
Damage-Associated Molecular Patterns, a Class of Potential Psoriasis Drug Targets




Abstract
1. Introduction
2. Immune Regulation in Psoriasis
2.1. Innate Immune Regulation in Psoriasis
2.2. Adaptive Immune Regulation in Psoriasis
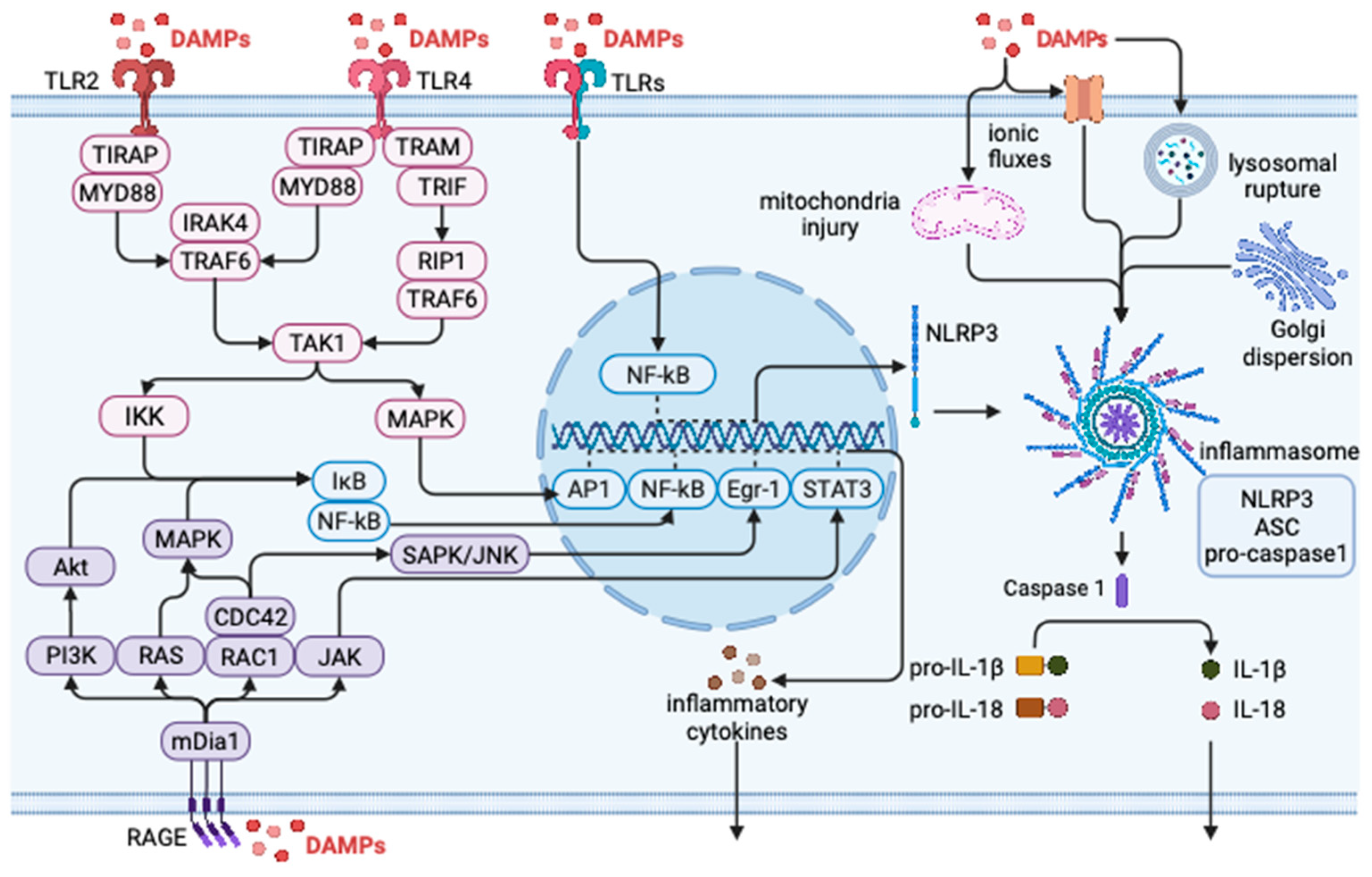
3. DAMPs and Inflammation
3.1. DAMP Regulates Inflammatory Response through TLRs
3.2. DAMP Regulates Inflammatory Response through NLRs
3.3. DAMP Regulates Inflammatory Response through RAGE
4. DAMPs in Psoriasis
4.1. HMGB1 and Psoriasis
4.2. S100 Protein and Psoriasis
4.3. HSP and Psoriasis
4.4. Other DAMPs and Psoriasis
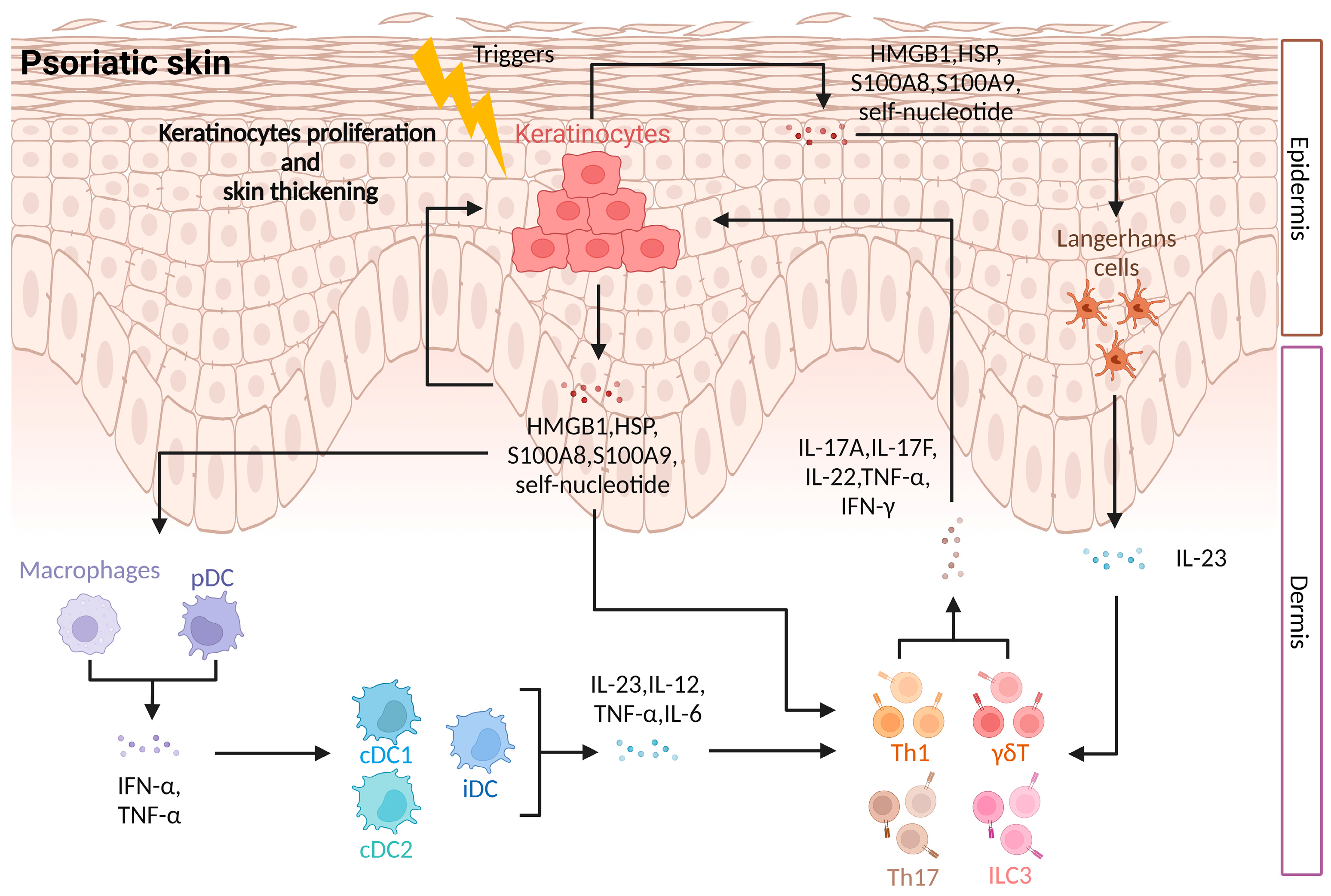
5. DAMPs Regulate the Immune Mechanism of Psoriasis
5.1. Activation of Innate Immunity
5.2. Regulation of Adaptive Immunity
5.3. Induction of Physiological Dysfunction of Keratinocytes
6. Future Directions and Prospects for Diagnosis and Treatment
6.1. Utilizing DAMP as a Diagnostic Marker
6.2. Directly Inhibiting or Using Antibodies against DAMP
6.3. Inhibiting DAMP Receptors or Signaling Pathways
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/publications/i/item/9789241565189 (accessed on 26 October 2016).
- Mou, Y.; Li, F.; Xu, Y.; Jin, X.; Dong, S.; Xia, J. Global trends in the incidence of psoriasis from 1990 to 2019. Eur. J. Dermatol. 2022, 32, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, T.D.; Schupp, C.W.; Armstrong, A.W. Psoriasis prevalence among adults in the United States. J. Am. Acad. Dermatol. 2014, 70, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.W.; Weinstein, G.D. Treatment of psoriasis. N. Engl. J. Med. 1995, 332, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Schon, M.P.; Boehncke, W.H. Psoriasis. N. Engl. J. Med. 2005, 352, 1899–1912. [Google Scholar] [CrossRef]
- Griffiths, C.E.; Barker, J.N. Pathogenesis and clinical features of psoriasis. Lancet 2007, 370, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Ljubenovic, M.; Lazarevic, V.; Golubovic, M.; Binic, I. Integrative Approach to Psoriasis Vulgaris. Holist. Nurs. Pract. 2018, 32, 133–139. [Google Scholar] [CrossRef]
- Nair, R.P.; Stuart, P.E.; Nistor, I.; Hiremagalore, R.; Chia, N.V.C.; Jenisch, S.; Weichenthal, M.; Abecasis, G.R.; Lim, H.W.; Christophers, E.; et al. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am. J. Hum. Genet. 2006, 78, 827–851. [Google Scholar] [CrossRef]
- Bos, J.D.; De Rie, M.A. The pathogenesis of psoriasis: Immunological facts and speculations. Immunol. Today 1999, 20, 40–46. [Google Scholar] [CrossRef]
- Hawkes, J.E.; Yan, B.Y.; Chan, T.C.; Krueger, J.G. Discovery of the IL-23/IL-17 Signaling Pathway and the Treatment of Psoriasis. J. Immunol. 2018, 201, 1605–1613. [Google Scholar] [CrossRef]
- Papp, K.A.; Tyring, S.; Lahfa, M.; Prinz, J.; Griffiths, C.E.; Nakanishi, A.M.; Zitnik, R.; van de Kerkhof, P.C.; Melvin, L.; Etanercept Psoriasis Study Group. A global phase III randomized controlled trial of etanercept in psoriasis: Safety, efficacy, and effect of dose reduction. Br. J. Dermatol. 2005, 152, 1304–1312. [Google Scholar] [CrossRef]
- Langley, R.G.; Tsai, T.F.; Flavin, S.; Song, M.; Randazzo, B.; Wasfi, Y.; Jiang, J.; Li, S.; Puig, L. Efficacy and safety of guselkumab in patients with psoriasis who have an inadequate response to ustekinumab: Results of the randomized, double-blind, phase III NAVIGATE trial. Br. J. Dermatol. 2018, 178, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Papp, K.; Gottlieb, A.; Jarell, A.; Reich, K.; Maari, C.; Gordon, K.B.; Ferris, L.K.; Langley, R.G.; Tada, Y.; et al. A head-to-head comparison of ixekizumab vs. guselkumab in patients with moderate-to-severe plaque psoriasis: 12-week efficacy, safety and speed of response from a randomized, double-blinded trial. Br. J. Dermatol. 2020, 182, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.; Strober, B.; Menter, A.; Gordon, K.; Weglowska, J.; Puig, L.; Papp, K.; Spelman, L.; Toth, D.; Kerdel, F.; et al. Phase 3 Studies Comparing Brodalumab with Ustekinumab in Psoriasis. N. Engl. J. Med. 2015, 373, 1318–1328. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Kamata, M.; Tada, Y. Dendritic Cells and Macrophages in the Pathogenesis of Psoriasis. Front. Immunol. 2022, 13, 941071. [Google Scholar] [CrossRef]
- Takagi, H.; Arimura, K.; Uto, T.; Fukaya, T.; Nakamura, T.; Choijookhuu, N.; Hishikawa, Y.; Sato, K. Plasmacytoid dendritic cells orchestrate TLR7-mediated innate and adaptive immunity for the initiation of autoimmune inflammation. Sci. Rep. 2016, 6, 24477. [Google Scholar] [CrossRef]
- Kopfnagel, V.; Wagenknecht, S.; Harder, J.; Hofmann, K.; Kleine, M.; Buch, A.; Sodeik, B.; Werfel, T. RNase 7 Strongly Promotes TLR9-Mediated DNA Sensing by Human Plasmacytoid Dendritic Cells. J. Investig. Dermatol. 2018, 138, 872–881. [Google Scholar] [CrossRef]
- Zaba, L.C.; Cardinale, I.; Gilleaudeau, P.; Sullivan-Whalen, M.; Suarez-Farinas, M.; Fuentes-Duculan, J.; Novitskaya, I.; Khatcherian, A.; Bluth, M.J.; Lowes, M.A.; et al. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J. Exp. Med. 2007, 204, 3183–3194. [Google Scholar] [CrossRef]
- Zaba, L.C.; Fuentes-Duculan, J.; Eungdamrong, N.J.; Abello, M.V.; Novitskaya, I.; Pierson, K.C.; Gonzalez, J.; Krueger, J.G.; Lowes, M.A. Psoriasis is characterized by accumulation of immunostimulatory and Th1/Th17 cell-polarizing myeloid dendritic cells. J. Investig. Dermatol. 2009, 129, 79–88. [Google Scholar] [CrossRef]
- Segura, E.; Touzot, M.; Bohineust, A.; Cappuccio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 2013, 38, 336–348. [Google Scholar] [CrossRef]
- Atmatzidis, D.H.; Lambert, W.C.; Lambert, M.W. Langerhans cell: Exciting developments in health and disease. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. Epigenetic regulation of macrophage polarization and function. Trends Immunol. 2013, 34, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhu, L.; Tian, H.; Sun, H.X.; Wang, R.; Zhang, L.; Zhao, Y. IL-23-induced macrophage polarization and its pathological roles in mice with imiquimod-induced psoriasis. Protein Cell 2018, 9, 1027–1038. [Google Scholar] [CrossRef]
- Zhou, S.; Li, Q.; Wu, H.; Lu, Q. The pathogenic role of innate lymphoid cells in autoimmune-related and inflammatory skin diseases. Cell. Mol. Immunol. 2020, 17, 335–346. [Google Scholar] [CrossRef]
- Villanova, F.; Flutter, B.; Tosi, I.; Grys, K.; Sreeneebus, H.; Perera, G.K.; Chapman, A.; Smith, C.H.; Di Meglio, P.; Nestle, F.O. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J. Investig. Dermatol. 2014, 134, 984–991. [Google Scholar] [CrossRef]
- Pantelyushin, S.; Haak, S.; Ingold, B.; Kulig, P.; Heppner, F.L.; Navarini, A.A.; Becher, B. Rorgammat+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. J. Clin. Investig. 2012, 122, 2252–2256. [Google Scholar] [CrossRef]
- Austin, L.M.; Ozawa, M.; Kikuchi, T.; Walters, I.B.; Krueger, J.G. The majority of epidermal T cells in Psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: A type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J. Investig. Dermatol. 1999, 113, 752–759. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, H.X.; Duan, Y.Q.; Wang, W.Z.; Zhang, T.Z.; Li, J.W.; Tu, Y.T. Analysis of Th1/Th2 response pattern for erythrodermic psoriasis. J. Huazhong Univ. Sci. Technol. Med. Sci. 2014, 34, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Johansen, C.; Usher, P.A.; Kjellerup, R.B.; Lundsgaard, D.; Iversen, L.; Kragballe, K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br. J. Dermatol. 2009, 160, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Benham, H.; Norris, P.; Goodall, J.; Wechalekar, M.D.; FitzGerald, O.; Szentpetery, A.; Smith, M.; Thomas, R.; Gaston, H. Th17 and Th22 cells in psoriatic arthritis and psoriasis. Arthritis Res. Ther. 2013, 15, R136. [Google Scholar] [CrossRef]
- Harper, E.G.; Guo, C.; Rizzo, H.; Lillis, J.V.; Kurtz, S.E.; Skorcheva, I.; Purdy, D.; Fitch, E.; Iordanov, M.; Blauvelt, A. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: Implications for psoriasis pathogenesis. J. Investig. Dermatol. 2009, 129, 2175–2183. [Google Scholar] [CrossRef] [PubMed]
- Chiricozzi, A.; Nograles, K.E.; Johnson-Huang, L.M.; Fuentes-Duculan, J.; Cardinale, I.; Bonifacio, K.M.; Gulati, N.; Mitsui, H.; Guttman-Yassky, E.; Suarez-Farinas, M.; et al. IL-17 induces an expanded range of downstream genes in reconstituted human epidermis model. PLoS ONE 2014, 9, e90284. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.Y.; Jia, K.; Zhang, Y. IL-17 promotes keratinocyte proliferation via the downregulation of C/EBPalpha. Exp. Ther. Med. 2016, 11, 631–636. [Google Scholar] [CrossRef]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 2011, 12, 568–575. [Google Scholar] [CrossRef]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A.; et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef]
- Scheinecker, C.; Goschl, L.; Bonelli, M. Treg cells in health and autoimmune diseases: New insights from single cell analysis. J. Autoimmun. 2020, 110, 102376. [Google Scholar] [CrossRef]
- Andersson, J.; Tran, D.Q.; Pesu, M.; Davidson, T.S.; Ramsey, H.; O’Shea, J.J.; Shevach, E.M. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J. Exp. Med. 2008, 205, 1975–1981. [Google Scholar] [CrossRef]
- Shevach, E.M. Regulatory T cells in autoimmmunity. Annu. Rev. Immunol. 2000, 18, 423–449. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H.; Gyulai, R.; Toichi, E.; Garaczi, E.; Shimada, S.; Stevens, S.R.; McCormick, T.S.; Cooper, K.D. Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: Mechanism underlying unrestrained pathogenic effector T cell proliferation. J. Immunol. 2005, 174, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.H.; Homey, B.; Cao, W.; Wang, Y.H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Morizane, S.; Gallo, R.L. Antimicrobial peptides in the pathogenesis of psoriasis. J. Dermatol. 2012, 39, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta 2010, 1805, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. 2020, 15, 493–518. [Google Scholar] [CrossRef]
- Wheeler, D.S.; Chase, M.A.; Senft, A.P.; Poynter, S.E.; Wong, H.R.; Page, K. Extracellular Hsp72, an endogenous DAMP, is released by virally infected airway epithelial cells and activates neutrophils via Toll-like receptor (TLR)-4. Respir. Res. 2009, 10, 31. [Google Scholar] [CrossRef]
- Barichello, T.; Generoso, J.S.; Goularte, J.A.; Collodel, A.; Pitcher, M.R.; Simoes, L.R.; Quevedo, J.; Dal-Pizzol, F. Does Infection-Induced Immune Activation Contribute to Dementia? Aging Dis. 2015, 6, 342–348. [Google Scholar] [CrossRef]
- Yu, L.; Wang, L.; Chen, S. Endogenous toll-like receptor ligands and their biological significance. J. Cell. Mol. Med. 2010, 14, 2592–2603. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Koblansky, A.A.; Ghosh, S. Recognition and signaling by toll-like receptors. Annu. Rev. Cell Dev. Biol. 2006, 22, 409–437. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Zhang, M.; Zhang, C.; Sang, H. Psoriasis-Like Inflammation Induced Renal Dysfunction through the TLR/NF-kappaB Signal Pathway. Biomed. Res. Int. 2020, 2020, 3535264. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Jiang, W.; Zhou, R. Control of Inflammasome Activation by Phosphorylation. Trends Biochem. Sci. 2018, 43, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Mathur, A.; Hayward, J.A.; Man, S.M. Molecular mechanisms of inflammasome signaling. J. Leukoc. Biol. 2018, 103, 233–257. [Google Scholar] [CrossRef]
- Luo, Q.; Zeng, J.; Li, W.; Lin, L.; Zhou, X.; Tian, X.; Liu, W.; Zhang, L.; Zhang, X. Silencing of miR-155 suppresses inflammatory responses in psoriasis through inflammasome NLRP3 regulation. Int. J. Mol. Med. 2018, 42, 1086–1095. [Google Scholar] [CrossRef]
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef]
- Kierdorf, K.; Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef]
- Lei, H.; Li, X.; Jing, B.; Xu, H.; Wu, Y. Human S100A7 Induces Mature Interleukin1alpha Expression by RAGE-p38 MAPK-Calpain1 Pathway in Psoriasis. PLoS ONE 2017, 12, e0169788. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Stegmann, F.; Gnanapragassam, V.S.; Lepenies, B. From structure to function—Ligand recognition by myeloid C-type lectin receptors. Comput. Struct. Biotechnol. J. 2022, 20, 5790–5812. [Google Scholar] [CrossRef] [PubMed]
- Iurescia, S.; Fioretti, D.; Rinaldi, M. The Innate Immune Signalling Pathways: Turning RIG-I Sensor Activation Against Cancer. Cancers 2020, 12, 3158. [Google Scholar] [CrossRef] [PubMed]
- Siskind, S.; Brenner, M.; Wang, P. TREM-1 Modulation Strategies for Sepsis. Front. Immunol. 2022, 13, 907387. [Google Scholar] [CrossRef] [PubMed]
- Rajaee, A.; Barnett, R.; Cheadle, W.G. Pathogen- and Danger-Associated Molecular Patterns and the Cytokine Response in Sepsis. Surg. Infect. 2018, 19, 107–116. [Google Scholar] [CrossRef]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef] [PubMed]
- Hoque, R.; Malik, A.F.; Gorelick, F.; Mehal, W.Z. Sterile inflammatory response in acute pancreatitis. Pancreas 2012, 41, 353–357. [Google Scholar] [CrossRef]
- Lee, B.; Song, Y.S.; Rhodes, C.; Goh, T.S.; Roh, J.S.; Jeong, H.; Park, J.; Lee, H.N.; Lee, S.G.; Kim, S.; et al. Protein phosphatase magnesium-dependent 1A induces inflammation in rheumatoid arthritis. Biochem. Biophys. Res. Commun. 2020, 522, 731–735. [Google Scholar] [CrossRef]
- Varhaug, K.N.; Vedeler, C.A.; Myhr, K.M.; Aarseth, J.H.; Tzoulis, C.; Bindoff, L.A. Increased levels of cell-free mitochondrial DNA in the cerebrospinal fluid of patients with multiple sclerosis. Mitochondrion 2017, 34, 32–35. [Google Scholar] [CrossRef]
- Boyapati, R.K.; Dorward, D.A.; Tamborska, A.; Kalla, R.; Ventham, N.T.; Doherty, M.K.; Whitfield, P.D.; Gray, M.; Loane, J.; Rossi, A.G.; et al. Mitochondrial DNA Is a Pro-Inflammatory Damage-Associated Molecular Pattern Released During Active IBD. Inflamm. Bowel Dis. 2018, 24, 2113–2122. [Google Scholar] [CrossRef]
- Goodwin, G.H.; Johns, E.W. The isolation and purification of the high mobility group (HMG) nonhistone chromosomal proteins. Methods Cell. Biol. 1977, 16, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Bustin, M.; Reeves, R. High-mobility-group chromosomal proteins: Architectural components that facilitate chromatin function. Prog. Nucleic Acid Res. Mol. Biol. 1996, 54, 35–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ward, M.F.; Sama, A.E.; Wang, H. Extracellular HMGB1 as a proinflammatory cytokine. J. Interferon Cytokine Res. 2004, 24, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Strohbuecker, L.; Lotfi, R.; Sucker, A.; Joosten, I.; Koenen, H.; Korber, A. High mobility group box 1 is increased in the sera of psoriatic patients with disease progression. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Yamaguchi, Y.; Watanabe, Y.; Takamura, N.; Aihara, M. Increased level of high mobility group box 1 in the serum and skin in patients with generalized pustular psoriasis. J. Dermatol. 2020, 47, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, H.; Zheng, H.; Zhou, X.; Shen, G.; Teng, X.; Liu, X.; Zhang, J.; Wei, X.; Hu, Z.; et al. Autophagy-based unconventional secretion of HMGB1 by keratinocytes plays a pivotal role in psoriatic skin in fl ammation. Autophagy 2021, 17, 529–552. [Google Scholar] [CrossRef]
- Zhang, W.; Guo, S.; Li, B.; Liu, L.; Ge, R.; Cao, T.; Wang, H.; Gao, T.; Wang, G.; Li, C. Proinflammatory effect of high-mobility group protein B1 on keratinocytes: An autocrine mechanism underlying psoriasis development. J. Pathol. 2017, 241, 392–404. [Google Scholar] [CrossRef]
- Strohbuecker, L.; Koenen, H.; van Rijssen, E.; van Cranenbroek, B.; Fasse, E.; Joosten, I.; Korber, A.; Bergmann, C. Increased dermal expression of chromatin-associated protein HMGB1 and concomitant T-cell expression of the DNA RAGE in patients with psoriasis vulgaris. Psoriasis 2019, 9, 7–17. [Google Scholar] [CrossRef]
- Chen, T.; Fu, L.X.; Guo, Z.P.; Yin, B.; Cao, N.; Qin, S. Involvement of high mobility group box-1 in imiquimod-induced psoriasis-like mice model. J. Dermatol. 2017, 44, 573–581. [Google Scholar] [CrossRef]
- Kuroiwa, Y.; Takakusagi, Y.; Kusayanagi, T.; Kuramochi, K.; Imai, T.; Hirayama, T.; Ito, I.; Yoshida, M.; Sakaguchi, K.; Sugawara, F. Identification and characterization of the direct interaction between methotrexate (MTX) and high-mobility group box 1 (HMGB1) protein. PLoS ONE 2013, 8, e63073. [Google Scholar] [CrossRef]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 proteins in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118677. [Google Scholar] [CrossRef] [PubMed]
- Schafer, B.W.; Heizmann, C.W. The S100 family of EF-hand calcium-binding proteins: Functions and pathology. Trends Biochem. Sci. 1996, 21, 134–140. [Google Scholar] [CrossRef]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed]
- Buchau, A.S.; Gallo, R.L. Innate immunity and antimicrobial defense systems in psoriasis. Clin. Dermatol. 2007, 25, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.S.; Wong, J.; Polyak, K.; Aronzon, D.; Enerback, C. Detection of psoriasin/S100A7 in the sera of patients with psoriasis. Br. J. Dermatol. 2009, 160, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Benoit, S.; Toksoy, A.; Ahlmann, M.; Schmidt, M.; Sunderkotter, C.; Foell, D.; Pasparakis, M.; Roth, J.; Goebeler, M. Elevated serum levels of calcium-binding S100 proteins A8 and A9 reflect disease activity and abnormal differentiation of keratinocytes in psoriasis. Br. J. Dermatol. 2006, 155, 62–66. [Google Scholar] [CrossRef]
- Defrene, J.; Berrazouane, S.; Esparza, N.; Page, N.; Cote, M.F.; Gobeil, S.; Aoudjit, F.; Tessier, P.A. Deletion of S100a8 and S100a9 Enhances Skin Hyperplasia and Promotes the Th17 Response in Imiquimod-Induced Psoriasis. J. Immunol. 2021, 206, 505–514. [Google Scholar] [CrossRef]
- Christmann, C.; Zenker, S.; Martens, L.; Hubner, J.; Loser, K.; Vogl, T.; Roth, J. Interleukin 17 Promotes Expression of Alarmins S100A8 and S100A9 During the Inflammatory Response of Keratinocytes. Front. Immunol. 2020, 11, 599947. [Google Scholar] [CrossRef]
- Grantham, H.J.; Hussain, A.B.; Reynolds, N.J. Serum S100A8/A9 May Act as Biomarker of Atherosclerosis Severity in Psoriasis. J. Investig. Dermatol. 2022, 142, 2848–2850. [Google Scholar] [CrossRef]
- Schonthaler, H.B.; Guinea-Viniegra, J.; Wculek, S.K.; Ruppen, I.; Ximenez-Embun, P.; Guio-Carrion, A.; Navarro, R.; Hogg, N.; Ashman, K.; Wagner, E.F. S100A8-S100A9 protein complex mediates psoriasis by regulating the expression of complement factor C3. Immunity 2013, 39, 1171–1181. [Google Scholar] [CrossRef]
- Tsan, M.F.; Gao, B. Heat shock protein and innate immunity. Cell. Mol. Immunol. 2004, 1, 274–279. [Google Scholar] [PubMed]
- Asea, A.; Rehli, M.; Kabingu, E.; Boch, J.A.; Bare, O.; Auron, P.E.; Stevenson, M.A.; Calderwood, S.K. Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 2002, 277, 15028–15034. [Google Scholar] [CrossRef]
- Basu, S.; Binder, R.J.; Ramalingam, T.; Srivastava, P.K. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 2001, 14, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Bayramgurler, D.; Ozkara, S.K.; Apaydin, R.; Ercin, C.; Bilen, N. Heat shock proteins 60 and 70 expression of cutaneous lichen planus: Comparison with normal skin and psoriasis vulgaris. J. Cutan. Pathol. 2004, 31, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Boehncke, W.H.; Dahlke, A.; Zollner, T.M.; Sterry, W. Differential expression of heat shock protein 70 (HSP70) and heat shock cognate protein 70 (HSC70) in human epidermis. Arch. Dermatol. Res. 1994, 287, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Czajkowski, R.; Kaszewski, S.; Tadrowski, T.; Grzanka, D.; Ziandarska, J.; Drewa, G.; Marek-Jozefowicz, L. Does HSP90 play an important role in psoriasis? Postep. Dermatol. Alergol. 2021, 38, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Conrad, C.; Dudli, C.; Kielhorn, E.; Nickoloff, B.J.; Nestle, F.O. Activation of dendritic antigen-presenting cells expressing common heat shock protein receptor CD91 during induction of psoriasis. Br. J. Dermatol. 2005, 152, 1211–1218. [Google Scholar] [CrossRef]
- Curry, J.L.; Qin, J.Z.; Bonish, B.; Carrick, R.; Bacon, P.; Panella, J.; Robinson, J.; Nickoloff, B.J. Innate immune-related receptors in normal and psoriatic skin. Arch. Pathol. Lab. Med. 2003, 127, 178–186. [Google Scholar] [CrossRef]
- Lv, J.; Zhou, D.; Wang, Y.; Sun, W.; Zhang, C.; Xu, J.; Yang, H.; Zhou, T.; Li, P. Effects of luteolin on treatment of psoriasis by repressing HSP90. Int. Immunopharmacol. 2020, 79, 106070. [Google Scholar] [CrossRef]
- Xiang, X.; Tu, C.; Li, Q.; Wang, W.; Huang, X.; Zhao, Z.; Xiong, H.; Mei, Z. Oxymatrine ameliorates imiquimod-induced psoriasis pruritus and inflammation through inhibiting heat shock protein 90 and heat shock protein 60 expression in keratinocytes. Toxicol. Appl. Pharmacol. 2020, 405, 115209. [Google Scholar] [CrossRef]
- Stenderup, K.; Rosada, C.; Gavillet, B.; Vuagniaux, G.; Dam, T.N. Debio 0932, a new oral Hsp90 inhibitor, alleviates psoriasis in a xenograft transplantation model. Acta Derm. Venereol. 2014, 94, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.S.; Thuesen, K.K.H.; Bregnhoj, A.; Moldovan, L.I.; Kristensen, L.S.; Grek, C.L.; Ghatnekar, G.S.; Iversen, L.; Johansen, C. The HSP90 inhibitor RGRN-305 exhibits strong immunomodulatory effects in human keratinocytes. Exp. Dermatol. 2021, 30, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xue, X.; Tang, W.; Su, L.; Zhang, L.; Zhang, Y. Cytosolic DNA-Mediated STING-Dependent Inflammation Contributes to the Progression of Psoriasis. J. Investig. Dermatol. 2022, 142, 898–906.e4. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Ogawa, Y.; Kinoshita, M.; Ihara, T.; Shimada, S.; Koizumi, S.; Kawamura, T. Mechanical stretch-induced ATP release from keratinocytes triggers Koebner phenomenon in psoriasis. J. Dermatol. Sci 2021, 103, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Chamilos, G.; Lande, R.; Gregorio, J.; Meller, S.; Facchinetti, V.; Homey, B.; Barrat, F.J.; Zal, T.; Gilliet, M. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J. Exp. Med. 2009, 206, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Dong, Y.; Hu, H.; Wang, Q.; Guo, S.; Fu, D.; Song, X.; Kalvakolanu, D.V.; Tian, Z. IL-33 contributes to disease severity in Psoriasis-like models of mouse. Cytokine 2019, 119, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Seifarth, F.G.; Lax, J.E.; Harvey, J.; DiCorleto, P.E.; Husni, M.E.; Chandrasekharan, U.M.; Tytell, M. Topical heat shock protein 70 prevents imiquimod-induced psoriasis-like inflammation in mice. Cell Stress Chaperones 2018, 23, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Messmer, D.; Yang, H.; Telusma, G.; Knoll, F.; Li, J.; Messmer, B.; Tracey, K.J.; Chiorazzi, N. High mobility group box protein 1: An endogenous signal for dendritic cell maturation and Th1 polarization. J. Immunol. 2004, 173, 307–313. [Google Scholar] [CrossRef]
- Dumitriu, I.E.; Bianchi, M.E.; Bacci, M.; Manfredi, A.A.; Rovere-Querini, P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J. Leukoc. Biol. 2007, 81, 84–91. [Google Scholar] [CrossRef]
- Xia, C.; Braunstein, Z.; Toomey, A.C.; Zhong, J.; Rao, X. S100 Proteins as an Important Regulator of Macrophage Inflammation. Front. Immunol. 2017, 8, 1908. [Google Scholar] [CrossRef]
- Sprenkeler, E.G.G.; Zandstra, J.; van Kleef, N.D.; Goetschalckx, I.; Verstegen, B.; Aarts, C.E.M.; Janssen, H.; Tool, A.T.J.; van Mierlo, G.; van Bruggen, R.; et al. S100A8/A9 Is a Marker for the Release of Neutrophil Extracellular Traps and Induces Neutrophil Activation. Cells 2022, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Killeen, M.E.; Ferris, L.; Kupetsky, E.A.; Falo, L., Jr.; Mathers, A.R. Signaling through purinergic receptors for ATP induces human cutaneous innate and adaptive Th17 responses: Implications in the pathogenesis of psoriasis. J. Immunol. 2013, 190, 4324–4336. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, L.; Bulek, K.; Martin, B.N.; Zepp, J.A.; Kang, Z.; Liu, C.; Herjan, T.; Misra, S.; Carman, J.A.; et al. The psoriasis-associated D10N variant of the adaptor Act1 with impaired regulation by the molecular chaperone hsp90. Nat. Immunol. 2013, 14, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.M.; Pappu, B.P.; Peng, J.; Martinez, G.J.; Zhang, Y.; Chung, Y.; Ma, L.; Yang, X.O.; Nurieva, R.I.; Tian, Q.; et al. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity 2010, 32, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Han, J.; Zhang, Y.; Wang, X.; Fan, H.; Hua, F.; Yao, R. TLR4-RelA-miR-30a signal pathway regulates Th17 differentiation during experimental autoimmune encephalomyelitis development. J. Neuroinflamm. 2019, 16, 183. [Google Scholar] [CrossRef] [PubMed]
- Wieten, L.; Broere, F.; van der Zee, R.; Koerkamp, E.K.; Wagenaar, J.; van Eden, W. Cell stress induced HSP are targets of regulatory T cells: A role for HSP inducing compounds as anti-inflammatory immuno-modulators? FEBS Lett. 2007, 581, 3716–3722. [Google Scholar] [CrossRef]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. Secukinumab in plaque psoriasis--results of two phase 3 trials. N. Engl. J. Med. 2014, 371, 326–338. [Google Scholar] [CrossRef]
- Batycka-Baran, A.; Hattinger, E.; Zwicker, S.; Summer, B.; Zack Howard, O.M.; Thomas, P.; Szepietowski, J.C.; Ruzicka, T.; Prinz, J.C.; Wolf, R. Leukocyte-derived koebnerisin (S100A15) and psoriasin (S100A7) are systemic mediators of inflammation in psoriasis. J. Dermatol. Sci. 2015, 79, 214–221. [Google Scholar] [CrossRef]
- Xue, J.; Suarez, J.S.; Minaai, M.; Li, S.; Gaudino, G.; Pass, H.I.; Carbone, M.; Yang, H. HMGB1 as a therapeutic target in disease. J. Cell. Physiol. 2021, 236, 3406–3419. [Google Scholar] [CrossRef]
- Yu, J.J.; Zhang, C.S.; Coyle, M.E.; Du, Y.; Zhang, A.L.; Guo, X.; Xue, C.C.; Lu, C. Compound glycyrrhizin plus conventional therapy for psoriasis vulgaris: A systematic review and meta-analysis of randomized controlled trials. Curr. Med. Res. Opin. 2017, 33, 279–287. [Google Scholar] [CrossRef]
- Chen, R.C.; Yi, P.P.; Zhou, R.R.; Xiao, M.F.; Huang, Z.B.; Tang, D.L.; Huang, Y.; Fan, X.G. The role of HMGB1-RAGE axis in migration and invasion of hepatocellular carcinoma cell lines. Mol. Cell. Biochem. 2014, 390, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Chen, M.; Xiao, C.; Yang, W.; Qin, Q.; Tan, Q.; Liang, Z.; Liao, X.; Mao, A.; Wei, C. Triptolide Suppresses Growth of Breast Cancer by Targeting HMGB1 in Vitro and in Vivo. Biol. Pharm. Bull. 2019, 42, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Sack, U.; Walther, W.; Scudiero, D.; Selby, M.; Aumann, J.; Lemos, C.; Fichtner, I.; Schlag, P.M.; Shoemaker, R.H.; Stein, U. S100A4-induced cell motility and metastasis is restricted by the Wnt/beta-catenin pathway inhibitor calcimycin in colon cancer cells. Mol. Biol. Cell 2011, 22, 3344–3354. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| DAMP | Psoriasis Patients | Experimental Psoriasis |
|---|---|---|
| HMGB1 | It is increased in the serum and skin of psoriasis patients [76,79]. | Its inhibitor, antibody, or deficiency reduces inflammation of IMQ-induced mice [77,78,80]. |
| S100s | S100A7: It is strongly expressed in psoriatic lesions and decreased in the serum of patients with psoriasis [86]. S100A8/A9: It is increased in the serum and lesion skin of psoriasis patients [87]. | S100A8/A9: Serum S100A8/A9 levels may act as biomarker of atherosclerosis severity in psoriasis; deletion of them enhances inflammation in imiquimod-induced psoriasis mice [88,90]. |
| HSPs | HSP60: The mean IRIDI scores for its expression in the basal, suprabasal, and superficial epidermal layers of psoriasis are higher than those of normal skin [95]. HSP70: The expression of it has no obvious differences between lesion and normal skin [96]. HSP90: The expression of it increases with the frequency of exacerbations of psoriasis throughout the year [97]. | HSP70: It prevents imiquimod-induced, psoriasis-like inflammation in mice [108]. HSP90: Its inhibitor alleviates psoriasis in a xenograft transplantation model [103]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Gong, B.; Chen, Z.; Song, J.; Xu, N.; Weng, Z. Damage-Associated Molecular Patterns, a Class of Potential Psoriasis Drug Targets. Int. J. Mol. Sci. 2024, 25, 771. https://doi.org/10.3390/ijms25020771
Gao Y, Gong B, Chen Z, Song J, Xu N, Weng Z. Damage-Associated Molecular Patterns, a Class of Potential Psoriasis Drug Targets. International Journal of Molecular Sciences. 2024; 25(2):771. https://doi.org/10.3390/ijms25020771
Chicago/Turabian StyleGao, Yaqi, Bishuang Gong, Zhenxing Chen, Jierong Song, Na Xu, and Zhuangfeng Weng. 2024. "Damage-Associated Molecular Patterns, a Class of Potential Psoriasis Drug Targets" International Journal of Molecular Sciences 25, no. 2: 771. https://doi.org/10.3390/ijms25020771
APA StyleGao, Y., Gong, B., Chen, Z., Song, J., Xu, N., & Weng, Z. (2024). Damage-Associated Molecular Patterns, a Class of Potential Psoriasis Drug Targets. International Journal of Molecular Sciences, 25(2), 771. https://doi.org/10.3390/ijms25020771