
CRISPR/Cas9 as a New Antiviral Strategy for Treating Hepatitis Viral Infections

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
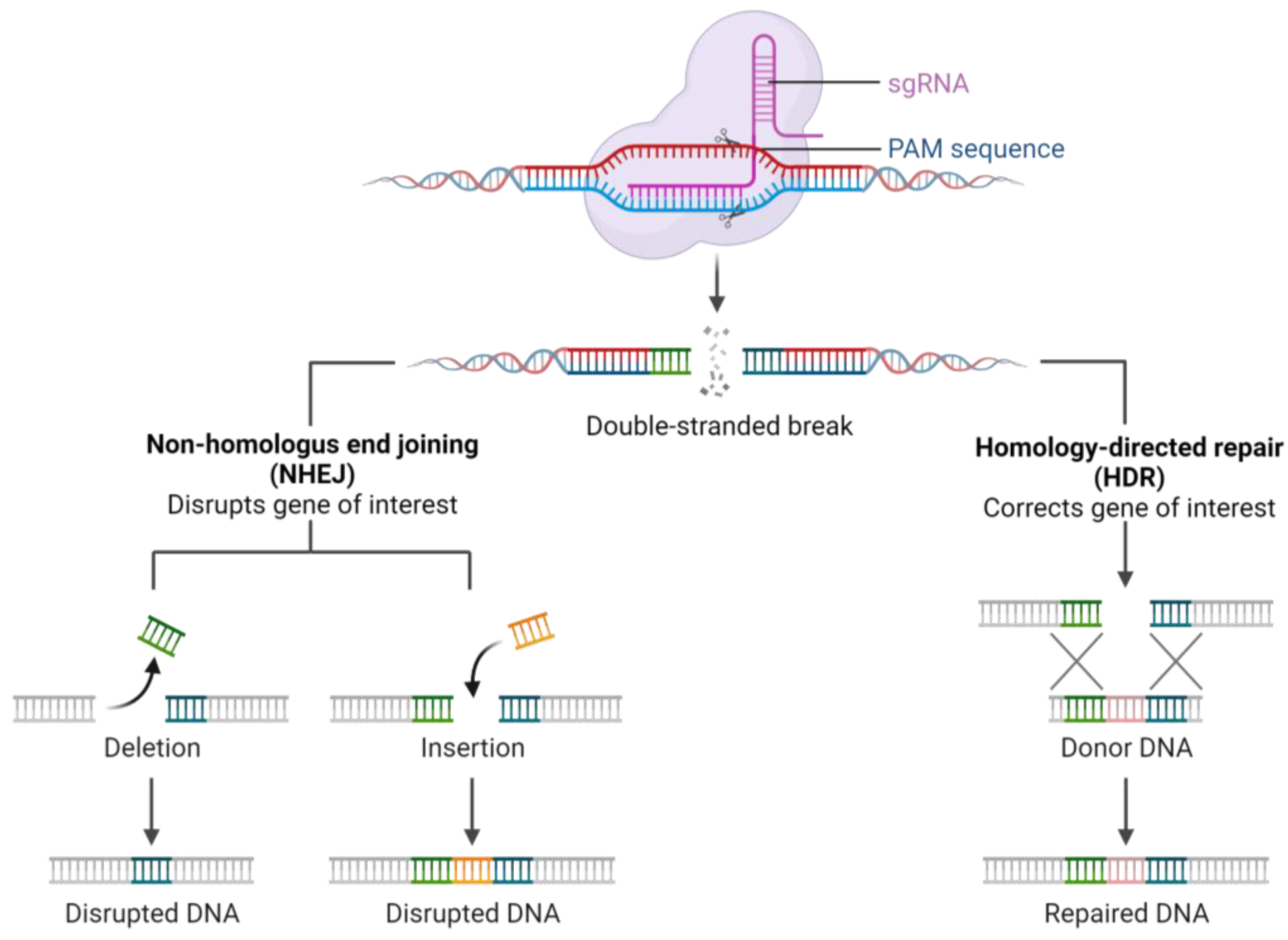
2. CRISPR Technology
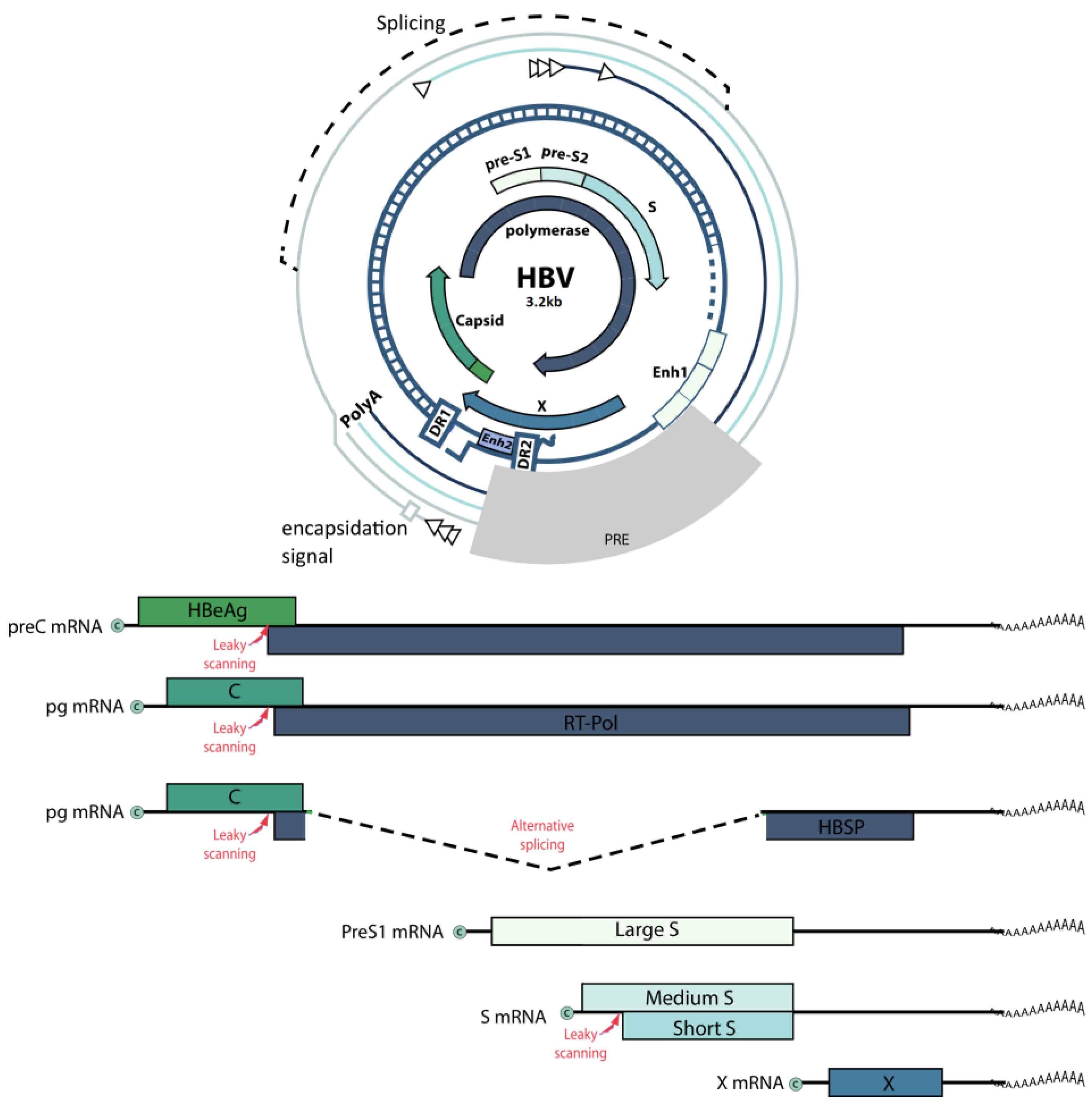
3. Hepatitis B Virus
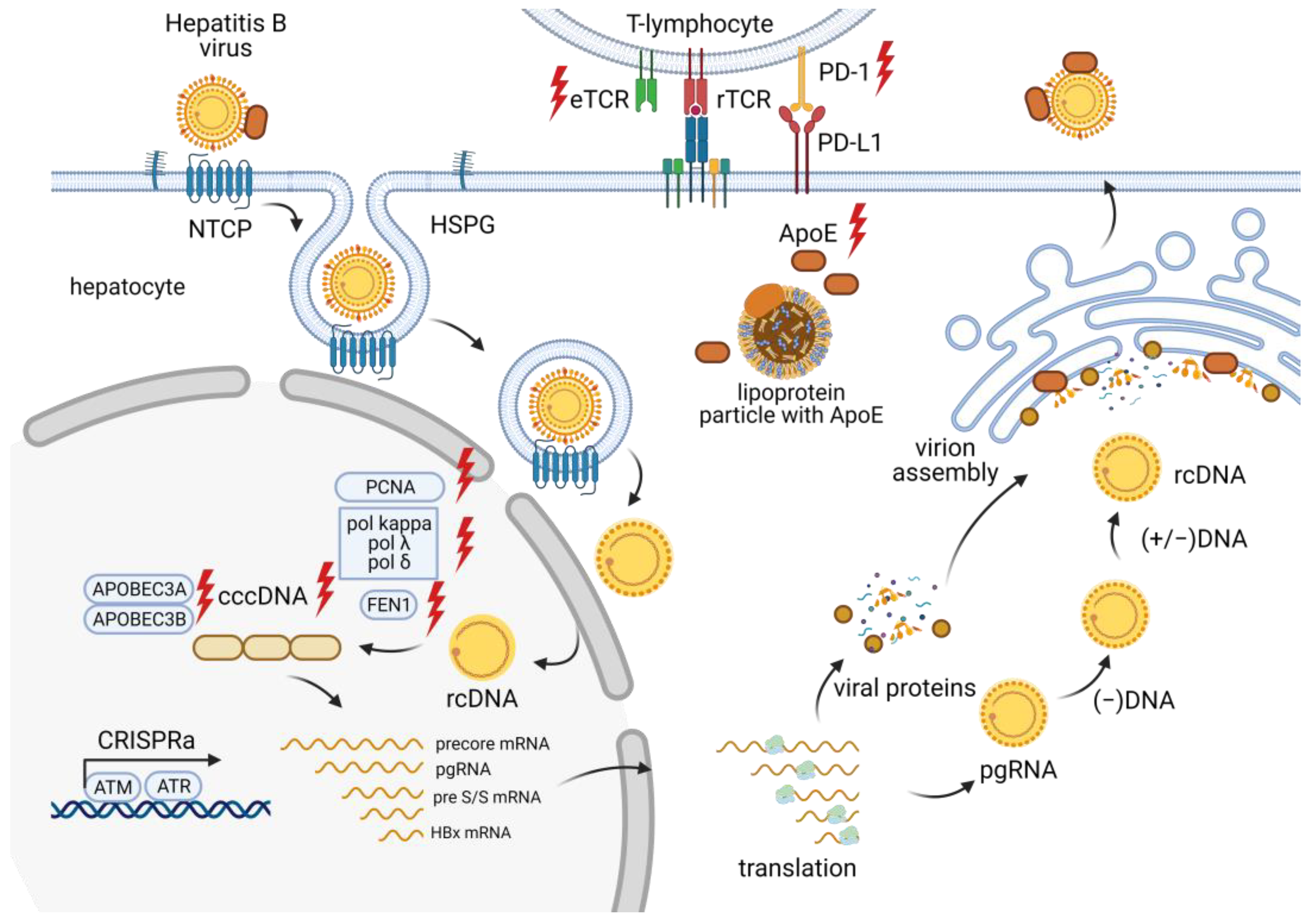
3.1. Targeting the CRISPR/Cas System to the HBV Genome
3.2. Host Factors Mediating HBV Infection
3.2.1. Apolipoprotein E
3.2.2. Cellular Kinases
3.2.3. Cellular Endonucleases
3.2.4. Host Antiviral Factors
3.2.5. Non-Coding RNAs
3.2.6. Cellular Polymerases Essential for HBV Replication
3.3. Host Factors Mediating HCC Development during HBV Infection
3.4. Modification of T Lymphocytes as a Strategy to Suppress HBV Infection
3.5. Epigenetic Editing of cccDNA
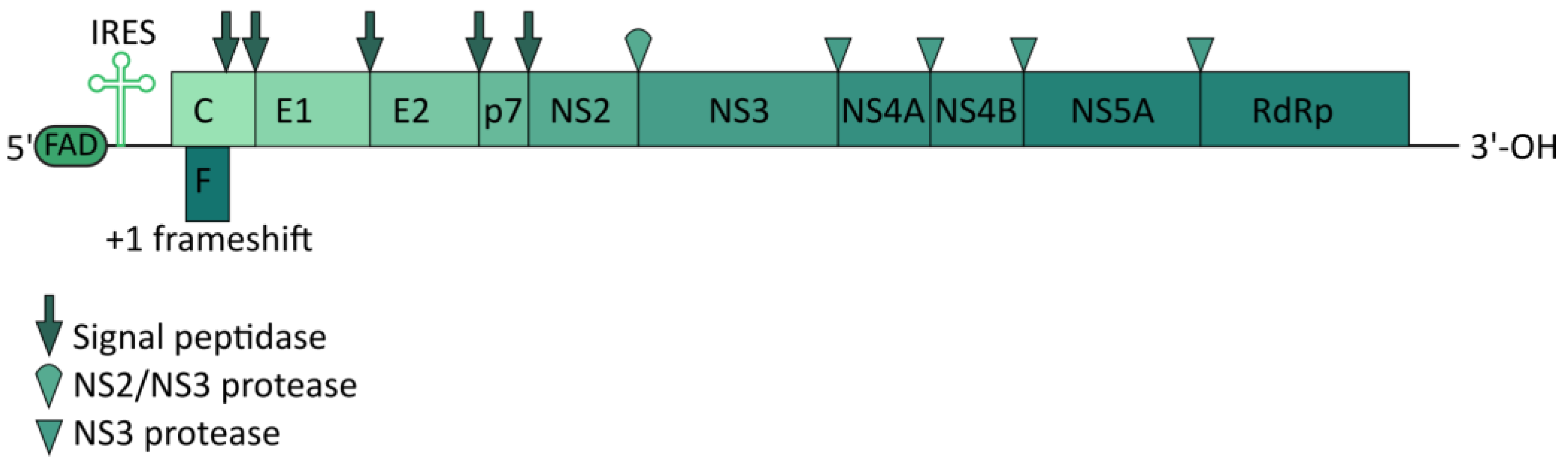
4. Hepatitis C Virus
4.1. Targeting the CRISPR/Cas System to the HCV Genome
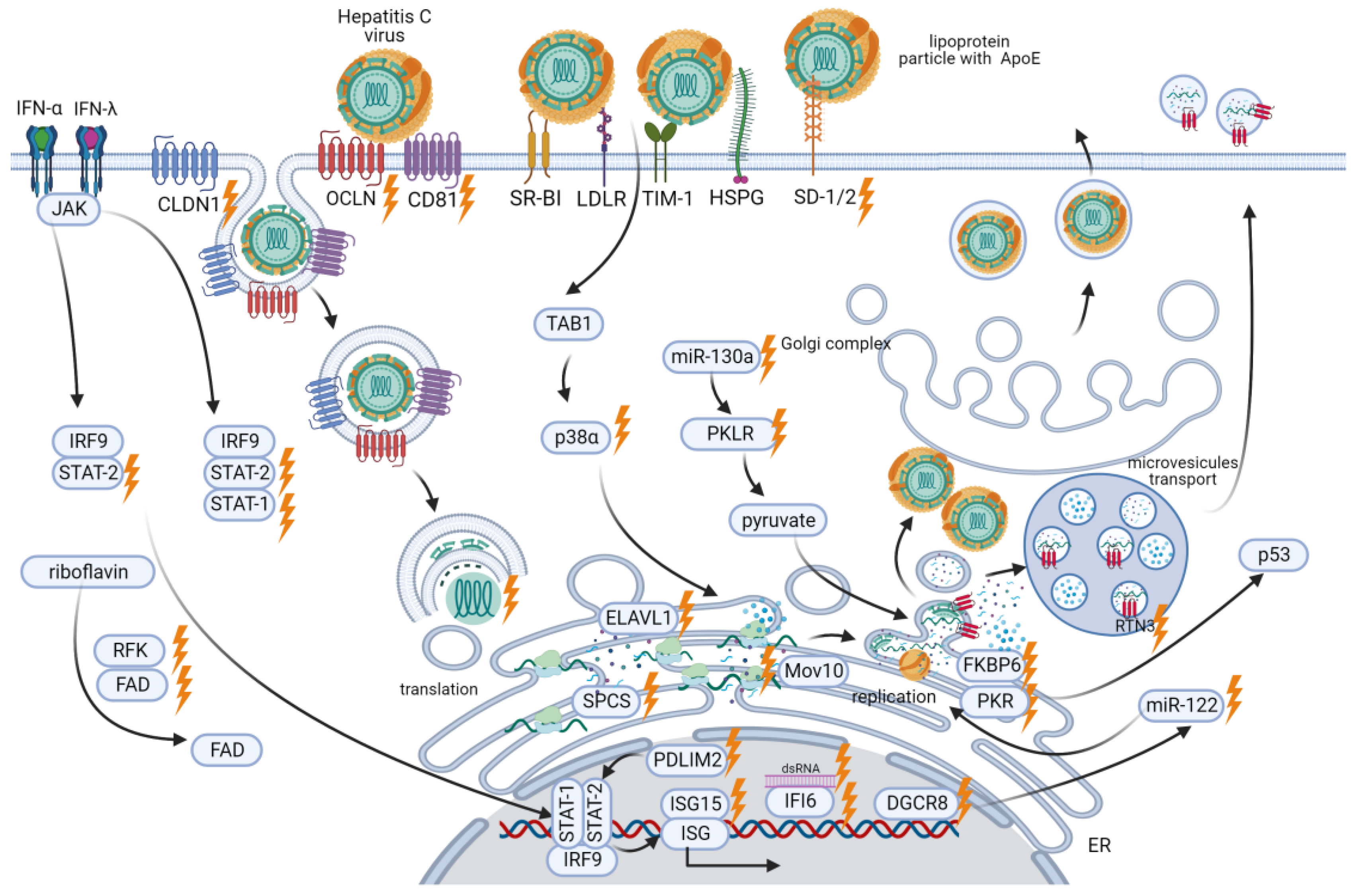
4.2. Host Factors Mediating HCV Infection
4.2.1. Factors Enabling HCV Entry into Cells
4.2.2. SPCS1
4.2.3. FKBP6
4.2.4. Non-Coding RNAs
4.2.5. RTN3
4.2.6. p38α
4.2.7. P-Body Protein Mov10
4.2.8. Host Antiviral Factors
4.2.9. PKLR
4.2.10. CRISPR Screening of Cellular Factors Essential for HCV
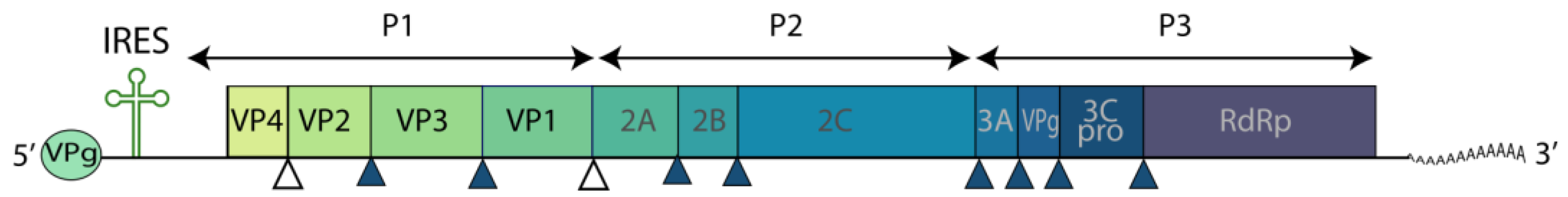
5. Hepatitis A Virus
5.1. Host Factors Mediating HAV Infection
5.1.1. Factors Enabling HAV Entry into Cells
5.1.2. Factors of the Innate Immune Response
5.1.3. GRP78
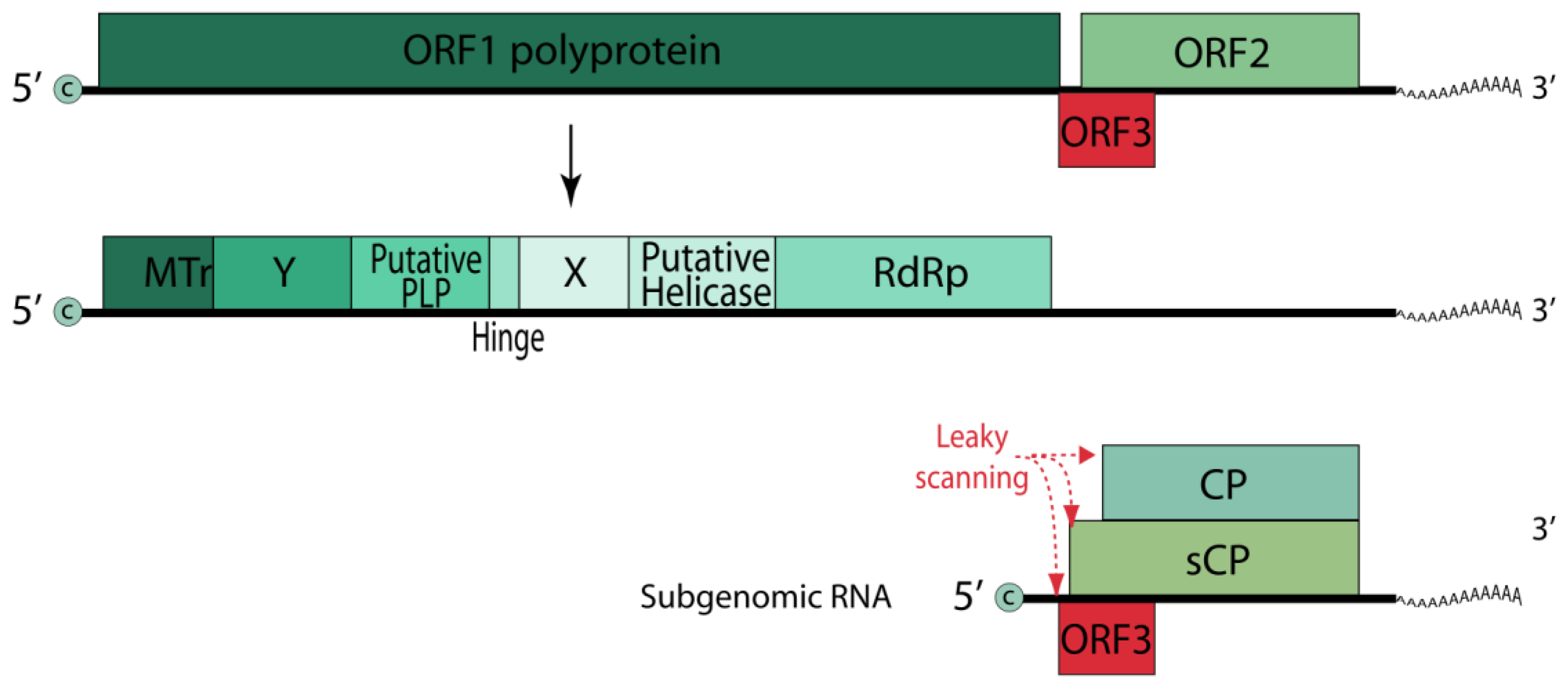
6. Hepatitis E Virus
BISPR
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hepatitis A. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-a (accessed on 20 February 2021).
- Hepatitis B. Available online: https://www.who.int/en/news-room/fact-sheets/detail/hepatitis-b (accessed on 26 January 2021).
- Hepatitis C. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed on 1 February 2021).
- Hepatitis E. Available online: https://www.who.int/en/news-room/fact-sheets/detail/hepatitis-e (accessed on 19 February 2021).
- Zeuzem, S. Interferon-Based Therapy for Chronic Hepatitis C: Current and Future Perspectives. Nat. Clin. Pract. Gastroenterol. Hepatol. 2008, 5, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.J.; Block, T.M.; McMahon, B.J.; Ghany, M.G.; Urban, S.; Guo, J.T.; Locarnini, S.; Zoulim, F.; Chang, K.M.; Lok, A.S. Present and Future Therapies of Hepatitis B: From Discovery to Cure. Hepatology 2015, 62, 1893–1908. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Juarez, A.; Vallejo, N.; Lopez-Lopez, P.; Díaz-Mareque, A.I.; Frias, M.; Vallejo, A.; Caballero-Gómez, J.; Rodríguez-Velasco, M.; Molina, E.; Aguilera, A. Ribavirin as a First Treatment Approach for Hepatitis e Virus Infection in Transplant Recipient Patients. Microorganisms 2020, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Fried, M.W.; Shiffman, M.L.; Reddy, K.R.; Smith, C.; Marinos, G.; Gonçales, F.L.; Häussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; et al. Peginterferon Alfa-2a plus Ribavirin for Chronic Hepatitis C Virus Infection. N. Engl. J. Med. 2002, 347, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, I.; Qujeq, D.; Yousefi, T.; Ferns, G.A.; Maniati, M.; Vaghari-Tabari, M. CRISPR/Cas9 Gene Editing: A New Therapeutic Approach in the Treatment of Infection and Autoimmunity. IUBMB Life 2020, 72, 1603–1621. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.; Díez-Villaseñor, C.S.; García-Martínez, J.; Soria, E. Intervening Sequences of Regularly Spaced Prokaryotic Repeats Derive from Foreign Genetic Elements. J. Mol. Evol. 2005, 60, 174–182. Available online: https://link.springer.com/article/10.1007/s00239-004-0046-3 (accessed on 12 December 2023). [CrossRef]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Ehrlich, S.D. Clustered Regularly Interspaced Short Palindrome Repeats (CRISPRs) Have Spacers of Extrachromosomal Origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [CrossRef]
- CRISPR Elements in Yersinia Pestis Acquire New Repeats by Preferential Uptake of Bacteriophage DNA, and Provide Additional Tools for Evolutionary Studies|Microbiology Society. Available online: https://www.microbiologyresearch.org/content/journal/micro/10.1099/mic.0.27437-0 (accessed on 12 December 2023).
- CRISPR Interference Limits Horizontal Gene Transfer in Staphylococci by Targeting DNA|Science. Available online: https://www.science.org/doi/10.1126/science.1165771 (accessed on 12 December 2023).
- Evolutionary Classification of CRISPR–Cas Systems: A Burst of Class 2 and Derived Variants|Nature Reviews Microbiology. Available online: https://www.nature.com/articles/s41579-019-0299-x (accessed on 12 December 2023).
- Cas9–crRNA Ribonucleoprotein Complex Mediates Specific DNA Cleavage for Adaptive Immunity in Bacteria PNAS. Available online: https://www.pnas.org/doi/full/10.1073/pnas.1208507109 (accessed on 12 December 2023).
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA Maturation by Trans-Encoded Small RNA and Host Factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef]
- Systematic Prediction of Genes Functionally Linked to CRISPR-Cas Systems by Gene Neighborhood Analysis|PNAS. Available online: https://www.pnas.org/doi/full/10.1073/pnas.1803440115 (accessed on 12 December 2023).
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Almendros, C. Short Motif Sequences Determine the Targets of the Prokaryotic CRISPR Defence System. Microbiology 2009, 155, 733–740. [Google Scholar] [CrossRef]
- Genome Engineering of Drosophila with the CRISPR RNA-Guided Cas9 Nuclease|Genetics | Oxford Academic. Available online: https://academic.oup.com/genetics/article/194/4/1029/5935344?login=true (accessed on 12 December 2023).
- Multiplex Genome Engineering Using CRISPR/Cas Systems|Science. Available online: https://www.science.org/doi/10.1126/science.1231143 (accessed on 12 December 2023).
- Schaefer, S. Hepatitis B Virus Taxonomy and Hepatitis B Virus Genotypes. World J. Gastroenterol. 2007, 13, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Dane, D.S.; Cameron, C.H.; Briggs, M. Virus-like particles in serum of patients with australia-antigen-associated hepatitis. Lancet 1970, 295, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D.; Prince, A.M. Hepatitis B Virus Infection—Natural History and Clinical Consequences. N. Engl. J. Med. 2004, 350, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.S.; Clayton, D.A.; Greenman, R.L. DNA of a Human Hepatitis B Virus Candidate. J. Virol. 1974, 14, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.; O’Connell, A.; Millman, I. Genome of Hepatitis B Virus: Restriction Enzyme Cleavage and Structure of DNA Extracted from Dane Particles. Proc. Natl. Acad. Sci. USA 1975, 72, 4597–4601. [Google Scholar] [CrossRef] [PubMed]
- Quasdorff, M.; Protzer, U. Control of Hepatitis B Virus at the Level of Transcription. J. Viral Hepat. 2010, 17, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Molecular Biology of Hepatitis B Virus Infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef]
- Block, T.M.; Rawat, S.; Brosgart, C.L. Chronic Hepatitis B: A Wave of New Therapies on the Horizon. Antivir. Res. 2015, 121, 69–81. [Google Scholar] [CrossRef]
- Virology. Getting Rid of a Persistent Troublemaker to Cure Hepatitis—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/24626921/ (accessed on 14 January 2021).
- Sakuma, T.; Masaki, K.; Abe-Chayama, H.; Mochida, K.; Yamamoto, T.; Chayama, K. Highly Multiplexed CRISPR-Cas9-Nuclease and Cas9-Nickase Vectors for Inactivation of Hepatitis B Virus. Genes Cells 2016, 21, 1253–1262. [Google Scholar] [CrossRef]
- Dong, C.; Qu, L.; Wang, H.; Wei, L.; Dong, Y.; Xiong, S. Targeting Hepatitis B Virus cccDNA by CRISPR/Cas9 Nuclease Efficiently Inhibits Viral Replication. Antivir. Res. 2015, 118, 110–117. [Google Scholar] [CrossRef]
- Ramanan, V.; Shlomai, A.; Cox, D.B.T.; Schwartz, R.E.; Michailidis, E.; Bhatta, A.; Scott, D.A.; Zhang, F.; Rice, C.M.; Bhatia, S.N. CRISPR/Cas9 Cleavage of Viral DNA Efficiently Suppresses Hepatitis B Virus. Sci. Rep. 2015, 5, 10833. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sheng, C.; Wang, S.; Yang, L.; Liang, Y.; Huang, Y.; Liu, H.; Li, P.; Yang, C.; Yang, X.; et al. Removal of Integrated Hepatitis B Virus DNA Using CRISPR-Cas9. Front. Cell. Infect. Microbiol. 2017, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Schiwon, M.; Ehrke-Schulz, E.; Oswald, A.; Bergmann, T.; Michler, T.; Protzer, U.; Ehrhardt, A. One-Vector System for Multiplexed CRISPR/Cas9 against Hepatitis B Virus cccDNA Utilizing High-Capacity Adenoviral Vectors. Mol. Ther. Nucleic Acids 2018, 12, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Scott, T.; Moyo, B.; Nicholson, S.; Maepa, M.B.; Watashi, K.; Ely, A.; Weinberg, M.S.; Arbuthnot, P. SsAAVs Containing Cassettes Encoding SaCas9 and Guides Targeting Hepatitis B Virus Inactivate Replication of the Virus in Cultured Cells. Sci. Rep. 2017, 7, 7401. [Google Scholar] [CrossRef] [PubMed]
- Karimova, M.; Beschorner, N.; Dammermann, W.; Chemnitz, J.; Indenbirken, D.; Bockmann, J.H.; Grundhoff, A.; Luth, S.; Buchholz, F.; Wiesch, J.S.Z.; et al. CRISPR/Cas9 Nickase-Mediated Disruption of Hepatitis B Virus Open Reading Frame S and X. Sci. Rep. 2015, 5, 13734. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Hua, L.; Liu, Y.H.; Gao, L.C.; Fu, J.; Wan, D.Y.; Dong, L.H.; Song, H.F.; Gao, X. Harnessing the Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)/CRISPR-Associated Cas9 System to Disrupt the Hepatitis B Virus. Gene Ther. 2015, 22, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, R.; Chen, S.; Guo, D.; Chen, Y. Inhibition of Hepatitis B Virus by the CRISPR/Cas9 System via Targeting the Conserved Regions of the Viral Genome. J. Gen. Virol. 2015, 96, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhang, X.; Ge, Q.; Yuan, C.; Chu, L.; Liang, H.-F.; Liao, Z.; Liu, Q.; Zhang, Z.; Zhang, B. CRISPR/Cas9-Mediated Knockout of HBsAg Inhibits Proliferation and Tumorigenicity of HBV-Positive Hepatocellular Carcinoma Cells. J. Cell. Biochem. 2018, 119, 8419–8431. [Google Scholar] [CrossRef]
- Tseng, T.C.; Liu, C.J.; Yang, H.C.; Su, T.H.; Wang, C.C.; Chen, C.L.; Hsu, C.A.; Fang-Tzu Kuo, S.; Liu, C.H.; Chen, P.J.; et al. Serum Hepatitis B Surface Antigen Levels Help Predict Disease Progression in Patients with Low Hepatitis B Virus Loads. Hepatology 2013, 57, 441–450. [Google Scholar] [CrossRef]
- Kostyusheva, A.P.; Kostyushev, D.S.; Brezgin, S.A.; Zarifyan, D.N.; Volchkova, E.V.; Chulanov, V.P. Small Molecular Inhibitors of DNA Double Strand Break Repair Pathways Increase the ANTI-HBV Activity of CRISPR/Cas9. Mol. Biol. 2019, 53, 311–323. [Google Scholar] [CrossRef]
- Kostyushev, D.; Kostyusheva, A.; Brezgin, S.; Zarifyan, D.; Utkina, A.; Goptar, I.; Chulanov, V. Suppressing the NHEJ Pathway by DNA-PKcs Inhibitor NU7026 Prevents Degradation of HBV cccDNA Cleaved by CRISPR/Cas9. Sci. Rep. 2019, 9, 1847. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, R.; Zhang, R.; Ding, S.; Zhang, T.; Yuan, Q.; Guan, G.; Chen, X.; Zhang, T.; Zhuang, H.; et al. The gRNA-miRNA-gRNA Ternary Cassette Combining CRISPR/Cas9 with RNAi Approach Strongly Inhibits Hepatitis B Virus Replication. Theranostics 2017, 7, 3090. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, T.; Fukuhara, T.; Ono, C.; Yamamoto, S.; Uemura, K.; Okamoto, T.; Sugiyama, M.; Motooka, D.; Nakamura, S.; Ikawa, M.; et al. Suppression of HBV Replication by the Expression of Nickase-and Nuclease Dead-Cas9. Sci. Rep. 2017, 7, 6122. [Google Scholar] [CrossRef] [PubMed]
- Kostyushev, D.; Kostyusheva, A.; Brezgin, S.; Ponomareva, N.; Zakirova, N.F.; Egorshina, A.; Yanvarev, D.V.; Bayurova, E.; Sudina, A.; Goptar, I.; et al. Depleting Hepatitis B Virus Relaxed Circular DNA Is Necessary for Resolution of Infection by CRISPR-Cas9. Mol. Ther. Nucleic Acids 2023, 31, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Apolipoprotein E: Cholesterol Transport Protein with Expanding Role in Cell Biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Luo, G.G. Human Apolipoprotein e Promotes Hepatitis B Virus Infection and Production. PLoS Pathog. 2019, 15, e1007874. [Google Scholar] [CrossRef] [PubMed]
- Turnell, A.S.; Grand, R.J. DNA Viruses and the Cellular DNA-Damage Response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef]
- Wang, J.; Jia, J.; Chen, R.; Ding, S.; Xu, Q.; Zhang, T.; Chen, X.; Liu, S.; Lu, F. RFX1 Participates in Doxorubicin-Induced Hepatitis B Virus Reactivation. Cancer Med. 2018, 7, 2021–2033. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Kostyusheva, A.; Brezgin, S.; Bayurova, E.; Gordeychuk, I.; Isaguliants, M.; Goptar, I.; Urusov, F.; Nikiforova, A.; Volchkova, E.; Kostyushev, D.; et al. ATM and ATR Expression Potentiates HBV Replication and Contributes to Reactivation of HBV Infection upon DNA Damage. Viruses 2019, 11, 997. [Google Scholar] [CrossRef]
- Gao, W.; Hu, J. Formation of Hepatitis B Virus Covalently Closed Circular DNA: Removal of Genome-Linked Protein. J. Virol. 2007, 81, 6164–6174. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Jiang, D.; Zhou, T.; Cuconati, A.; Block, T.M.; Guo, J.-T. Characterization of the Intracellular Deproteinized Relaxed Circular DNA of Hepatitis B Virus: An Intermediate of Covalently Closed Circular DNA Formation. J. Virol. 2007, 81, 12472–12484. [Google Scholar] [CrossRef]
- Kitamura, K.; Que, L.; Shimadu, M.; Koura, M.; Ishihara, Y.; Wakae, K.; Nakamura, T.; Watashi, K.; Wakita, T.; Muramatsu, M. Flap Endonuclease 1 Is Involved in cccDNA Formation in the Hepatitis B Virus. PLoS Pathog. 2018, 14, e1007124. [Google Scholar] [CrossRef]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and Nonhepatotoxic Degradation of Nuclear Hepatitis B Virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Brezgin, S.; Kostyusheva, A.; Bayurova, E.; Gordeychuk, I.; Isaguliants, M.; Goptar, I.; Nikiforova, A.; Smirnov, V.; Volchkova, E.; Glebe, D.; et al. Replenishment of Hepatitis B Virus cccDNA Pool Is Restricted by Baseline Expression of Host Restriction Factors in Vitro. Microorganisms 2019, 7, 533. [Google Scholar] [CrossRef] [PubMed]
- Kostyushev, D.S.; Zueva, A.P.; Brezgin, S.A.; Lipatnikov, A.D.; Simirskii, V.N.; Glebe, D.; Volchkova, E.V.; Shipulin, G.A.; Chulanov, V.P. Overexpression of DNA-Methyltransferases in Persistency of cccDNA Pool in Chronic Hepatitis B. Ter. Arkhiv 2017, 89, 21–26. [Google Scholar] [CrossRef]
- Park, S.Y.; Jeong, M.S.; Han, C.W.; Yu, H.S.; Jang, S.B. Structural and Functional Insight into Proliferating Cell Nuclear Antigen. J. Microbiol. Biotechnol. 2016, 26, 637–647. [Google Scholar] [CrossRef]
- Stoimenov, I.; Lagerqvist, A. The PCNA Pseudogenes in the Human Genome. BMC Res. Notes 2012, 5, 87. [Google Scholar] [CrossRef]
- Feng, J.; Yang, G.; Liu, Y.; Gao, Y.; Zhao, M.; Bu, Y.; Yuan, H.; Yuan, Y.; Yun, H.; Sun, M.; et al. LncRNA PCNAP1 Modulates Hepatitis b Virus Replication and Enhances Tumor Growth of Liver Cancer. Theranostics 2019, 9, 5227–5245. [Google Scholar] [CrossRef]
- Sarfaraz, N.; Somarowthu, S.; Bouchard, M.J. The Interplay of Long Noncoding RNAs and Hepatitis B Virus. J. Med. Virol. 2023, 95, e28058. [Google Scholar] [CrossRef]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA Polymerase κ Is a Key Cellular Factor for the Formation of Covalently Closed Circular DNA of Hepatitis B Virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Sheraz, M.; McGrane, M.; Chang, J.; Guo, J.T. DNA Polymerase Alpha Is Essential for Intracellular Amplification of Hepatitis B Virus Covalently Closed Circular DNA. PLoS Pathog. 2019, 15, e1007742. [Google Scholar] [CrossRef]
- El-Serag, H.B. Epidemiology of Viral Hepatitis and Hepatocellular Carcinoma. Gastroenterology 2012, 142, 1264. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P. Reversibility of Hepatitis B Virus Cirrhosis after Therapy: Who and Why? Liver Int. 2015, 35, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.F. Prevention of Hepatitis B Virus-Related Hepatocellular Carcinoma. Gastroenterology 2004, 127, S303–S309. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Q.; Zeng, Z.; Wu, P.; Jiang, Y.; Luo, T.; Ji, X.; Zhang, Q.; Hao, Y.; Chen, L. Increased Expression of G9A Contributes to Carcinogenesis and Indicates Poor Prognosis in Hepatocellular Carcinoma. Oncol. Lett. 2018, 15, 9757–9765. [Google Scholar] [CrossRef]
- Scheer, S.; Zaph, C. The Lysine Methyltransferase G9a in Immune Cell Differentiation and Function. Front. Immunol. 2017, 8, 429. [Google Scholar] [CrossRef]
- Wei, L.; Chiu, D.K.C.; Tsang, F.H.C.; Law, C.T.; Cheng, C.L.H.; Au, S.L.K.; Lee, J.M.F.; Wong, C.C.L.; Ng, I.O.L.; Wong, C.M. Histone Methyltransferase G9a Promotes Liver Cancer Development by Epigenetic Silencing of Tumor Suppressor Gene RARRES3. J. Hepatol. 2017, 67, 758–769. [Google Scholar] [CrossRef]
- Jeng, K.S.; Jeng, C.J.; Jeng, W.J.; Chang, C.F.; Sheen, I.S. Role of C-X-C Chemokine Ligand 12/C-X-C Chemokine Receptor 4 in the Progression of Hepatocellular Carcinoma (Review). Oncol. Lett. 2017, 14, 1905–1910. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, W.; Ding, Y.; Guo, X.; Yuan, Y.; Li, D. CRISPR/Cas9-Mediated Genome Engineering of CXCR4 Decreases the Malignancy of Hepatocellular Carcinoma Cells In Vitro and In Vivo. Oncol. Rep. 2017, 37, 3565–3571. [Google Scholar] [CrossRef]
- He, J.; Zhang, W.; Li, A.; Chen, F.; Luo, R. Knockout of NCOA5 Impairs Proliferation and Migration of Hepatocellular Carcinoma Cells by Suppressing Epithelial-to-Mesenchymal Transition. Biochem. Biophys. Res. Commun. 2018, 500, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Iwagami, Y.; Huang, C.K.; Olsen, M.J.; Thomas, J.M.; Jang, G.; Kim, M.; Lin, Q.; Carlson, R.I.; Wagner, C.E.; Dong, X.; et al. Aspartate β-Hydroxylase Modulates Cellular Senescence through Glycogen Synthase Kinase 3β in Hepatocellular Carcinoma. Hepatology 2016, 63, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tang, J.; Cai, X.; Huang, Y.; Gao, Q.; Liang, L.; Tian, L.; Yang, Y.; Zheng, Y.; Hu, Y.; et al. HBx Mutations Promote Hepatoma Cell Migration through the Wnt/β-Catenin Signaling Pathway. Cancer Sci. 2016, 107, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Yao, M.; Fang, M.; Pan, L.; Wang, L.; Yang, J.; Dong, Z.; Yao, D. Oncogenic WNt3A: A Candidate Specific Marker and Novel Molecular Target for Hepatocellular Carcinoma. J. Cancer 2019, 10, 5862–5873. [Google Scholar] [CrossRef] [PubMed]
- Kawai-Kitahata, F.; Asahina, Y.; Tanaka, S.; Kakinuma, S.; Murakawa, M.; Nitta, S.; Watanabe, T.; Otani, S.; Taniguchi, M.; Goto, F.; et al. Comprehensive Analyses of Mutations and Hepatitis B Virus Integration in Hepatocellular Carcinoma with Clinicopathological Features. J. Gastroenterol. 2016, 51, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.H.; Huang, C.C.; Lin, P.R.; Chang, H.W.; Ger, L.P.; Lin, Y.W.; Changchien, C.S.; Lee, C.M.; Tai, M.H. Expression and Prognostic Role of Tumor Suppressor Gene PTEN/MMAC1/TEP1 in Hepatocellular Carcinoma. Cancer 2003, 97, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qi, X.; Zeng, Z.; Wang, L.; Wang, J.; Zhang, T.; Xu, Q.; Shen, C.; Zhou, G.; Yang, S.; et al. CRISPR/Cas9-Mediated P53 and Pten Dual Mutation Accelerates Hepatocarcinogenesis in Adult Hepatitis B Virus Transgenic Mice. Sci. Rep. 2017, 7, 2796. [Google Scholar] [CrossRef]
- Kim, C.M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx Gene of Hepatitis B Virus Induces Liver Cancer in Transgenic Mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef]
- Tang, H.; Oishi, N.; Kaneko, S.; Murakami, S. Molecular Functions and Biological Roles of Hepatitis B Virus x Protein. Cancer Sci. 2006, 97, 977–983. [Google Scholar] [CrossRef]
- Stengel, K.; Zheng, Y. Cdc42 in Oncogenic Transformation, Invasion, and Tumorigenesis. Cell. Signal. 2011, 23, 1415–1423. [Google Scholar] [CrossRef]
- Xu, Y.; Qi, Y.; Luo, J.; Yang, J.; Xie, Q.; Deng, C.; Su, N.; Wei, W.; Shi, D.; Xu, F.; et al. Hepatitis B Virus X Protein Stimulates Proliferation, Wound Closure and Inhibits Apoptosis of HuH-7 Cells via CDC42. Int. J. Mol. Sci. 2017, 18, 586. [Google Scholar] [CrossRef] [PubMed]
- Callegari, E.; Gramantieri, L.; Domenicali, M.; D’Abundo, L.; Sabbioni, S.; Negrini, M. MicroRNAs in Liver Cancer: A Model for Investigating Pathogenesis and Novel Therapeutic Approaches. Cell Death Differ. 2015, 22, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.J.; Deng, Y.L.; Liang, H.F.; Jaoude, J.C.; Liu, F.Y. Hepatitis B Virus X Protein Promotes CREB-Mediated Activation of miR-3188 and Notch Signaling in Hepatocellular Carcinoma. Cell Death Differ. 2017, 24, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Mueller, H.; Wildum, S.; Luangsay, S.; Walther, J.; Lopez, A.; Tropberger, P.; Ottaviani, G.; Lu, W.; Parrott, N.J.; Zhang, J.D.; et al. A Novel Orally Available Small Molecule That Inhibits Hepatitis B Virus Expression. J. Hepatol. 2018, 68, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhou, C.; Jiang, M.; Wang, Y.; Wang, J.; Cheng, Z.; Wang, M.; Liu, Y.; Liang, C.; Wang, J.; et al. Discovery of RG7834: The First-in-Class Selective and Orally Available Small Molecule Hepatitis B Virus Expression Inhibitor with Novel Mechanism of Action. J. Med. Chem. 2018, 61, 10619–10634. [Google Scholar] [CrossRef]
- Hyrina, A.; Jones, C.; Chen, D.; Clarkson, S.; Cochran, N.; Feucht, P.; Hoffman, G.; Lindeman, A.; Russ, C.; Sigoillot, F.; et al. A Genome-Wide CRISPR Screen Identifies ZCCHC14 as a Host Factor Required for Hepatitis B Surface Antigen Production. Cell Rep. 2019, 29, 2970–2978.e6. [Google Scholar] [CrossRef]
- Brechot, C.; Pourcel, C.; Louise, A.; Rain, B.; Tiollais, P. Presence of Integrated Hepatitis B Virus DNA Sequences in Cellular DNA of Human Hepatocellular Carcinoma. Nature 1980, 286, 533–535. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular Carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef]
- Ye, B.; Liu, X.; Li, X.; Kong, H.; Tian, L.; Chen, Y. T-Cell Exhaustion in Chronic Hepatitis B Infection: Current Knowledge and Clinical Significance. Cell Death Dis. 2015, 6, e1694. [Google Scholar] [CrossRef]
- Kuscu, C.; Parlak, M.; Tufan, T.; Yang, J.; Szlachta, K.; Wei, X.; Mammadov, R.; Adli, M. CRISPR-STOP: Gene Silencing through Base-Editing-Induced Nonsense Mutations. Nat. Methods 2017, 14, 710–712. [Google Scholar] [CrossRef]
- Billon, P.; Bryant, E.E.; Joseph, S.A.; Nambiar, T.S.; Hayward, S.B.; Rothstein, R.; Ciccia, A. CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol. Cell 2017, 67, 1068–1079.e4. [Google Scholar] [CrossRef] [PubMed]
- Preece, R.; Pavesi, A.; Gkazi, S.A.; Stegmann, K.A.; Georgiadis, C.; Tan, Z.M.; Aw, J.Y.J.; Maini, M.K.; Bertoletti, A.; Qasim, W. CRISPR-Mediated Base Conversion Allows Discriminatory Depletion of Endogenous T Cell Receptors for Enhanced Synthetic Immunity. Mol. Ther. Methods Clin. Dev. 2020, 19, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Bunse, T.; Kosinska, A.D.; Michler, T.; Protzer, U. PD-L1 Silencing in Liver Using siRNAs Enhances Efficacy of Therapeutic Vaccination for Chronic Hepatitis B. Biomolecules 2022, 12, 470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, N.; Zhang, P.; Li, F.; Yang, C.; Zhu, Q.; Han, Q.; Lv, Y.; Zhou, Z.; Liu, Z. PD-1 mRNA Expression Is Associated with Clinical and Viral Profile and PD1 3′-Untranslated Region Polymorphism in Patients with Chronic HBV Infection. Immunol. Lett. 2014, 162, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Qiang, R.; Lu, J.; Tuo, X.; Yang, X.; Li, X. Enhanced Antiviral Benefit of Combination Therapy with Anti-HBV and Anti-PD1 gRNA/Cas9 Produces a Synergistic Antiviral Effect in HBV Infection. Mol. Immunol. 2021, 130, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Hensel, K.O.; Rendon, J.C.; Navas, M.C.; Rots, M.G.; Postberg, J. Virus–Host Interplay in Hepatitis B Virus Infection and Epigenetic Treatment Strategies. FEBS J. 2017, 284, 3550–3572. [Google Scholar] [CrossRef] [PubMed]
- Laufer, B.I.; Singh, S.M. Strategies for Precision Modulation of Gene Expression by Epigenome Editing: An Overview. Epigenet. Chromatin 2015, 8, 34. [Google Scholar] [CrossRef]
- Belema, M.; Meanwell, N.A. Discovery of Daclatasvir, a Pan-Genotypic Hepatitis C Virus NS5A Replication Complex Inhibitor with Potent Clinical Effect. J. Med. Chem. 2014, 5057–5071. Available online: https://pubs.acs.org/doi/10.1021/jm500335h (accessed on 27 October 2023). [CrossRef]
- Cholongitas, E.; Papatheodoridis, G.V. Sofosbuvir: A Novel Oral Agent for Chronic Hepatitis C. Ann. Gastroenterol. 2014, 27, 331–337. [Google Scholar]
- Daclatasvir, Sofosbuvir with or without Ribavirin for 24 Weeks in Hepatitis C Genotype 3 Cirrhosis: A Real-Life Study. Available online: https://www.elsevier.es/en-revista-annals-hepatology-16-pdf-S1665268119300456 (accessed on 27 October 2023).
- Robertson, B.; Myers, G.; Howard, C.; Brettin, T.; Bukh, J.; Gaschen, B.; Gojobori, T.; Maertens, G.; Mizokami, M.; Nainan, O.; et al. Classification, Nomenclature, and Database Development for Hepatitis C Virus (HCV) and Related Viruses: Proposals for Standardization. Arch. Virol. 1998, 143, 2493–2503. [Google Scholar] [CrossRef]
- Dubuisson, J.; Cosset, F.L. Virology and Cell Biology of the Hepatitis C Virus Life Cycle—An Update. J. Hepatol. 2014, 61, S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Chang, K.M. Hepatitis C Virus: Virology and Life Cycle. Clin. Mol. Hepatol. 2013, 19, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Moriishi, K.; Matsuura, Y. Host Factors Involved in the Replication of Hepatitis C Virus. Rev. Med. Virol. 2007, 17, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.U.; Iman, K.; Khalid, M.F.; Salman, H.M.; Shafi, T.; Rafi, M.; Javaid, N.; Hussain, R.; Ahmad, F.; Shahzad-Ul-Hussan, S.; et al. Evolution of Efficacious Pangenotypic Hepatitis C Virus Therapies. Med. Res. Rev. 2019, 39, 1091–1136. [Google Scholar] [CrossRef]
- Liu, Z.; Mao, X.; Yu, K.; Suo, C.; Jin, L.; Zhang, T.; Chen, X. Prevalence of HCV Resistance-associated Substitutions among Treatment-failure Patients Receiving Direct-acting Antiviral Agents. J. Viral Hepat. 2020, 27, 585–592. [Google Scholar] [CrossRef]
- Powdrill, M.H.; Tchesnokov, E.P.; Kozak, R.A.; Russell, R.S.; Martin, R.; Svarovskaia, E.S.; Mo, H.; Kouyos, R.D.; Götte, M. Contribution of a Mutational Bias in Hepatitis C Virus Replication to the Genetic Barrier in the Development of Drug Resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 20509–20513. [Google Scholar] [CrossRef]
- Ashraf, M.U.; Salman, H.M.; Khalid, M.F.; Khan, M.H.F.; Anwar, S.; Afzal, S.; Idrees, M.; Chaudhary, S.U. CRISPR-Cas13a Mediated Targeting of Hepatitis C Virus Internal-Ribosomal Entry Site (IRES) as an Effective Antiviral Strategy. Biomed. Pharmacother. 2021, 136, 111239. [Google Scholar] [CrossRef]
- Price, A.A.; Sampson, T.R.; Ratner, H.K.; Grakoui, A.; Weiss, D.S. Cas9-Mediated Targeting of Viral RNA in Eukaryotic Cells. Proc. Natl. Acad. Sci. USA 2015, 112, 6164–6169. [Google Scholar] [CrossRef]
- Wang, J.; Qiao, L.; Hou, Z.; Luo, G. TIM-1 Promotes Hepatitis C Virus Cell Attachment and Infection. J. Virol. 2017, 91, 10-1128. [Google Scholar] [CrossRef]
- Jiang, J.; Wu, X.; Tang, H.; Luo, G. Apolipoprotein E Mediates Attachment of Clinical Hepatitis C Virus to Hepatocytes by Binding to Cell Surface Heparan Sulfate Proteoglycan Receptors. PLoS ONE 2013, 8, e67982. [Google Scholar] [CrossRef]
- Fan, H.; Qiao, L.; Kang, K.-D.; Fan, J.; Wei, W.; Luo, G. Attachment and Postattachment Receptors Important for Hepatitis C Virus Infection and Cell-to-Cell Transmission. J. Virol. 2017, 91, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic Dissection of Flaviviridae Host Factors through Genome-Scale CRISPR Screens. Nature 2016, 535, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Randall, G.; Higginbottom, A.; Monk, P.; Rice, C.M.; McKeating, J.A. CD81 Is Required for Hepatitis C Virus Glycoprotein-Mediated Viral Infection. J. Virol. 2004, 78, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, W.; Shen, L.; Turner, J.R.; Coyne, C.B.; Wang, T. Tight Junction Proteins Claudin-1 and Occludin Control Hepatitis C Virus Entry and Are Downregulated during Infection to Prevent Superinfection. J. Virol. 2009, 83, 2011–2014. [Google Scholar] [CrossRef]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; De Jong, Y.P.; Rice, C.M. Human Occludin Is a Hepatitis C Virus Entry Factor Required for Infection of Mouse Cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef]
- Shirasago, Y.; Shimizu, Y.; Tanida, I.; Suzuki, T.; Suzuki, R.; Sugiyama, K.; Wakita, T.; Hanada, K.; Yagi, K.; Kondoh, M.; et al. Occludin-Knockout Human Hepatic Huh7.5.1-8-Derived Cells Are Completely Resistant to Hepatitis C Virus Infection. Biol. Pharm. Bull. 2016, 39, 839–848. [Google Scholar] [CrossRef]
- Meertens, L.; Bertaux, C.; Cukierman, L.; Cormier, E.; Lavillette, D.; Cosset, F.-L.; Dragic, T. The Tight Junction Proteins Claudin-1, -6, and -9 Are Entry Cofactors for Hepatitis C Virus. J. Virol. 2008, 82, 3555–3560. [Google Scholar] [CrossRef]
- Hopcraft, S.E.; Evans, M.J. Selection of a Hepatitis C Virus with Altered Entry Factor Requirements Reveals a Genetic Interaction between the E1 Glycoprotein and Claudins. Hepatology 2015, 62, 1059–1069. [Google Scholar] [CrossRef]
- Kalemera, M.D.; Capella-Pujol, J.; Chumbe, A.; Underwood, A.; Bull, R.A.; Schinkel, J.; Sliepen, K.; Grove, J. Optimized Cell Systems for the Investigation of Hepatitis C Virus E1E2 Glycoproteins. J. Gen. Virol. 2020, 102, 001512. [Google Scholar] [CrossRef]
- Zhang, R.; Miner, J.J.; Gorman, M.J.; Rausch, K.; Ramage, H.; White, J.P.; Zuiani, A.; Zhang, P.; Fernandez, E.; Zhang, Q.; et al. A CRISPR Screen Defines a Signal Peptide Processing Pathway Required by Flaviviruses. Nature 2016, 535, 164–168. [Google Scholar] [CrossRef]
- Geller, R.; Taguwa, S.; Frydman, J. Broad Action of Hsp90 as a Host Chaperone Required for Viral Replication. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Kawakami, K.; Yokoe, H.; Yoshimura, K.; Matsuda, M.; Yasumoto, J.; Maekawa, S.; Yamashita, A.; Tanaka, T.; Ikeda, M.; et al. Involvement of FKBP6 in Hepatitis C Virus Replication. Sci. Rep. 2015, 5, 16699. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Fraser, P. No-Nonsense Functions for Long Noncoding RNAs. Cell 2011, 145, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Duan, X.; Holmes, J.A.; Li, W.; Lee, S.H.; Tu, Z.; Zhu, C.; Salloum, S.; Lidofsky, A.; Schaefer, E.A.; et al. A Long Noncoding RNA Regulates Hepatitis C Virus Infection Through Interferon Alpha–Inducible Protein 6. Hepatology 2019, 69, 1004–1019. [Google Scholar] [CrossRef] [PubMed]
- Shehab-Eldeen, S.; Essa, A.; Arafat, E.S.; Sleem, A.S.; Alhosary, A.A.; Darwish, E.; Essa, A.; Al-Omair, O.A.; Al-Khoufi, E.A.; Al Abdulqader, A.K.; et al. Serum LINC00152 and UCA1 in HCV-Induced Hepatocellular Carcinoma: Clinical Significance and Prognostic Value. Biol. Targets Ther. 2023, 17, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Bukong, T.N.; Momen-Heravi, F.; Kodys, K.; Bala, S.; Szabo, G. Exosomes from Hepatitis C Infected Patients Transmit HCV Infection and Contain Replication Competent Viral RNA in Complex with Ago2-miR122-HSP90. PLoS Pathog. 2014, 10, e1004424. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Abosmaha, E.; Coffin, C.S.; Labonté, P.; Bukong, T.N. Reticulon-3 Modulates the Incorporation of Replication Competent Hepatitis C Virus Molecules for Release inside Infectious Exosomes. PLoS ONE 2020, 15, e0239153. [Google Scholar] [CrossRef]
- Cheng, Y.; Sun, F.; Wang, L.; Gao, M.; Xie, Y.; Sun, Y.; Liu, H.; Yuan, Y.; Yi, W.; Huang, Z.; et al. Virus-Induced P38 MAPK Activation Facilitates Viral Infection. Theranostics 2020, 10, 12223–12240. [Google Scholar] [CrossRef]
- Liu, D.; Ndongwe, T.P.; Puray-Chavez, M.; Casey, M.C.; Izumi, T.; Pathak, V.K.; Tedbury, P.R.; Sarafianos, S.G. Effect of P-body Component Mov10 on HCV Virus Production and Infectivity. FASEB J. 2020, 34, 9433–9449. [Google Scholar] [CrossRef]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient Genome Modification by CRISPR-Cas9 Nickase with Minimal off-Target Effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef]
- Domingues, P.; Bamford, C.G.G.; Boutell, C.; McLauchlan, J. Inhibition of Hepatitis C Virus RNA Replication by ISG15 Does Not Require Its Conjugation to Protein Substrates by the HERC5 E3 Ligase. J. Gen. Virol. 2015, 96, 3236–3242. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, S.; Takeuchi, K.; Chihara, K.; Honjoh, C.; Kato, Y.; Yoshiki, H.; Hotta, H.; Sada, K. STAT1 Is Essential for the Inhibition of Hepatitis C Virus Replication by Interferon-λ but Not by Interferon-α. Sci. Rep. 2016, 6, 38336. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 Mutations and Hepatocellular Carcinoma: Insights into the Etiology and Pathogenesis of Liver Cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef] [PubMed]
- Narbus, C.M.; Israelow, B.; Sourisseau, M.; Michta, M.L.; Hopcraft, S.E.; Zeiner, G.M.; Evans, M.J. HepG2 Cells Expressing MicroRNA miR-122 Support the Entire Hepatitis C Virus Life Cycle. J. Virol. 2011, 85, 12087–12092. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.K.; Midkiff, B.R.; Israelow, B.; Evans, M.J.; Lanford, R.E.; Walker, C.M.; Lemon, S.M.; McGiverna, D.R. Hepatitis C Virus Indirectly Disrupts DNA Damage-Induced P53 Responses by Activating Protein Kinase R. mBio 2017, 8, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.A.; Berry-Wynne, K.M.; Dos Santos, T.; Addison, W.R.; McFarlane, N.; Hobman, T.; Tyrrell, D.L. HCV and Flaviviruses Hijack Cellular Mechanisms for Nuclear STAT2 Degradation: Up-Regulation of PDLIM2 Suppresses the Innate Immune Response. PLoS Pathog. 2019, 15, e1007949. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Li, S.; Holmes, J.A.; Tu, Z.; Li, Y.; Cai, D.; Liu, X.; Li, W.; Yang, C.; Jiao, B.; et al. MicroRNA 130a Regulates Both Hepatitis C Virus and Hepatitis B Virus Replication through a Central Metabolic Pathway. J. Virol. 2018, 92, 2009–2026. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.M.; Steitz, J.A. HuR and mRNA Stability. Cell. Mol. Life Sci. 2001, 58, 266–277. [Google Scholar] [CrossRef]
- Averhoff, F.M.; Khudyakov, Y.; Nelson, N.P. Hepatitis A Vaccines. In Plotkin’s Vaccines; Elsevier: Amsterdam, The Netherlands, 2018; pp. 319–341.e15. [Google Scholar]
- Lemon, S.M.; Robertson, B.H. Taxonomic Classification of Hepatitis A Virus. In Viral Hepatitis and Liver Disease; Springer Japan: Tokyo, Japan, 1994; pp. 50–53. [Google Scholar]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A Pathogenic Picornavirus Acquires an Envelope by Hijacking Cellular Membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef]
- Stuart, D.I.; Ren, J.; Wang, X.; Rao, Z.; Fry, E.E. Hepatitis a Virus Capsid Structure. Cold Spring Harb. Perspect. Med. 2019, 9, a031807. [Google Scholar] [CrossRef]
- McKnight, K.L.; Lemon, S.M. Hepatitis A Virus Genome Organization and Replication Strategy. Cold Spring Harb. Perspect. Med. 2018, 8, a033480. [Google Scholar] [CrossRef] [PubMed]
- Costafreda, M.I.; Kaplan, G. HAVCR1 (CD365) and Its Mouse Ortholog Are Functional Hepatitis A Virus (HAV) Cellular Receptors That Mediate HAV Infection. J. Virol. 2018, 92, e02065-17. [Google Scholar] [CrossRef] [PubMed]
- Costafreda, M.I.; Abbasi, A.; Lu, H.; Kaplan, G. Exosome Mimicry by a HAVCR1–NPC1 Pathway of Endosomal Fusion Mediates Hepatitis A Virus Infection. Nat. Microbiol. 2020, 5, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Su, H.-L.; Liao, C.-L.; Lin, Y.-L. Japanese Encephalitis Virus Infection Initiates Endoplasmic Reticulum Stress and an Unfolded Protein Response. J. Virol. 2002, 76, 4162–4171. [Google Scholar] [CrossRef]
- Tardif, K.D.; Waris, G.; Siddiqui, A. Hepatitis C Virus, ER Stress, and Oxidative Stress. Trends Microbiol. 2005, 13, 159–163. [Google Scholar] [CrossRef] [PubMed]
- He, B. Viruses, Endoplasmic Reticulum Stress, and Interferon Responses. Cell Death Differ. 2006, 13, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Kanda, T.; Haga, Y.; Sasaki, R.; Nakamura, M.; Wu, S.; Nakamoto, S.; Shirasawa, H.; Okamoto, H.; Yokosuka, O. Glucose-Regulated Protein 78 Is an Antiviral against Hepatitis a Virus Replication. Exp. Ther. Med. 2017, 13, 3305–3308. [Google Scholar] [CrossRef]
- Zhu, F.C.; Zhang, J.; Zhang, X.F.; Zhou, C.; Wang, Z.Z.; Huang, S.J.; Wang, H.; Yang, C.L.; Jiang, H.M.; Cai, J.P.; et al. Efficacy and Safety of a Recombinant Hepatitis e Vaccine in Healthy Adults: A Large-Scale, Randomised, Double-Blind Placebo-Controlled, Phase 3 Trial. Lancet 2010, 376, 895–902. [Google Scholar] [CrossRef]
- Doceul, V.; Bagdassarian, E.; Demange, A.; Pavio, N. Zoonotic Hepatitis E Virus: Classification, Animal Reservoirs and Transmission Routes. Viruses 2016, 8, 270. [Google Scholar] [CrossRef]
- Kenney, S.P.; Meng, X.J. Hepatitis E Virus Genome Structure and Replication Strategy. Cold Spring Harb. Perspect. Med. 2019, 9, a031724. [Google Scholar] [CrossRef]
- Barriocanal, M.; Carnero, E.; Segura, V.; Fortes, P. Long Non-Coding RNA BST2/BISPR Is Induced by IFN and Regulates the Expression of the Antiviral Factor Tetherin. Front. Immunol. 2014, 5, 655. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, J.; Jia, X.; McNatt, M.W.; Zang, T.; Pan, B.; Meng, W.; Wang, H.W.; Bieniasz, P.D.; Xiong, Y. Structural Insight into the Mechanisms of Enveloped Virus Tethering by Tetherin. Proc. Natl. Acad. Sci. USA 2010, 107, 18428–18432. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, D.; Joshi, P.; Panda, S.K. Hepatitis E Virus (HEV) Egress: Role of BST2 (Tetherin) and Interferon Induced Long Noncoding RNA (lncRNA) BISPR. PLoS ONE 2017, 12, e0187334. [Google Scholar] [CrossRef] [PubMed]
- ViralZone: A Knowledge Resource to Understand Virus Diversity|Nucleic Acids Research|’Oxford Academic. Available online: https://academic.oup.com/nar/article/39/suppl_1/D576/2508477?login=true (accessed on 14 December 2023).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartosh, U.I.; Dome, A.S.; Zhukova, N.V.; Karitskaya, P.E.; Stepanov, G.A. CRISPR/Cas9 as a New Antiviral Strategy for Treating Hepatitis Viral Infections. Int. J. Mol. Sci. 2024, 25, 334. https://doi.org/10.3390/ijms25010334
Bartosh UI, Dome AS, Zhukova NV, Karitskaya PE, Stepanov GA. CRISPR/Cas9 as a New Antiviral Strategy for Treating Hepatitis Viral Infections. International Journal of Molecular Sciences. 2024; 25(1):334. https://doi.org/10.3390/ijms25010334
Chicago/Turabian StyleBartosh, Ulyana I., Anton S. Dome, Natalya V. Zhukova, Polina E. Karitskaya, and Grigory A. Stepanov. 2024. "CRISPR/Cas9 as a New Antiviral Strategy for Treating Hepatitis Viral Infections" International Journal of Molecular Sciences 25, no. 1: 334. https://doi.org/10.3390/ijms25010334
APA StyleBartosh, U. I., Dome, A. S., Zhukova, N. V., Karitskaya, P. E., & Stepanov, G. A. (2024). CRISPR/Cas9 as a New Antiviral Strategy for Treating Hepatitis Viral Infections. International Journal of Molecular Sciences, 25(1), 334. https://doi.org/10.3390/ijms25010334