Mol. Sci. 2020, 21, 6507. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Xie, M.; Gademann, F.; Cao, J.; Wang, P.; Guo, Y.; Zhang, L.; Su, T.; Zhang, J.; Chen, J. Protective effects of human iPS-derived retinal pigmented epithelial cells on retinal degenerative disease. Stem Cell Res. Ther. 2020, 11, 98. [Google Scholar] [CrossRef]
- Martin-Lopez, M.; Gonzalez-Munoz, E.; Gomez-Gonzalez, E.; Sanchez-Pernaute, R.; Marquez-Rivas, J.; Fernandez-Munoz, B. Modeling chronic cervical spinal cord injury in aged rats for cell therapy studies. J. Clin. Neurosci. 2021, 94, 76–85. [Google Scholar] [CrossRef]
- Gargioli, C.; Coletta, M.; De Grandis, F.; Cannata, S.M.; Cossu, G. PlGF-MMP-9-expressing cells restore microcirculation and efficacy of cell therapy in aged dystrophic muscle. Nat. Med. 2008, 14, 973–978. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Liu, D.; Zhu, M.; Zhang, Y.; Diao, Y. Crossing the blood-brain barrier with AAV vectors. Metab. Brain Dis. 2021, 36, 45–52. [Google Scholar] [CrossRef]
- Issa, S.S.; Shaimardanova, A.A.; Solovyeva, V.V.; Rizvanov, A.A. Various AAV Serotypes and Their Applications in Gene Therapy: An Overview. Cells 2023, 12, 785. [Google Scholar] [CrossRef] [PubMed]
- Shaimardanova, A.A.; Chulpanova, D.S.; Mullagulova, A.I.; Afawi, Z.; Gamirova, R.G.; Solovyeva, V.V.; Rizvanov, A.A. Gene and Cell Therapy for Epilepsy: A Mini Review. Front. Mol. Neurosci. 2022, 15, 868531. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, T.; Zhu, J. Gene Therapy Strategies Targeting Aging-Related Diseases. Aging Dis. 2023, 14, 398–417. [Google Scholar] [CrossRef]
- Shaimardanova, A.A.; Chulpanova, D.S.; Solovyeva, V.V.; Issa, S.S.; Mullagulova, A.I.; Titova, A.A.; Mukhamedshina, Y.O.; Timofeeva, A.V.; Aimaletdinov, A.M.; Nigmetzyanov, I.R.; et al. Increasing beta-hexosaminidase A activity using genetically modified mesenchymal stem cells. Neural Regen. Res. 2024, 19, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Bijlani, S.; Pang, K.M.; Sivanandam, V.; Singh, A.; Chatterjee, S. The Role of Recombinant AAV in Precise Genome Editing. Front. Genome Ed. 2021, 3, 799722. [Google Scholar] [CrossRef]
- Vila, L.; Roca, C.; Elias, I.; Casellas, A.; Lage, R.; Franckhauser, S.; Bosch, F. AAV-mediated Sirt1 overexpression in skeletal muscle activates oxidative capacity but does not prevent insulin resistance. Mol. Ther. Methods Clin. Dev. 2016, 5, 16072. [Google Scholar] [CrossRef] [PubMed]
- Adu-Agyeiwaah, Y.; Vieira, C.P.; Asare-Bediako, B.; Li Calzi, S.; DuPont, M.; Floyd, J.; Boye, S.; Chiodo, V.; Busik, J.V.; Grant, M.B. Intravitreal Administration of AAV2-SIRT1 Reverses Diabetic Retinopathy in a Mouse Model of Type 2 Diabetes. Transl. Vis. Sci. Technol. 2023, 12, 20. [Google Scholar] [CrossRef]
- Gao, P.; You, M.; Li, L.; Zhang, Q.; Fang, X.; Wei, X.; Zhou, Q.; Zhang, H.; Wang, M.; Lu, Z.; et al. Salt-Induced Hepatic Inflammatory Memory Contributes to Cardiovascular Damage Through Epigenetic Modulation of SIRT3. Circulation 2022, 145, 375–391. [Google Scholar] [CrossRef]
- Diao, Z.; Ji, Q.; Wu, Z.; Zhang, W.; Cai, Y.; Wang, Z.; Hu, J.; Liu, Z.; Wang, Q.; Bi, S.; et al. SIRT3 consolidates heterochromatin and counteracts senescence. Nucleic Acids Res. 2021, 49, 4203–4219. [Google Scholar] [CrossRef]
- Ling, W.; Krager, K.; Richardson, K.K.; Warren, A.D.; Ponte, F.; Aykin-Burns, N.; Manolagas, S.C.; Almeida, M.; Kim, H.N. Mitochondrial Sirt3 contributes to the bone loss caused by aging or estrogen deficiency. JCI Insight 2021, 6, e146728. [Google Scholar] [CrossRef]
- Song, S.; Ding, Y.; Dai, G.L.; Zhang, Y.; Xu, M.T.; Shen, J.R.; Chen, T.T.; Chen, Y.; Meng, G.L. Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharmacol. Sin. 2021, 42, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Qin, H.; Zhou, M.; Zhang, T.; Zhang, Y.; Ding, H.; Xu, L.; Song, J. Knockdown of SIRT3 perturbs protective effects of irisin against bone loss in diabetes and periodontitis. Free Radic. Biol. Med. 2023, 200, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Finigan, A.; Figg, N.L.; Uryga, A.K.; Bennett, M.R. SIRT6 Protects Smooth Muscle Cells From Senescence and Reduces Atherosclerosis. Circ. Res. 2021, 128, 474–491. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Huang, Y.; Zhu, L.; Yang, K.; Liang, K.; Tan, J.; Yu, B. SIRT6 promotes angiogenesis and hemorrhage of carotid plaque via regulating HIF-1alpha and reactive oxygen species. Cell Death Dis. 2021, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.J.; Vasu, V.; Griffin, D.K. Telomere Biology and Human Phenotype. Cells 2019, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Jaijyan, D.K.; Selariu, A.; Cruz-Cosme, R.; Tong, M.; Yang, S.; Stefa, A.; Kekich, D.; Sadoshima, J.; Herbig, U.; Tang, Q.; et al. New intranasal and injectable gene therapy for healthy life extension. Proc. Natl. Acad. Sci. USA 2022, 119, e2121499119. [Google Scholar] [CrossRef]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.W.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Sui, S.; Hua, Y.; Zhao, A.; Tian, X.; Wang, R.; Guo, W.; Yu, W.; Zou, K.; et al. Targeting HMGB3/hTERT axis for radioresistance in cervical cancer. J. Exp. Clin. Cancer Res. 2020, 39, 243. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Imura, A.; Urakawa, I.; Shimada, T.; Murakami, J.; Aono, Y.; Hasegawa, H.; Yamashita, T.; Nakatani, K.; Saito, Y.; et al. Establishment of sandwich ELISA for soluble alpha-Klotho measurement: Age-dependent change of soluble alpha-Klotho levels in healthy subjects. Biochem. Biophys. Res. Commun. 2010, 398, 513–518. [Google Scholar] [CrossRef]
- Gao, D.; Wang, S.; Lin, Y.; Sun, Z. In vivo AAV delivery of glutathione reductase gene attenuates anti-aging gene klotho deficiency-induced kidney damage. Redox Biol. 2020, 37, 101692. [Google Scholar] [CrossRef]
- Chen, K.; Wang, S.; Sun, Z. In Vivo Cardiac-specific Expression of Adenylyl Cyclase 4 Gene Protects against Klotho Deficiency-induced Heart Failure. Transl. Res. 2022, 244, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Roig-Soriano, J.; Sanchez-de-Diego, C.; Esandi-Jauregui, J.; Verdes, S.; Abraham, C.R.; Bosch, A.; Ventura, F.; Chillon, M. Differential toxicity profile of secreted and processed alpha-Klotho expression over mineral metabolism and bone microstructure. Sci. Rep. 2023, 13, 4211. [Google Scholar] [CrossRef] [PubMed]
- Chuchana, P.; Mausset-Bonnefont, A.L.; Mathieu, M.; Espinoza, F.; Teigell, M.; Toupet, K.; Ripoll, C.; Djouad, F.; Noel, D.; Jorgensen, C.; et al. Secreted alpha-Klotho maintains cartilage tissue homeostasis by repressing NOS2 and ZIP8-MMP13 catabolic axis. Aging 2018, 10, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Luo, X.; Zeng, C.; Li, S.; Ma, M.; Wu, Y. Klotho ameliorated cognitive deficits in a temporal lobe epilepsy rat model by inhibiting ferroptosis. Brain Res. 2021, 1772, 147668. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Luo, X.; Ye, L.; Huang, H.; Wu, Y. Klotho alleviates NLRP3 inflammasome-mediated neuroinflammation in a temporal lobe epilepsy rat model by activating the Nrf2 signaling pathway. Epilepsy Behav. 2022, 128, 108509. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension 2009, 54, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Roig-Soriano, J.; Grinan-Ferre, C.; Espinosa-Parrilla, J.F.; Abraham, C.R.; Bosch, A.; Pallas, M.; Chillon, M. AAV-mediated expression of secreted and transmembrane alphaKlotho isoforms rescues relevant aging hallmarks in senescent SAMP8 mice. Aging Cell 2022, 21, e13581. [Google Scholar] [CrossRef]
- Fan, J.; Wang, S.; Chen, K.; Sun, Z. Aging impairs arterial compliance via Klotho-mediated downregulation of B-cell population and IgG levels. Cell Mol. Life Sci. 2022, 79, 494. [Google Scholar] [CrossRef]
- Sewell, P.E.; Ediriweera, D.; Gomez Rios, E.; Guadarrama, O.A.; Eusebio, Y.; Gonzalez, L.; Parrish, E.L. Safety Study of AAV hTert and Klotho Gene Transfer Therapy for Dementia. J. Regen. Biol. Med. 2021, 3, 1–15. [Google Scholar] [CrossRef]
- Clemens, Z.; Sivakumar, S.; Pius, A.; Sahu, A.; Shinde, S.; Mamiya, H.; Luketich, N.; Cui, J.; Dixit, P.; Hoeck, J.D.; et al. The biphasic and age-dependent impact of klotho on hallmarks of aging and skeletal muscle function. eLife 2021, 10, e61138. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, V.; Jambrina, C.; Casana, E.; Sacristan, V.; Munoz, S.; Darriba, S.; Rodo, J.; Mallol, C.; Garcia, M.; Leon, X.; et al. FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol. Med. 2018, 10, e8791. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Horvath, T.L.; Mangelsdorf, D.J.; Kliewer, S.A.; Dixit, V.D. Prolongevity hormone FGF21 protects against immune senescence by delaying age-related thymic involution. Proc. Natl. Acad. Sci. USA 2016, 113, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Villarroya, J.; Gallego-Escuredo, J.M.; Delgado-Angles, A.; Cairo, M.; Moure, R.; Gracia Mateo, M.; Domingo, J.C.; Domingo, P.; Giralt, M.; Villarroya, F. Aging is associated with increased FGF21 levels but unaltered FGF21 responsiveness in adipose tissue. Aging Cell 2018, 17, e12822. [Google Scholar] [CrossRef] [PubMed]
- Davidsohn, N.; Pezone, M.; Vernet, A.; Graveline, A.; Oliver, D.; Slomovic, S.; Punthambaker, S.; Sun, X.; Liao, R.; Bonventre, J.V.; et al. A single combination gene therapy treats multiple age-related diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 23505–23511. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, M.; Kumar, S.; Sharife, H.; Volinsky, E.; Gileles-Hillel, A.; Licht, T.; Permyakova, A.; Hinden, L.; Azar, S.; Friedmann, Y.; et al. Counteracting age-related VEGF signaling insufficiency promotes healthy aging and extends life span. Science 2021, 373, eabc8479. [Google Scholar] [CrossRef]
- Qi, X.; Francelin, C.; Mitter, S.; Boye, S.L.; Gu, H.; Quigley, J.; Grant, M.B.; Boulton, M.E. beta-secretase 1 overexpression by AAV-mediated gene delivery prevents retina degeneration in a mouse model of age-related macular degeneration. Mol. Ther. 2023, 31, 2042–2055. [Google Scholar] [CrossRef]
- Logan, C.; Lyzogubov, V.; Bora, N.; Bora, P. Role of Adiponectin Peptide I (APNp1) in Age-Related Macular Degeneration. Biomolecules 2022, 12, 1232. [Google Scholar] [CrossRef]
- Choudhary, M.; Ildefonso, C.J.; Lewin, A.S.; Malek, G. Gene Delivery of a Caspase Activation and Recruitment Domain Improves Retinal Pigment Epithelial Function and Modulates Inflammation in a Mouse Model with Features of Dry Age-Related Macular Degeneration. J. Ocul. Pharmacol. Ther. 2022, 38, 359–371. [Google Scholar] [CrossRef]
- Millington-Ward, S.; Chadderton, N.; Finnegan, L.K.; Post, I.J.M.; Carrigan, M.; Gardiner, T.; Peixoto, E.; Maloney, D.; Humphries, M.M.; Stitt, A.; et al. AAV-mediated gene therapy improving mitochondrial function provides benefit in age-related macular degeneration models. Clin. Transl. Med. 2022, 12, e952. [Google Scholar] [CrossRef]
- Molinari, C.; Morsanuto, V.; Ruga, S.; Notte, F.; Farghali, M.; Galla, R.; Uberti, F. The Role of BDNF on Aging-Modulation Markers. Brain Sci. 2020, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Swartzlander, D.B.; Ghosh, A.; Fryer, J.D.; Wang, B.; Zheng, H. Clusterin secreted from astrocyte promotes excitatory synaptic transmission and ameliorates Alzheimer’s disease neuropathology. Mol. Neurodegener. 2021, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.T.; Baumann, T.L.; Brown, L.; Kruegel, B.R.; Ostrove, J.M.; Herzog, C.D. Advancing neurotrophic factors as treatments for age-related neurodegenerative diseases: Developing and demonstrating “clinical proof-of-concept” for AAV-neurturin (CERE-120) in Parkinson’s disease. Neurobiol. Aging 2013, 34, 35–61. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Zhao, J.; Zhang, J.; Fang, Z.; Deng, J.; Fan, Z.; Guo, Y.; Han, J.; Hou, W.; Dong, H.; et al. MD2 contributes to the pathogenesis of perioperative neurocognitive disorder via the regulation of alpha5GABA(A) receptors in aged mice. J. Neuroinflamm. 2021, 18, 204. [Google Scholar] [CrossRef]
- Klein, R.L.; Hirko, A.C.; Meyers, C.A.; Grimes, J.R.; Muzyczka, N.; Meyer, E.M. NGF gene transfer to intrinsic basal forebrain neurons increases cholinergic cell size and protects from age-related, spatial memory deficits in middle-aged rats. Brain Res. 2000, 875, 144–151. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitaeva, K.V.; Solovyeva, V.V.; Blatt, N.L.; Rizvanov, A.A. Eternal Youth: A Comprehensive Exploration of Gene, Cellular, and Pharmacological Anti-Aging Strategies. Int. J. Mol. Sci. 2024, 25, 643. https://doi.org/10.3390/ijms25010643
Kitaeva KV, Solovyeva VV, Blatt NL, Rizvanov AA. Eternal Youth: A Comprehensive Exploration of Gene, Cellular, and Pharmacological Anti-Aging Strategies. International Journal of Molecular Sciences. 2024; 25(1):643. https://doi.org/10.3390/ijms25010643
Chicago/Turabian StyleKitaeva, Kristina V., Valeriya V. Solovyeva, Nataliya L. Blatt, and Albert A. Rizvanov. 2024. "Eternal Youth: A Comprehensive Exploration of Gene, Cellular, and Pharmacological Anti-Aging Strategies" International Journal of Molecular Sciences 25, no. 1: 643. https://doi.org/10.3390/ijms25010643
APA StyleKitaeva, K. V., Solovyeva, V. V., Blatt, N. L., & Rizvanov, A. A. (2024). Eternal Youth: A Comprehensive Exploration of Gene, Cellular, and Pharmacological Anti-Aging Strategies. International Journal of Molecular Sciences, 25(1), 643. https://doi.org/10.3390/ijms25010643