Abstract
Abnormal activation of receptor tyrosine kinases (RTKs) contributes to tumorigenesis, while protein tyrosine phosphatases (PTPs) contribute to tumor control. One of the most representative PTPs is Src homology region 2 (SH2) domain-containing phosphatase 1 (SHP-1), which is associated with either an increased or decreased survival rate depending on the cancer type. Hypermethylation in the promoter region of PTPN6, the gene for the SHP-1 protein, is a representative epigenetic regulation mechanism that suppresses the expression of SHP-1 in tumor cells. SHP-1 comprises two SH2 domains (N-SH2 and C-SH2) and a catalytic PTP domain. Intramolecular interactions between the N-SH2 and PTP domains inhibit SHP-1 activity. Opening of the PTP domain by a conformational change in SHP-1 increases enzymatic activity and contributes to a tumor control phenotype by inhibiting the activation of the Janus kinase/signal transducer and activator of transcription (JAK/STAT3) pathway. Although various compounds that increase SHP-1 activation or expression have been proposed as tumor therapeutics, except sorafenib and its derivatives, few candidates have demonstrated clinical significance. In some cancers, SHP-1 expression and activation contribute to a tumorigenic phenotype by inducing a tumor-friendly microenvironment. Therefore, developing anticancer drugs targeting SHP-1 must consider the effect of SHP-1 on both cell biological mechanisms of SHP-1 in tumor cells and the tumor microenvironment according to the target cancer type. Furthermore, the use of combination therapies should be considered.
1. Introduction
Protein tyrosine kinases (RTKs), particularly receptor tyrosine kinases, are well known for their contribution to tumorigenesis. RTKs mediate intercellular communication, controlling a wide range of biological functions, including cell division, cell motility, and differentiation. Mutations responsible for abnormal activation of genes encoding RTKs, such as EGFR, HER2/ErbB2, and MET, have been identified in various cancer types [1]. Many studies have been conducted on how they contribute to cancer development, and the RTKs have been proposed as targets for developing tumor therapies [2,3]. RTKs are activated by receptor-specific ligands, such as growth factors, leading to receptor dimerization or oligomerization. Dimerization induces activation of the intracellular tyrosine kinase domain through trans-autophosphorylation.
In contrast to RTKs, phosphatases are enzymes that dephosphorylate substrate proteins, and among them, protein tyrosine phosphatase (PTP) is thought to inhibit RTK function by dephosphorylating proteins phosphorylated by RTK. However, not all PTPs inhibit PTK-mediated tumorigenesis, and PTPs that contribute to tumorigenesis were recently reported [4,5,6]. Among the PTPs, Src homology region 2 (SH2) domain-containing phosphatase 1 (SHP-1) is the first known SHP and is involved in cell cycle control, cancer cell migration and invasion, and apoptosis induction. In this review, we summarize the regulation of SHP-1 expression, the relationship between SHP-1 expression and tumors, the regulation of SHP-1-mediated cell signaling, the function of SHP-1 in the tumor microenvironment, and the use of SHP-1 in the development of tumor therapeutics.
2. Epigenetic Regulation of SHP-1 Expression
PTPN6 encodes SHP-1, a nonreceptor tyrosine phosphatase. PTPN6 mutations have been reported in several cancers; the total incidence of such mutations is 2.7% (uterine carcinosarcoma, 7.01%; testicular germ cell carcinoma, 6.04%; ovarian cancer, 5.82%; and melanoma, 5.18%). Phosphatase mutations occur most frequently (42.27%); however, there are few reports on their correlation with the SHP-1 function, which is scarce [7]. Two proteins of different sizes can be synthesized from different translation initiation codons located in exon 1 and of the PTPN6 gene, located on human chromosome 12p13 [8,9]. The two proteins show similar enzymatic activity [8,10]. Promoter 1, located upstream of exon 1, is activated in epithelial-origin cells [11], and promoter 2, located upstream of exon 2, is activated in hematopoietic cells [12]. Promoter 1 is activated by various transcription factors, including NFκB, upstream stimulatory factor 1 (USF-1), or NF-Y [11,13]; promoter 2 can be activated by NFκB p65 and PU.1 in hematopoietic cells (Figure 1A) [14,15].
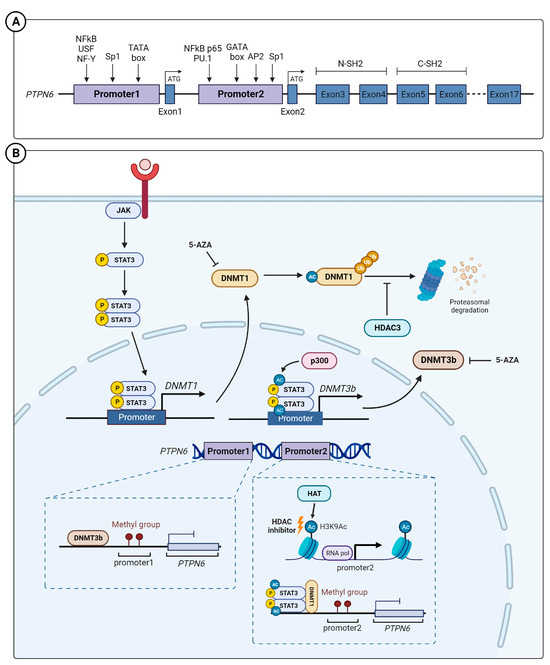
Figure 1.
PTPN6 gene structure and regulation of SHP-1 expression. (A) Schematic diagram of the SHP-1 gene (PTPN6). The PTPN6 has 17 exons; promoters 1 and promoter 2 are located in exon 1 and exon 2, respectively. Promoter 1 and 2 are mainly active in epithelial and hematopoietic cells, respectively. (B) The epigenetic regulatory mechanism regulating SHP-1 expression. JAK induces phosphorylation of STAT3 (pSTAT3), and the pSTAT3 forms a dimer and induces the transcription of DNMT3B.The methylation of the SHP-1 promoter region via DNMT and H3L9 acetylation (Ac) via histone acetyltransferases can down- and upregulate SHP-1 expression, respectively. USF—upstream stimulatory factor; DNMT—DNA methyltransferases; HDAC—histone deacetylase; 5-AZA—5-Aza-2′-deoxycytidine.
Epigenetic silencing affects SHP-1 expression in tumor cells. DNA methylation is a crucial epigenetic mechanism in regulating gene expression, with reports that hypomethylated DNA is associated with tumorigenesis and tumor development [16]. Reports suggest the transcriptional repression of the PTNP6 promoter by hypermethylated CpG islands during the regulation of SHP-1 expression, which shows tumor suppressor activity in hematological malignancies [17,18,19], esophageal squamous cell carcinoma [20], and gastric adenocarcinoma [21,22]. To date, three types of DNA methyltransferases (DNMT) have been identified: DNMT1, DNMT2, and DNMT3a/b. [23]. Among them, DNMT1 controls SHP-1 expression by inducing aberrant methylation on promoter 2 of PTPN6 in chronic myelogenous leukemia cells [24]. SHP-1 expression has an inverse relationship with DNMT1 and STAT3; its expression decreases when the activation of DNMT1 and STAT3 in tumor cells increases [25]. This is because activated STAT3 induces DNMT1 expression [26]. STAT3-DNMT1 interaction [27], which requires STAT3 acetylation [28], mediates DNMT1-mediated epigenetic gene silencing [28]. Therefore, the activated STAT3 inhibits SHP-1 expression via DNMT1 [27]. In carcinoma-associated fibroblasts (CAFs), which mediate the initiation of a pro-invasive tumor microenvironment, p300-histone acetyltransferase acetylates STAT3, which in turn upregulates and activates DNMT3b DNA methyltransferase. DNMT3b represses SHP-1 expression via CpG motif methylation of the PTPN6 promoter. Consistently, in human lung and head and neck carcinoma, STAT3 acetylation and phosphorylation are inversely correlated with SHP-1 expression [29].
In addition to DNA methylation, histone acetylation is another representative epigenetic gene expression regulation mechanism. In a cohort of 37 patients with diffuse large B-cell lymphoma (DLBL), hypermethylation of the P2 promoter of PTPN6 was only observed in 57% of patients. When treated with a DNA methyltransferase inhibitor (5-aza-deoxycytidine) and histone deacetylase (HDAC) inhibitor (LBH589), the inhibition of PTPN6 expression in DLBL cells was reversed. LBH589 induces SHP-1 expression by increasing the H3K9Ac mark within the PTPN6 P2 promoter [30]. Although LBH589 induces SHP-1 expression in chronic myeloid leukemia, HDAC does not directly combine with the PTPN6 promoter [31]. HDAC3 and DNMT1 expression are increased in hypertrophy cell models and high-fat diet rat models. It was reported that HDAC3-mediated DNMT1 deacetylation causes an increase in DNMT1 stability. Therefore, an increase in DNMT1 suppresses SHP-1 expression (Figure 1B) [32]. Although promoter methylation is a relatively well-established epigenetic gene expression regulation mechanism of SHP-1, more precise mechanisms for HDAC-mediated regulation need to be investigated.
3. SHP-1 Expression in Tumors
SHP-1 expression may be regarded as a prognostic marker associated with decreased and increased tumor pathological symptoms. The association between SHP-1 expression and patient survival differs based on tumor type (Table 1). High levels of SHP-1 expression are related to high survival rates in patients with tumors, including breast cancer, esophageal squamous cell carcinoma, hepatocellular carcinoma, and prostate cancer [20,33,34,35,36,37]. Additionally, there is a report on the antitumor function of SHP-1 in gastric cancer, although it is not associated with SHP-1 expression [38]. The expression of colon cancer-associated transcript (CCAT5) was upregulated in ascites-derived gastric cancer cells, and increased expression of CCAT5 was found to be associated with poor patient prognosis. CCAT5 binds to the C-end domain of STAT3 and inhibits STAT3Y705 dephosphorylation mediated by SHP-1, thereby inducing STAT3 nuclear entry and metastatic activation, which promotes gastric cancer progression. In contrast, few reports suggest that SHP-1 expression and tumor patient survival are inversely correlated in patients with acute myeloid leukemia, colorectal cancer, and glioblastoma [39,40,41]. Therefore, although SHP-1 is generally regarded as a phosphatase with a tumor suppressor function, its role in tumor prognosis is probably dependent on the cancer type. Although the mechanism of STAT3 suppression by SHP-1 as a tumor suppressor mechanism is well-established, the SHP-1-mediated tumor-friendly molecular mechanism is not.

Table 1.
Clinicopathological role of SHP-1 in tumors.
4. The Function of SHP-1 and Tumors
4.1. SHP-1 Structure and Its Activity
SHP-1 comprises two N-terminal SH2 domains (N-SH2 and C-SH2), a PTP domain, and a C-terminal tail containing several phosphorylation sites and a nuclear localization signal [42]. A structural study on SHP-1 lacking the C-terminal tail revealed that the intramolecular interaction of the N-SH2 domain with the PTP domain forms an autoinhibitory form of SHP-1, preventing the N-SH2 domain from exposing Cys455 of the active site and blocking substrate access to the active site. This autoinhibitory form is further stabilized by hydrogen bonding and salt bridge formation between the N-SH2 domain and PTP domain residues. The C-SH2 domain has been proposed as flexible and mobile and might play a role in sensing phosphopeptides, thereby weakening the autoinhibitory interaction between the SH2 and PTP domains (Figure 2A) [43].
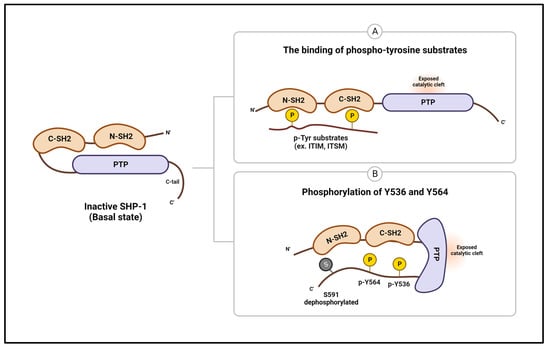
Figure 2.
Protein structures and regulation of SHP-1. The enzymatic activity of SHP-1 is inhibited by intramolecular interaction between the N-SH2 and PTP domains. (A) The binding of the SH2 domain by tyrosine-phosphorylated substrates and (B) phosphorylation of tyrosine residues in the C-terminal tail causes a conformational change that opens the phosphatase active site and contributes to phosphatase activation. ITIM—immunoreceptor tyrosine-based inhibitory motif; ITSM—immunoreceptor tyrosine-based switch motif.
Furthermore, a recent study elucidating the structure of the full-length and active forms of SHP-1 has shown that when the two SH2 domains bind to a ligand, the flexible C-SH2 domain rotates, causing the N-SH2 domain to rearrange and detach from the ligand’s active site. In addition, newly identified interactions between the N-SH2 and PTP domains and between the two SH2 domains further stabilized the open conformation of SHP-1 [44]. SHP-1 activity can also be regulated by phosphorylation, and three phosphorylation sites have been discovered to date: Tyr536, Tyr564, and Ser591. Tyr536 and Tyr564 are phosphorylated by Src family kinases, leading to increased SHP-1 activity. Tyr536 phosphorylation increases SHP-1 activity by inducing interaction with the N-SH2 domain and inhibiting interaction with the PTPase domain. On the other hand, Tyr564 phosphorylation indirectly increases PTPase activity by binding to the C-SH2 domain [45]. Additionally, upon stimulation by cellular activation signals, protein kinase C phosphorylates C-terminal Ser591 of SHP-1, thereby inhibiting its phosphatase activity (Figure 2B) [46,47]. Thus, additional biochemical and molecular biology studies are necessary to clarify how the C-terminal tail regulates SHP-1 activity and function and how it interacts with the other three domains.
4.2. Antitumor Activity of SHP-1
Tyrosine phosphorylation of proteins is a reversible posttranslational modification regulated by tyrosine kinases and PTPs. A common feature of cancer progression is the abnormal activation of tyrosine kinases due to an imbalance between phosphorylation and dephosphorylation. The Janus kinase (JAK)/signal transducer and activator of transcription (STAT3) pathway is a representative tyrosine phosphorylation-mediated oncogenic signaling pathway.
STAT3 forms a dimer by phosphorylating tyrosine residues (Tyr705), and the dimerized STAT3 moves to the nucleus, where it acts as a transcription factor [48,49,50,51]. Unregulated activation of STAT3 occurs in various cancers and contributes to tumorigenesis [52,53]. As upstream tyrosine kinases of STAT3, JAKs transmit the signals from receptors/ligand interactions to STAT3. Various ligands, such as IL-6, IFNs, PDGF, TGF, IGF, and EGF, activate STAT3 via their receptor-associated JAK activation [54,55,56,57,58,59,60]. The IL6/JAK2/STAT3 signaling axis is best studied in the context of tumor metastasis and tumor pathology since it induces epithelial–mesenchymal transition (EMT), resulting in tumor metastasis by promoting the expression of EMT-inducing transcription factors, such as Snail, ZEB1, JunB, and Twist1 [51,61,62,63,64]. Therefore, regulating the JAK/STAT3 signaling axis may be an effective target in tumor therapy. SHP-1 is a representative tyrosine phosphatase associated with tumor suppression and is a negative regulator of receptor-related signaling of three families: growth factor receptors with tyrosine kinase activity [65,66,67], cytokine receptors [60,68,69] and receptors involved in the immune response [70,71,72,73]. Generally, SHP-1-mediated inhibition of the JAK/STAT3 pathway is inversely correlated with tumor progression, aggressiveness, and metastasis [74,75,76]. SHP-1 binding to erythropoietin receptor (EPOR) inhibits erythropoietin (EPO)-mediated cell proliferation by inducing JAK2 dephosphorylation [69,77]. Furthermore, SHP-1 acts to dephosphorylate JAK1 in the IFN-α receptor signaling pathway [68]. SHP-1 silences the JAK/STAT pathway by inducing the dephosphorylation of both JAK and STAT3, and the loss of SHP-1 expression enhances JAK/STAT3 signaling in large cell lymphoma [78].
SHP-1 was reported to exert tyrosine phosphatase activity that directly downregulated pSTAT3 (Tyr705) and was a potent inhibitor of EMT in HCC and colorectal cancer (CRC) (Figure 3) [79,80]. Studies on several small molecules showing anticancer efficacy more clearly suggested SHP-1-mediated inhibition of the JAK/STAT3 signaling axis. Treatment with small molecules, such as 1′-acetoxychavicol acetate (ACA) [81], plumbagin [82], and allicin [83], significantly inhibited STAT3 through induction of SHP-1 in several types of cancer cells, including breast cancer, gastric cancer, and cholangiocarcinoma.
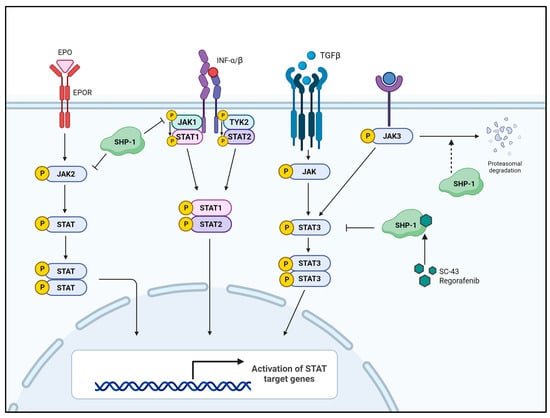
Figure 3.
SHP-1-mediated inhibition of the JAK/STAT signaling pathway. Several growth factors and cytokines activate their associated receptors, which, in turn, activate JAK. Activated JAK then activates STAT through phosphorylation and moves the activated STAT (p-STAT) to the nucleus, upregulating the expression of STAT-related genes. SHP-1 directly dephosphorylates STAT3 or its upstream JAKs, thereby inhibiting cell proliferation, survival, migration, and invasion. EPO—erythropoietin; EPOR—erythropoietin receptor; TYK2—tyrosine kinase 2.
4.3. Association of SHP-1 with Tumorigenesis
The molecular mechanisms underlying the function of SHP-1 as a protein associated with tumorigenesis are not as well understood as those underlying the function of SHP-1 as a tumor suppressor. However, SHP-1 has been suggested to be associated with pro-tumorigenesis in some cancers. P53 inhibits SHP-1 expression, which reduces the Inhibition of SHP-1 expression by p53 reduces the proliferation of breast cancer cells by inducing trkA-Tyr674/Tyr675 phosphorylation [84]. Altered SHP-1 expression leads to changes in some components of the cell cycle. In ovarian cancer, where SHP-1 expression levels are high, inhibiting SHP-1 expression gradually reduces tumor growth by increasing the intracellular levels of Cdk2/p27 Kip1 and Cdk2/SHP-1 complex [85], which is opposite to the mechanism used by SHP-1 to inhibit cell division.
Additionally, SHP-1 deficiency in prostate cancer resulted in p27 accumulation, CDK6 reduction, retinoblastoma protein hypophosphorylation, cyclin E-CDK2 inhibition, and cycle arrest in phase G1 [86]. A common cause of radiotherapy failure in many tumors is radiation resistance, and the degree of radiosensitivity varies among tumor cells. SHP-1 has been found to reduce radiosensitization, and SHP-1 overexpression in the nasopharyngeal carcinoma cell line CNE-2 caused radiation resistance, which, in turn, reduced apoptosis by enhancing DNA double-strand break repair and increasing cell cycle arrest in phase S [87,88,89]. The molecular mechanisms underlying the double-edged sword of SHP-1’s effect on tumorigenesis remain poorly understood, suggesting differential protein expression pools in various cancers and differences in oncogenic signaling.
4.4. SHP-1-Related Small Molecules for Tumor Therapy
SHP-1 is a candidate molecular target for anticancer drug development because it regulates tumor growth and progression by downregulating JAK/STAT3 signaling. Natural products have recently received considerable attention regarding their potential as antitumor drugs, and several small molecules that inhibit STAT3 activity by inducing SHP-1 have been proposed as anticancer drug candidates (Table 2). To date, these small molecules have only been observed to have limited effects at the cellular level or in animal models. The Food and Drug Administration has approved two SHP-1-related anticancer drugs: sorafenib and regorafenib (Figure 4). Sorafenib is a multikinase inhibitor that promotes apoptosis by targeting STAT3 signaling in a variety of carcinomas, including pancreatic cancer and glioblastoma [90,91]. Sorafenib also increases the enzymatic activity of SHP-1 through direct interaction between the N-SH2 and the PTP domains in HCC cells, thereby downregulating STAT3 activity. Among the sorafenib derivatives, SC-43 and SC-40 were reported as more potent SHP-1 agonists than sorafenib and showed therapeutic potential for HCC treatment [92]. Additionally, SC-43 was confirmed to act as an SHP-1 agonist in cholangiocarcinoma [93], CRC [94], and breast cancer [95]. SC-60, another sorafenib derivative, also had an anticancer effect by increasing SHP-1 activity in HCC and triple-negative breast cancer (TNBC) [93,96,97]. Regorafenib is a multiple protein kinases inhibitor that is very similar to sorafenib, and it enhances SHP-1 activity in HCC and CRC to promote apoptosis by inhibiting STAT3 signaling [98,99]. SC-78, a derivative of regorafenib, also inhibits tumor growth and metastasis in TNBC by interfering with the paracrine and autocrine pathways of VEGF-A through the SHP-1/STAT3 signaling axis [100].
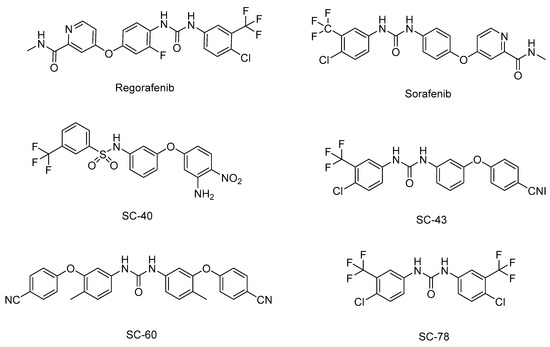
Figure 4.
Chemical structures of SHP-1 agonists, regorafenib, and sorafenib and their derivatives.
Although SHP-1 is generally known as a tumor suppressor, its expression is upregulated in some high-grade breast cancers [101] and ovarian cancers [102]. Substantial inhibitors of SHP-1 phosphatase activity have been developed and are undergoing preclinical and clinical studies at present, including NSC-87877, sodium stibogluconate (SSG), tyrosine phosphatase inhibitor 1, and suramin, but only a few have shown antitumor activity in experimental tumor models [93]. SSG has undergone Phase I trials for both malignant melanoma (NCT00498979) and advanced malignancies (NCT00629200), but no significant effect on tumor development was reported [103,104].
Table 2.
Natural compound-mediated STAT3 inhibition through SHP-1 induction.
Table 2.
Natural compound-mediated STAT3 inhibition through SHP-1 induction.
| Compound | Source | Cancer Type | Remarks | Ref |
|---|---|---|---|---|
| Guggulsterone | Commiphora mukul | Myeloma | Guggulsterone suppressed the expression of STAT3-associated antiapoptotic gene products and enhanced the anticancer effects of bortezomib. | [105,106] |
| Morin | Moraceae family | Myeloma | The number and position of hydroxyl groups in the B ring of flavonols are important inhibitors of STAT3 activation. | [107] |
| Genipin | Gardenia | Myeloma | Genipin effectively enhanced the cytotoxic effects of anticancer drugs such as bortezomib, thalidomide, and paclitaxel. | [108] |
| Capillarisin (CPS) | Artemisia capillaries | Myeloma | CPS induced cell cycle arrest in the sub-G1 phase and enhanced the anticancer effects of bortezomib. | [109] |
| Ergosterol peroxide (EP) | Ganoderma lucidum | Myeloma | EP inhibited the growth of U266 cells inoculated into female BALB/c mice and effectively reduced STAT3 activity and CD34 expression. | [110] |
| Icariside II | Epimedium koreanum | Myeloid leukemia | SHP-1 inhibition using siRNA significantly blocked icariside II-induced STAT3 inactivation and apoptosis in U937 cells. | [111] |
| Honokiol (HNK) | Magnolia officinalis | Myeloid leukemia | HNK induces the expression of SHP-1 by increasing the expression of its related transcription factor, PU.1. | [112] |
| Zerumbone | Zingiber zerumbet | Renal cell carcinoma | Zerumbone inhibited growth of human RCC xenograft tumors and STAT3 activation in athymic nu/nu mice. | [113,114] |
| α-mangostin (α-MGT) | Mangosteen | Hepatocellular carcinoma | α-MGT exhibited anti-HCC effects by inhibiting SHP-1 degradation induced by the ubiquitin–proteasome pathway. | [115] |
| Emodin | Rheum palmatum | Hepato cellular carcinoma (HCC) | Emodin inhibited human HCC orthotopic tumor growth and STAT3 activation in athymic male nu/nu mice. | [116] |
| Plumbagin | Plumbago zeylanica | Gastric cancer | Plumbagin not only induced apoptosis but also inhibited gastric cancer cell proliferation, migration, and invasion. | [82] |
| Allicin | Garlic | Cholangiocarcinoma(CCA) | Allicin inhibited CCA cell migration, invasion, and EMT and induced cell death. It also attenuated CCA tumor growth in a nude mouse model. | [83] |
| 1′-acetoxychavicol acetate (ACA) | Languas galanga | Breast cancer | ACA potently inhibited osteolysis in a mouse breast cancer skeletal metastasis model through the SHP-1/STAT3/MMPs signaling pathway. | [81] |
| Pectolinarigenin | Cirsium chanroenicum | Osteosarcoma | Pectolinarigenin interfered with the STAT3/DNMT1/HDAC1 complex formation at the SHP-1 promoter. | [117] |
4.5. The Function of SHP-1 and SHP-2 in the Tumor Microenvironment
SHP-1 affects not only tumor cells but also the tumor microenvironment. From the perspective of tumor treatment, the antitumor effect of SHP-1 on tumor cells is expected, while in the tumor immune environment, the function of SHP-1 is generally tumor-friendly. SHP-1 is expressed in all mature hematopoietic lineages. Notably, its regulatory mechanism related to T-cell activation is thought to be closely related to tumor targeting activity and tumor treatment in the tumor microenvironment. SHP-1 limits antigen-specific T-cell activation by dephosphorylating the T-cell receptor (TCR) ζ chain or downstream adapter proteins, such as lymphocyte-cell-specific protein-tyrosine kinase (Lck), ZAP70, Vav family proteins, and PI3K [42,118]. CD8+ T-cells from SHP-1 deficient motheaten mice [119] showed more stable and longer-lasting immunological synapses with antigen-presenting cells (APC), which reduced the T-cell activation threshold, thereby increasing the activation of T cells with low antigen specificity, leading to effective tumor suppression through tumor-specific effector T cells [120,121]. Knockout of SHP-1 in CD133 chimeric antigen receptor (CAR) T-cells significantly enhanced the cytolytic effect on CD133+ glioma cell lines by CAR T-cells and increased secretion of TNF-α, IL-2 and IFN-γ [122]. CAR T-cells currently approved by the US FDA possess a TCR-derived ζ chain as an intracellular activation domain in addition to a co-stimulatory (4-1BB or CD28) domain [123]. CARs containing CD3δ, CD3ε, or CD3γ cytoplasmic tails have outperformed conventional ζ CAR T-cells in vivo. Making CARs mutated to phenylalanine on the intracellular domain N-terminal tyrosine of CD3γ and CD3δ will only phosphorylate at the C-terminal tyrosine (BBγFY and BBδFY), and SHP-1 will preferentially bind to CARs containing that single phosphorylated BBδ, fine-tuning and balancing T-cell activation to prevent exhaustion and dysfunction [124]. SHP-1 is strongly activated on CD8+ nonlytic tumor-infiltrating lymphocytes (TILs), co-localizes with Lck on nonlytic TILs, and inhibiting SHP-1 on lytic TILs overcomes the inhibition of TIL cytolysis by tumors. Contact between nonlytic TILs and tumor cells activates SHP-1, which rapidly dephosphorylates the Lck activation motif (Tyr394), thereby inhibiting effector phase function [125].
Immune checkpoints suppress autoimmunity and contribute to maintaining immune homeostasis by limiting T-cell activation. The exhausted T cells express inhibitory receptors such as PD-1 as immune checkpoint molecules. Some tumor cells use immune checkpoints to acquire immune tolerance to tumor-specific T cells [126]. Strategies blocking the interaction between programmed cell death protein 1 (PD-1) and PD ligand (PD-L)1/2, or between cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and CD80/CD86 have been developed to overcome the immune checkpoints in tumor tissue. SHP-1 is recruited to the cytoplasmic tail of PD-1 by binding to immunoreceptor tyrosine-based switch motifs (ITSM), which then induces dephosphorylation and inactivation of proximal signaling molecules activated through TCRs [127,128]. Lymphocyte-activating gene 3 (LAG3) associates with SHP-1/2, and LAG3/PD-1 collaboration limits CD8+ T-cell signaling, which dampened antitumor immunity in a murine ovarian cancer model [129]. Recently, it has also been reported that PD-1 interacts more effectively with SHP-2 than SHP-1. B and T-lymphocyte attenuator (BTLA), on the other hand, has been reported to interact more effectively with SHP-1 than SHP-2 due to an interaction between the immunoreceptor tyrosine-based inhibitory motif (ITIM) and the N-terminus of SHP-1 [130,131]. PD-1 interacts primarily with SHP-2 but also with SHP-1 in the absence of SHP-2, and both PD-1-SHP-1 and PD-1-SHP-2 complexes attenuate TCR and CD28 signaling pathways [132]. Although the interaction between SHP-1/SHP-2 and PD-1 is thought to contribute to T cell exhaustion, the CD4cre Ptpn6/11fl/fl mice do not improve T cell-mediated tumor control. Depleting these phosphatases from the polyclonal T-cell compartment does not improve tumor control, suggesting that caution should be taken when considering their inhibition for immunotherapeutic approaches [133].
The function of regulatory T-cells (Tregs) in immune tolerance in the tumor microenvironment has been well studied, but the function of SHP-1 in the tumor microenvironment in relation to Tregs is paradoxical. Loss of SHP-1, a negative regulator of TCR signaling, renders naïve CD4+ and CD8+ T-cells resistant to Treg-mediated suppression in a T-cell-specific manner (Figure 5) [134]. On the other hand, loss of SHP-1 expression in Tregs significantly increases their capacity, and specific pharmacological inhibition of SHP-1 enzymatic activity via the anticancer drug SSG considerably increased Treg suppressive activity both in vivo and ex vivo [135]. Therefore, SHP-1 function differs according to the cell population, and the ability to control SHP-1 expression or function in different cell populations would be advantageous for tumor control.
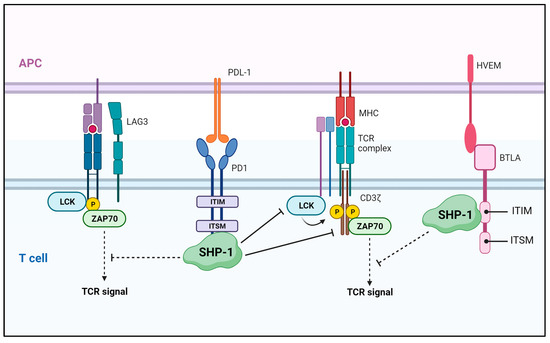
Figure 5.
SHP-1-mediated regulation in the tumor microenvironment. SHP-1 blocks T cell activation by negatively regulating TCR signaling through binding to the ITSM site and ITIM domain of the coinhibitory molecules, PD-1 and BTLA, respectively. APC—antigen-presenting cell; LAG—lymphocyte-activating gene 3; LCK—lymphocyte-cell-specific protein-tyrosine kinase; ZAP70—zeta-chain-associated protein kinase 70; PD1—programmed cell death protein 1; HVEM—herpes virus entry mediator; BTLA—B and T-lymphocyte attenuator.
5. Conclusions
With some exceptions, SHP-1-mediated inhibition of RTK signaling in cancer cells is generally associated with antitumor effects, and small molecules that increase SHP-1 expression and induce its activity may still be important candidate anticancer therapeutics. However, the clinical development of drugs that increase SHP-1 function or expression is extremely limited. Even if a drug candidate exerts a tumor-suppressing function specific to tumor cells, it is difficult to judge its overall effectiveness because it also suppresses the immune environment within the tumor tissue. In particular, it inhibits the activity of cytotoxic T lymphocytes, which display specific cytotoxicity against tumor cells. Therefore, when developing a tumor treatment targeting SHP-1, the function of SHP-1 in the type of cancer to be treated must first be determined, and the development of an effective combination treatment must also be considered. For example, for carcinomas with antitumor effects mediated by SHP-1, the combination of an SHP-1 agonist with SHP-1 knockdown or the CD3-mutated [124] CAR T-cell therapy may be considered.
Author Contributions
The manuscript was designed, written, and reviewed by K.D.K., K.W.L., and S.L., J.Y.K. contributed to writing the revised manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2021R1A2C1004847 and 2021R1A5A8029490).
Conflicts of Interest
The authors declare no conflict of interest.
References
- McDonell, L.M.; Kernohan, K.D.; Boycott, K.M.; Sawyer, S.L. Receptor tyrosine kinase mutations in developmental syndromes and cancer: Two sides of the same coin. Hum. Mol. Genet. 2015, 24, R60–R66. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef] [PubMed]
- Sastry, S.K.; Elferink, L.A. Checks and balances: Interplay of RTKs and PTPs in cancer progression. Biochem. Pharmacol. 2011, 82, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Bollu, L.R.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer. Clin. Cancer Res. 2017, 23, 2136–2142. [Google Scholar] [CrossRef] [PubMed]
- Sivaganesh, V.; Sivaganesh, V.; Scanlon, C.; Iskander, A.; Maher, S.; Le, T.; Peethambaran, B. Protein Tyrosine Phosphatases: Mechanisms in Cancer. Int. J. Mol. Sci. 2021, 22, 12865. [Google Scholar] [CrossRef] [PubMed]
- Varone, A.; Spano, D.; Corda, D. Shp1 in Solid Cancers and Their Therapy. Front. Oncol. 2020, 10, 935. [Google Scholar] [CrossRef]
- Plutzky, J.; Neel, B.G.; Rosenberg, R.D.; Eddy, R.L.; Byers, M.G.; Jani-Sait, S.; Shows, T.B. Chromosomal localization of an SH2-containing tyrosine phosphatase (PTPN6). Genomics 1992, 13, 869–872. [Google Scholar] [CrossRef]
- Matsushita, M.; Tsuchiya, N.; Oka, T.; Yamane, A.; Tokunaga, K. New variations of human SHP-1. Immunogenetics 1999, 49, 577–579. [Google Scholar] [CrossRef]
- Walton, K.M.; Dixon, J.E. Protein tyrosine phosphatases. Annu. Rev. Biochem. 1993, 62, 101–120. [Google Scholar] [CrossRef]
- Xu, Y.; Banville, D.; Zhao, H.F.; Zhao, X.; Shen, S.H. Transcriptional activity of the SHP-1 gene in MCF7 cells is differentially regulated by binding of NF-Y factor to two distinct CCAAT-elements. Gene 2001, 269, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Banville, D.; Stocco, R.; Shen, S.H. Human protein tyrosine phosphatase 1C (PTPN6) gene structure: Alternate promoter usage and exon skipping generate multiple transcripts. Genomics 1995, 27, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Tsui, H.W.; Hasselblatt, K.; Martin, A.; Mok, S.C.; Tsui, F.W. Molecular mechanisms underlying SHP-1 gene expression. Eur. J. Biochem. 2002, 269, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Kydd, A.R.; Nakase, K.; Noonan, K.M.; Murakami, A.; Tao, H.; Dwyer, M.; Xu, C.; Zhu, Q.; Marasco, W.A. Negative regulation of the SH2-homology containing protein-tyrosine phosphatase-1 (SHP-1) P2 promoter by the HTLV-1 Tax oncoprotein. Blood 2007, 110, 2110–2120. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, P.; Zhang, Q.; Liu, X.; Kasprzycka, M.; Marzec, M.; Wasik, M.A. PU. 1 activates transcription of SHP-1 gene in hematopoietic cells. J. Biol. Chem. 2007, 282, 6316–6323. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, S.; Van Emburgh, B.O.; Robertson, K.D. DNA methylation in development and human disease. Mutat. Res. 2008, 647, 30–38. [Google Scholar] [CrossRef]
- Takeuchi, S.; Matsushita, M.; Zimmermann, M.; Ikezoe, T.; Komatsu, N.; Seriu, T.; Schrappe, M.; Bartram, C.R.; Koeffler, H.P. Clinical significance of aberrant DNA methylation in childhood acute lymphoblastic leukemia. Leuk. Res. 2011, 35, 1345–1349. [Google Scholar] [CrossRef][Green Version]
- Kucuk, C.; Hu, X.; Jiang, B.; Klinkebiel, D.; Geng, H.; Gong, Q.; Bouska, A.; Iqbal, J.; Gaulard, P.; McKeithan, T.W.; et al. Global promoter methylation analysis reveals novel candidate tumor suppressor genes in natural killer cell lymphoma. Clin. Cancer Res. 2015, 21, 1699–1711. [Google Scholar] [CrossRef]
- Ding, K.; Chen, X.; Wang, Y.; Liu, H.; Song, W.; Li, L.; Wang, G.; Song, J.; Shao, Z.; Fu, R. Plasma DNA methylation of p16 and shp1 in patients with B cell non-Hodgkin lymphoma. Int. J. Clin. Oncol. 2017, 22, 585–592. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, S.; Liu, X.; Liu, J. Aberrant promoter 2 methylation-mediated downregulation of protein tyrosine phosphatase, non-receptor type 6, is associated with progression of esophageal squamous cell carcinoma. Mol. Med. Rep. 2019, 19, 3273–3282. [Google Scholar] [CrossRef]
- Joo, M.K.; Park, J.J.; Yoo, H.S.; Lee, B.J.; Chun, H.J.; Lee, S.W.; Bak, Y.T. Epigenetic regulation and anti-tumorigenic effects of SH2-containing protein tyrosine phosphatase 1 (SHP1) in human gastric cancer cells. Tumor Biol. 2016, 37, 4603–4612. [Google Scholar] [CrossRef] [PubMed]
- Bernal, C.; Aguayo, F.; Villarroel, C.; Vargas, M.; Diaz, I.; Ossandon, F.J.; Santibanez, E.; Palma, M.; Aravena, E.; Barrientos, C.; et al. Reprimo as a potential biomarker for early detection in gastric cancer. Clin. Cancer Res. 2008, 14, 6264–6269. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Blumenthal, R.M. Mammalian DNA methyltransferases: A structural perspective. Structure 2008, 16, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, X.; Guo, X.; Liu, X.; Luo, J. DNA methyltransferase 1 mediated aberrant methylation and silencing of SHP-1 gene in chronic myelogenous leukemia cells. Leuk. Res. 2017, 58, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.; Raghunath, P.; Wasik, A.; Junkins-Hopkins, J.M.; Jones, D.; Zhang, Q.; Odum, N.; Wasik, M.A. Loss of SHP-1 tyrosine phosphatase expression correlates with the advanced stages of cutaneous T-cell lymphoma. Hum. Pathol. 2007, 38, 462–467. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.Y.; Woetmann, A.; Raghunath, P.N.; Odum, N.; Wasik, M.A. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood 2006, 108, 1058–1064. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.Y.; Marzec, M.; Raghunath, P.N.; Nagasawa, T.; Wasik, M.A. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 6948–6953. [Google Scholar] [CrossRef]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef]
- Witzig, T.E.; Hu, G.; Offer, S.M.; Wellik, L.E.; Han, J.J.; Stenson, M.J.; Dogan, A.; Diasio, R.B.; Gupta, M. Epigenetic mechanisms of protein tyrosine phosphatase 6 suppression in diffuse large B-cell lymphoma: Implications for epigenetic therapy. Leukemia 2014, 28, 147–154. [Google Scholar] [CrossRef][Green Version]
- Zhang, X.; Yang, L.; Liu, X.; Nie, Z.; Wang, X.; Pan, Y.; Luo, J. Research on the epigenetic regulation mechanism of the PTPN6 gene in advanced chronic myeloid leukaemia. Br. J. Haematol. 2017, 178, 728–738. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Gao, B.; Yang, Y.; Jia, S.B.; Ma, X.P.; Zhang, M.H.; Wang, L.J.; Ma, A.Q.; Zhang, Q.N. Histone deacetylase 3 suppresses the expression of SHP-1 via deacetylation of DNMT1 to promote heart failure. Life Sci. 2022, 292, 119552. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Huang, T.T.; Chu, P.Y.; Huang, C.T.; Lee, C.H.; Wang, W.L.; Lau, K.Y.; Tsai, W.C.; Chao, T.I.; Su, J.C.; et al. The tyrosine kinase inhibitor nintedanib activates SHP-1 and induces apoptosis in triple-negative breast cancer cells. Exp. Mol. Med. 2017, 49, e366. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Chen, K.F.; Chao, T.I.; Chu, P.Y.; Huang, C.T.; Huang, T.T.; Yang, H.P.; Wang, W.L.; Lee, C.H.; Lau, K.Y.; et al. Sequential combination of docetaxel with a SHP-1 agonist enhanced suppression of p-STAT3 signaling and apoptosis in triple negative breast cancer cells. J. Mol. Med. 2017, 95, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Geng, Q.; Xian, R.; Yu, Y.; Chen, F.; Li, R. SHP-1 acts as a tumor suppressor by interacting with EGFR and predicts the prognosis of human breast cancer. Cancer Biol. Med. 2021, 19, 468–485. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.Z.; Ding, K.; Wang, Z.R.; Ding, C.H.; Lei, S.J.; Liu, J.P.; Yin, C.; Hu, P.F.; Ding, J.; Chen, W.S.; et al. SHP-1 Acts as a Tumor Suppressor in Hepatocarcinogenesis and HCC Progression. Cancer Res. 2018, 78, 4680–4691. [Google Scholar] [CrossRef]
- Tassidis, H.; Brokken, L.J.; Jirstrom, K.; Ehrnstrom, R.; Ponten, F.; Ulmert, D.; Bjartell, A.; Harkonen, P.; Wingren, A.G. Immunohistochemical detection of tyrosine phosphatase SHP-1 predicts outcome after radical prostatectomy for localized prostate cancer. Int. J. Cancer 2010, 126, 2296–2307. [Google Scholar] [CrossRef]
- Liu, C.; Shen, A.; Song, J.; Cheng, L.; Zhang, M.; Wang, Y.; Liu, X. LncRNA-CCAT5-mediated crosstalk between Wnt/beta-Catenin and STAT3 signaling suggests novel therapeutic approaches for metastatic gastric cancer with high Wnt activity. Cancer Commun. 2023, 1–25. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, J.; Du, Z.; Huang, J.; Cheng, Y.; Yi, W.; Li, T.; Yang, J.; Chen, C. Comprehensive analysis of PTPN family expression and prognosis in acute myeloid leukemia. Front. Genet. 2022, 13, 1087938. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Y.; Huang, Y.; Yuan, X.; Cao, Z.; Zhao, Z. PTPN6-EGFR Protein Complex: A Novel Target for Colon Cancer Metastasis. J. Oncol. 2022, 2022, 7391069. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, J.; Zhang, M.; Liu, S.; Wang, T.; Wu, T.; Li, B.; Zhao, S.; Wang, H.; Li, L.; et al. Single-cell and bulk sequencing analyses reveal the immune suppressive role of PTPN6 in glioblastoma. Aging 2023, 15, 9822–9841. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, U. SHP-1 and SHP-2 in T cells: Two phosphatases functioning at many levels. Immunol. Rev. 2009, 228, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, L.; He, D.; Song, X.; Liang, X.; Zhao, Z.J.; Zhou, G.W. Crystal structure of human protein-tyrosine phosphatase SHP-1. J. Biol. Chem. 2003, 278, 6516–6520. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, L.; Song, X.; Mo, Y.; Komma, C.; Bellamy, H.D.; Zhao, Z.J.; Zhou, G.W. Crystal structure of human protein tyrosine phosphatase SHP-1 in the open conformation. J. Cell. Biochem. 2011, 112, 2062–2071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shen, K.; Lu, W.; Cole, P.A. The role of C-terminal tyrosine phosphorylation in the regulation of SHP-1 explored via expressed protein ligation. J. Biol. Chem. 2003, 278, 4668–4674. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kruhlak, M.J.; Hao, J.J.; Shaw, S. Rapid T cell receptor-mediated SHP-1 S591 phosphorylation regulates SHP-1 cellular localization and phosphatase activity. J. Leukoc. Biol. 2007, 82, 742–751. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jones, M.L.; Craik, J.D.; Gibbins, J.M.; Poole, A.W. Regulation of SHP-1 tyrosine phosphatase in human platelets by serine phosphorylation at its C terminus. J. Biol. Chem. 2004, 279, 40475–40483. [Google Scholar] [CrossRef]
- Guschin, D.; Rogers, N.; Briscoe, J.; Witthuhn, B.; Watling, D.; Horn, F.; Pellegrini, S.; Yasukawa, K.; Heinrich, P.; Stark, G.R.; et al. A major role for the protein tyrosine kinase JAK1 in the JAK/STAT signal transduction pathway in response to interleukin-6. EMBO J. 1995, 14, 1421–1429. [Google Scholar] [CrossRef]
- Cimica, V.; Chen, H.C.; Iyer, J.K.; Reich, N.C. Dynamics of the STAT3 transcription factor: Nuclear import dependent on Ran and importin-beta1. PLoS ONE 2011, 6, e20188. [Google Scholar] [CrossRef]
- Bournazou, E.; Bromberg, J. Targeting the tumor microenvironment: JAK-STAT3 signaling. JAKSTAT 2013, 2, e23828. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.A. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 2007, 251, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Roeser, J.C.; Leach, S.D.; McAllister, F. Emerging strategies for cancer immunoprevention. Oncogene 2015, 34, 6029–6039. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. Corrigendum: IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2017, 18, 1271. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Vignais, M.L.; Sadowski, H.B.; Watling, D.; Rogers, N.C.; Gilman, M. Platelet-derived growth factor induces phosphorylation of multiple JAK family kinases and STAT proteins. Mol. Cell. Biol. 1996, 16, 1759–1769. [Google Scholar] [CrossRef]
- Wang, J.; Xu, Z.; Wang, Z.; Du, G.; Lun, L. TGF-beta signaling in cancer radiotherapy. Cytokine 2021, 148, 155709. [Google Scholar] [CrossRef]
- Himpe, E.; Kooijman, R. Insulin-like growth factor-I receptor signal transduction and the Janus Kinase/Signal Transducer and Activator of Transcription (JAK-STAT) pathway. Biofactors 2009, 35, 76–81. [Google Scholar] [CrossRef]
- Beach, K.M.; Wang, J.; Otteson, D.C. Regulation of Stem Cell Properties of Muller Glia by JAK/STAT and MAPK Signaling in the Mammalian Retina. Stem Cells Int. 2017, 2017, 1610691. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Hong, J.; Du, W.; Lin, Y.W.; Ren, L.L.; Wang, Y.C.; Su, W.Y.; Wang, J.L.; Cui, Y.; Wang, Z.H.; et al. Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J. Biol. Chem. 2012, 287, 5819–5832. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Shen, J.; Fang, Z.; Qiao, L.; Feng, R.; Lin, X.; Li, S. Abnormally expressed JunB transactivated by IL-6/STAT3 signaling promotes uveal melanoma aggressiveness via epithelial-mesenchymal transition. Biosci. Rep. 2018, 38, BSR20180532. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.S.; Hui, C.C.; Pawson, T. SH2-containing phosphotyrosine phosphatase as a target of protein-tyrosine kinases. Science 1993, 259, 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.E.; Chang, S.; Trub, T.; Neel, B.G. Regulation of colony-stimulating factor 1 receptor signaling by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol. Cell. Biol. 1996, 16, 3685–3697. [Google Scholar] [CrossRef]
- Yi, T.; Ihle, J.N. Association of hematopoietic cell phosphatase with c-Kit after stimulation with c-Kit ligand. Mol. Cell. Biol. 1993, 13, 3350–3358. [Google Scholar] [CrossRef]
- David, M.; Chen, H.E.; Goelz, S.; Larner, A.C.; Neel, B.G. Differential regulation of the alpha/beta interferon-stimulated Jak/Stat pathway by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol. Cell. Biol. 1995, 15, 7050–7058. [Google Scholar] [CrossRef]
- Klingmuller, U.; Lorenz, U.; Cantley, L.C.; Neel, B.G.; Lodish, H.F. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell 1995, 80, 729–738. [Google Scholar] [CrossRef]
- Plas, D.R.; Johnson, R.; Pingel, J.T.; Matthews, R.J.; Dalton, M.; Roy, G.; Chan, A.C.; Thomas, M.L. Direct regulation of ZAP-70 by SHP-1 in T cell antigen receptor signaling. Science 1996, 272, 1173–1176. [Google Scholar] [CrossRef]
- Perez-Villar, J.J.; Whitney, G.S.; Bowen, M.A.; Hewgill, D.H.; Aruffo, A.A.; Kanner, S.B. CD5 negatively regulates the T-cell antigen receptor signal transduction pathway: Involvement of SH2-containing phosphotyrosine phosphatase SHP-1. Mol. Cell. Biol. 1999, 19, 2903–2912. [Google Scholar] [CrossRef] [PubMed]
- Daigle, I.; Yousefi, S.; Colonna, M.; Green, D.R.; Simon, H.U. Death receptors bind SHP-1 and block cytokine-induced anti-apoptotic signaling in neutrophils. Nat. Med. 2002, 8, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, I.; Hemmer, B.; Vergelli, M.; Martin, R.; Biddison, W.E.; Germain, R.N. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat. Immunol. 2003, 4, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Alicea-Velazquez, N.L.; Jakoncic, J.; Boggon, T.J. Structure-guided studies of the SHP-1/JAK1 interaction provide new insights into phosphatase catalytic domain substrate recognition. J. Struct. Biol. 2013, 181, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.J.; Harbor, P.; Tabrizi, M.; Yi, T.; Williams, B.R. Protein-tyrosine phosphatase Shp-1 is a negative regulator of IL-4- and IL-13-dependent signal transduction. J. Biol. Chem. 1998, 273, 33893–33896. [Google Scholar] [CrossRef] [PubMed]
- Migone, T.S.; Cacalano, N.A.; Taylor, N.; Yi, T.; Waldmann, T.A.; Johnston, J.A. Recruitment of SH2-containing protein tyrosine phosphatase SHP-1 to the interleukin 2 receptor; loss of SHP-1 expression in human T-lymphotropic virus type I-transformed T cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3845–3850. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Berrada, K.; Yang, W.; Tabrizi, M.; Platanias, L.C.; Yi, T. Direct association with and dephosphorylation of Jak2 kinase by the SH2-domain-containing protein tyrosine phosphatase SHP-1. Mol. Cell. Biol. 1996, 16, 6985–6992. [Google Scholar] [CrossRef]
- Han, Y.; Amin, H.M.; Franko, B.; Frantz, C.; Shi, X.; Lai, R. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood 2006, 108, 2796–2803. [Google Scholar] [CrossRef]
- Fan, L.C.; Shiau, C.W.; Tai, W.T.; Hung, M.H.; Chu, P.Y.; Hsieh, F.S.; Lin, H.; Yu, H.C.; Chen, K.F. SHP-1 is a negative regulator of epithelial-mesenchymal transition in hepatocellular carcinoma. Oncogene 2015, 34, 5252–5263. [Google Scholar] [CrossRef]
- Fan, L.C.; Teng, H.W.; Shiau, C.W.; Tai, W.T.; Hung, M.H.; Yang, S.H.; Jiang, J.K.; Chen, K.F. Regorafenib (Stivarga) pharmacologically targets epithelial-mesenchymal transition in colorectal cancer. Oncotarget 2016, 7, 64136–64147. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, L.; Chen, G.; Zhang, J.; Li, Z.; Lu, W.; Liu, M.; Pang, X. Small molecule 1’-acetoxychavicol acetate suppresses breast tumor metastasis by regulating the SHP-1/STAT3/MMPs signaling pathway. Breast Cancer Res. Treat. 2014, 148, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.K.; Park, J.J.; Kim, S.H.; Yoo, H.S.; Lee, B.J.; Chun, H.J.; Lee, S.W.; Bak, Y.T. Antitumorigenic effect of plumbagin by induction of SH2-containing protein tyrosine phosphatase 1 in human gastric cancer cells. Int. J. Oncol. 2015, 46, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhu, B.; Zhao, L.; Liu, Y.; Zhao, F.; Feng, J.; Jin, Y.; Sun, J.; Geng, R.; Wei, Y. Allicin Inhibits Proliferation and Invasion in Vitro and in Vivo via SHP-1-Mediated STAT3 Signaling in Cholangiocarcinoma. Cell Physiol. Biochem. 2018, 47, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Montano, X. Repression of SHP-1 expression by p53 leads to trkA tyrosine phosphorylation and suppression of breast cancer cell proliferation. Oncogene 2009, 28, 3787–3800. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Caron, D.; Savard, P.E.; Doillon, C.J.; Olivier, M.; Shink, E.; Lussier, J.G.; Faure, R.L. Protein tyrosine phosphatase inhibition induces anti-tumor activity: Evidence of Cdk2/p27 kip1 and Cdk2/SHP-1 complex formation in human ovarian cancer cells. Cancer Lett. 2008, 262, 265–275. [Google Scholar] [CrossRef]
- Rodriguez-Ubreva, F.J.; Cariaga-Martinez, A.E.; Cortes, M.A.; Romero-De Pablos, M.; Ropero, S.; Lopez-Ruiz, P.; Colas, B. Knockdown of protein tyrosine phosphatase SHP-1 inhibits G1/S progression in prostate cancer cells through the regulation of components of the cell-cycle machinery. Oncogene 2010, 29, 345–355. [Google Scholar] [CrossRef]
- Pan, X.; Mou, J.; Liu, S.; Sun, Z.; Meng, R.; Zhou, Z.; Wu, G.; Peng, G. SHP-1 overexpression increases the radioresistance of NPC cells by enhancing DSB repair, increasing S phase arrest and decreasing cell apoptosis. Oncol. Rep. 2015, 33, 2999–3005. [Google Scholar] [CrossRef][Green Version]
- Peng, G.; Cao, R.B.; Li, Y.H.; Zou, Z.W.; Huang, J.; Ding, Q. Alterations of cell cycle control proteins SHP-1/2, p16, CDK4 and cyclin D1 in radioresistant nasopharyngeal carcinoma cells. Mol. Med. Rep. 2014, 10, 1709–1716. [Google Scholar] [CrossRef][Green Version]
- Peng, G.; Cao, R.; Xue, J.; Li, P.; Zou, Z.; Huang, J.; Ding, Q. Increased expression of SHP-1 is associated with local recurrence after radiotherapy in patients with nasopharyngeal carcinoma. Radiol. Oncol. 2014, 48, 40–49. [Google Scholar] [CrossRef]
- Huang, S.; Sinicrope, F.A. Sorafenib inhibits STAT3 activation to enhance TRAIL-mediated apoptosis in human pancreatic cancer cells. Mol. Cancer Ther. 2010, 9, 742–750. [Google Scholar] [CrossRef]
- Yang, F.; Brown, C.; Buettner, R.; Hedvat, M.; Starr, R.; Scuto, A.; Schroeder, A.; Jensen, M.; Jove, R. Sorafenib induces growth arrest and apoptosis of human glioblastoma cells through the dephosphorylation of signal transducers and activators of transcription 3. Mol. Cancer Ther. 2010, 9, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Shiau, C.W.; Chen, P.J.; Chu, P.Y.; Huang, H.P.; Liu, C.Y.; Huang, J.W.; Chen, K.F. Discovery of novel Src homology region 2 domain-containing phosphatase 1 agonists from sorafenib for the treatment of hepatocellular carcinoma. Hepatology 2014, 59, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.H.; Chen, L.J.; Chen, Y.L.; Tsai, M.S.; Shiau, C.W.; Chao, T.I.; Liu, C.Y.; Kao, J.H.; Chen, K.F. Targeting SHP-1-STAT3 signaling: A promising therapeutic approach for the treatment of cholangiocarcinoma. Oncotarget 2017, 8, 65077–65089. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.C.; Teng, H.W.; Shiau, C.W.; Tai, W.T.; Hung, M.H.; Yang, S.H.; Jiang, J.K.; Chen, K.F. Pharmacological Targeting SHP-1-STAT3 Signaling Is a Promising Therapeutic Approach for the Treatment of Colorectal Cancer. Neoplasia 2015, 17, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Tseng, L.M.; Su, J.C.; Chang, K.C.; Chu, P.Y.; Tai, W.T.; Shiau, C.W.; Chen, K.F. Novel sorafenib analogues induce apoptosis through SHP-1 dependent STAT3 inactivation in human breast cancer cells. Breast Cancer Res. 2013, 15, R63. [Google Scholar] [CrossRef]
- Liu, C.Y.; Su, J.C.; Huang, T.T.; Chu, P.Y.; Huang, C.T.; Wang, W.L.; Lee, C.H.; Lau, K.Y.; Tsai, W.C.; Yang, H.P.; et al. Sorafenib analogue SC-60 induces apoptosis through the SHP-1/STAT3 pathway and enhances docetaxel cytotoxicity in triple-negative breast cancer cells. Mol. Oncol. 2017, 11, 266–279. [Google Scholar] [CrossRef]
- Tai, W.T.; Shiau, C.W.; Li, Y.S.; Chen, Y.L.; Chu, P.Y.; Huang, J.W.; Hsu, C.Y.; Hsu, Y.C.; Chen, P.J.; Chen, K.F. SC-60, a dimer-based sorafenib derivative, shows a better anti-hepatocellular carcinoma effect than sorafenib in a preclinical hepatocellular carcinoma model. Mol. Cancer Ther. 2014, 13, 27–36. [Google Scholar] [CrossRef]
- Tai, W.T.; Chu, P.Y.; Shiau, C.W.; Chen, Y.L.; Li, Y.S.; Hung, M.H.; Chen, L.J.; Chen, P.L.; Su, J.C.; Lin, P.Y.; et al. STAT3 mediates regorafenib-induced apoptosis in hepatocellular carcinoma. Clin. Cancer Res. 2014, 20, 5768–5776. [Google Scholar] [CrossRef]
- Fan, L.C.; Teng, H.W.; Shiau, C.W.; Lin, H.; Hung, M.H.; Chen, Y.L.; Huang, J.W.; Tai, W.T.; Yu, H.C.; Chen, K.F. SHP-1 is a target of regorafenib in colorectal cancer. Oncotarget 2014, 5, 6243–6251. [Google Scholar] [CrossRef]
- Su, J.C.; Mar, A.C.; Wu, S.H.; Tai, W.T.; Chu, P.Y.; Wu, C.Y.; Tseng, L.M.; Lee, T.C.; Chen, K.F.; Liu, C.Y.; et al. Disrupting VEGF-A paracrine and autocrine loops by targeting SHP-1 suppresses triple negative breast cancer metastasis. Sci. Rep. 2016, 6, 28888. [Google Scholar] [CrossRef]
- Insabato, L.; Amelio, I.; Quarto, M.; Zannetti, A.; Tolino, F.; de Mauro, G.; Cerchia, L.; Riccio, P.; Baumhoer, D.; Condorelli, G.; et al. Elevated expression of the tyrosine phosphatase SHP-1 defines a subset of high-grade breast tumors. Oncology 2009, 77, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.C.; Kwok, T.T.; Berkowitz, R.S.; Barrett, A.J.; Tsui, F.W. Overexpression of the protein tyrosine phosphatase, nonreceptor type 6 (PTPN6), in human epithelial ovarian cancer. Gynecol. Oncol. 1995, 57, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Pathak, M.K.; Lindner, D.J.; Ketterer, M.E.; Farver, C.; Borden, E.C. Anticancer activity of sodium stibogluconate in synergy with IFNs. J. Immunol. 2002, 169, 5978–5985. [Google Scholar] [CrossRef]
- Naing, A.; Reuben, J.M.; Camacho, L.H.; Gao, H.; Lee, B.N.; Cohen, E.N.; Verschraegen, C.; Stephen, S.; Aaron, J.; Hong, D.; et al. Phase I Dose Escalation Study of Sodium Stibogluconate (SSG), a Protein Tyrosine Phosphatase Inhibitor, Combined with Interferon Alpha for Patients with Solid Tumors. J. Cancer 2011, 2, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.S.; Sethi, G.; Sung, B.; Goel, A.; Ralhan, R.; Aggarwal, B.B. Guggulsterone, a farnesoid X receptor antagonist, inhibits constitutive and inducible STAT3 activation through induction of a protein tyrosine phosphatase SHP-1. Cancer Res. 2008, 68, 4406–4415. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, S.; Zarif, L.; Kuttikrishnan, S.; Prabhu, K.S.; Patil, K.; Nisar, S.; Abou-Saleh, H.; Merhi, M.; Dermime, S.; Bhat, A.A.; et al. Guggulsterone Induces Apoptosis in Multiple Myeloma Cells by Targeting High Mobility Group Box 1 via Janus Activated Kinase/Signal Transducer and Activator of Transcription Pathway. Cancers 2022, 14, 5621. [Google Scholar] [CrossRef]
- Gupta, S.C.; Phromnoi, K.; Aggarwal, B.B. Morin inhibits STAT3 tyrosine 705 phosphorylation in tumor cells through activation of protein tyrosine phosphatase SHP1. Biochem. Pharmacol. 2013, 85, 898–912. [Google Scholar] [CrossRef]
- Lee, J.C.; Ahn, K.S.; Jeong, S.J.; Jung, J.H.; Kwon, T.R.; Rhee, Y.H.; Kim, S.H.; Kim, S.Y.; Yoon, H.J.; Zhu, S.; et al. Signal transducer and activator of transcription 3 pathway mediates genipin-induced apoptosis in U266 multiple myeloma cells. J. Cell. Biochem. 2011, 112, 1552–1562. [Google Scholar] [CrossRef]
- Lee, J.H.; Chiang, S.Y.; Nam, D.; Chung, W.S.; Lee, J.; Na, Y.S.; Sethi, G.; Ahn, K.S. Capillarisin inhibits constitutive and inducible STAT3 activation through induction of SHP-1 and SHP-2 tyrosine phosphatases. Cancer Lett. 2014, 345, 140–148. [Google Scholar] [CrossRef]
- Rhee, Y.H.; Jeong, S.J.; Lee, H.J.; Lee, H.J.; Koh, W.; Jung, J.H.; Kim, S.H.; Sung-Hoon, K. Inhibition of STAT3 signaling and induction of SHP1 mediate antiangiogenic and antitumor activities of ergosterol peroxide in U266 multiple myeloma cells. BMC Cancer 2012, 12, 28. [Google Scholar] [CrossRef]
- Kang, S.H.; Jeong, S.J.; Kim, S.H.; Kim, J.H.; Jung, J.H.; Koh, W.; Kim, J.H.; Kim, D.K.; Chen, C.Y.; Kim, S.H. Icariside II induces apoptosis in U937 acute myeloid leukemia cells: Role of inactivation of STAT3-related signaling. PLoS ONE 2012, 7, e28706. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Yu, Z.; Wu, J.; Yu, K.; Hong, G.; Lu, Z.; Gao, S. Honokiol Inhibits Constitutive and Inducible STAT3 Signaling via PU.1-Induced SHP1 Expression in Acute Myeloid Leukemia Cells. Tohoku J. Exp. Med. 2015, 237, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Rajendran, P.; Li, F.; Kim, C.; Sikka, S.; Siveen, K.S.; Kumar, A.P.; Ahn, K.S.; Sethi, G. Abrogation of STAT3 signaling cascade by zerumbone inhibits proliferation and induces apoptosis in renal cell carcinoma xenograft mouse model. Mol. Carcinog. 2015, 54, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Sethi, G.; Ravish, A.; Mohan, A.K.; Pandey, V.; Lobie, P.E.; Basappa, S.; Basappa, B.; Ahn, K.S. Discovery of imidazopyridine-pyrazoline-hybrid structure as SHP-1 agonist that suppresses phospho-STAT3 signaling in human breast cancer cells. Chem. Biol. Interact. 2023, 386, 110780. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tan, Y.P.; Zhao, L.; Wang, L.; Fu, N.J.; Zheng, S.P.; Shen, X.F. Anticancer activity of dietary xanthone alpha-mangostin against hepatocellular carcinoma by inhibition of STAT3 signaling via stabilization of SHP1. Cell Death Dis. 2020, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, A.; Shanmugam, M.K.; Ong, T.H.; Li, F.; Perumal, E.; Chen, L.; Vali, S.; Abbasi, T.; Kapoor, S.; Ahn, K.S.; et al. Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3. Br. J. Pharmacol. 2013, 170, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, S.; Li, J.; Yin, F.; Hua, Y.; Wang, Z.; Wang, H.; Zuo, D.; Xu, J.; Cai, Z. Pectolinarigenin acts as a potential anti-osteosarcoma agent via mediating SHP-1/JAK2/STAT3 signaling. Biomed. Pharmacother. 2022, 153, 113323. [Google Scholar] [CrossRef]
- Kilgore, N.E.; Carter, J.D.; Lorenz, U.; Evavold, B.D. Cutting edge: Dependence of TCR antagonism on Src homology 2 domain-containing protein tyrosine phosphatase activity. J. Immunol. 2003, 170, 4891–4895. [Google Scholar] [CrossRef]
- Tsui, H.W.; Siminovitch, K.A.; de Souza, L.; Tsui, F.W. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat. Genet. 1993, 4, 124–129. [Google Scholar] [CrossRef]
- Sathish, J.G.; Dolton, G.; Leroy, F.G.; Matthews, R.J. Loss of Src homology region 2 domain-containing protein tyrosine phosphatase-1 increases CD8+ T cell-APC conjugate formation and is associated with enhanced in vivo CTL function. J. Immunol. 2007, 178, 330–337. [Google Scholar] [CrossRef]
- Snook, J.P.; Soedel, A.J.; Ekiz, H.A.; O’Connell, R.M.; Williams, M.A. Inhibition of SHP-1 Expands the Repertoire of Antitumor T Cells Available to Respond to Immune Checkpoint Blockade. Cancer Immunol. Res. 2020, 8, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, L.; Zhong, M.; Long, Y.; Yang, W.; Liu, T.; Huang, X.; Ma, X. CRISPR/Cas9-mediated knockout of intracellular molecule SHP-1 enhances tumor-killing ability of CD133-targeted CAR T cells in vitro. Exp. Hematol. Oncol. 2023, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Velasco Cardenas, R.M.; Brandl, S.M.; Melendez, A.V.; Schlaak, A.E.; Buschky, A.; Peters, T.; Beier, F.; Serrels, B.; Taromi, S.; Raute, K.; et al. Harnessing CD3 diversity to optimize CAR T cells. Nat. Immunol. 2023, 24, 2135–2149. [Google Scholar] [CrossRef] [PubMed]
- Monu, N.; Frey, A.B. Suppression of proximal T cell receptor signaling and lytic function in CD8+ tumor-infiltrating T cells. Cancer Res. 2007, 67, 11447–11454. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef]
- Sheppard, K.A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef]
- Huang, R.Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 2015, 6, 27359–27377. [Google Scholar] [CrossRef]
- Xu, X.; Masubuchi, T.; Cai, Q.; Zhao, Y.; Hui, E. Molecular features underlying differential SHP1/SHP2 binding of immune checkpoint receptors. Elife 2021, 10, e74276. [Google Scholar] [CrossRef]
- Xu, X.; Hou, B.; Fulzele, A.; Masubuchi, T.; Zhao, Y.; Wu, Z.; Hu, Y.; Jiang, Y.; Ma, Y.; Wang, H.; et al. PD-1 and BTLA regulate T cell signaling differentially and only partially through SHP1 and SHP2. J. Cell Biol. 2020, 219, e201905085. [Google Scholar] [CrossRef] [PubMed]
- Celis-Gutierrez, J.; Blattmann, P.; Zhai, Y.; Jarmuzynski, N.; Ruminski, K.; Gregoire, C.; Ounoughene, Y.; Fiore, F.; Aebersold, R.; Roncagalli, R.; et al. Quantitative Interactomics in Primary T Cells Provides a Rationale for Concomitant PD-1 and BTLA Coinhibitor Blockade in Cancer Immunotherapy. Cell Rep. 2019, 27, 3315–3330.e7. [Google Scholar] [CrossRef] [PubMed]
- Ventura, P.M.O.; Gakovic, M.; Fischer, B.A.; Spinelli, L.; Rota, G.; Pathak, S.; Khameneh, H.J.; Zenobi, A.; Thomson, S.; Birchmeier, W.; et al. Concomitant deletion of Ptpn6 and Ptpn11 in T cells fails to improve anticancer responses. EMBO Rep. 2022, 23, e55399. [Google Scholar] [CrossRef] [PubMed]
- Mercadante, E.R.; Lorenz, U.M. T Cells Deficient in the Tyrosine Phosphatase SHP-1 Resist Suppression by Regulatory T Cells. J. Immunol. 2017, 199, 129–137. [Google Scholar] [CrossRef]
- Iype, T.; Sankarshanan, M.; Mauldin, I.S.; Mullins, D.W.; Lorenz, U. The protein tyrosine phosphatase SHP-1 modulates the suppressive activity of regulatory T cells. J. Immunol. 2010, 185, 6115–6127. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).