
Distinct Signaling Pathways for Autophagy-Driven Cell Death and Survival in Adult Hippocampal Neural Stem Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Four Cellular Stress Conditions Trigger Autophagy in HCN Cells
2.2. 2DG and GL Did Not Affect Cell Viability, but IW and CORT Induced Cell Death Which Met Autophagic Cell Death Criteria
2.3. 2DG-Induced Autophagy Is Cytoprotective and Reduces IW- or CORT-Induced Cell Death
2.4. The Inhibition of MAPK Blocks 2DG-Induced Autophagy but Not IW- or CORT-Induced Autophagy
2.5. The Blockage of ERK and JNK Impedes the Protection Effects of 2DG
2.6. The Inhibition of GSK-3β Blocks IW- or CORT-Induced ACD but Not 2DG-Induced Autophagy
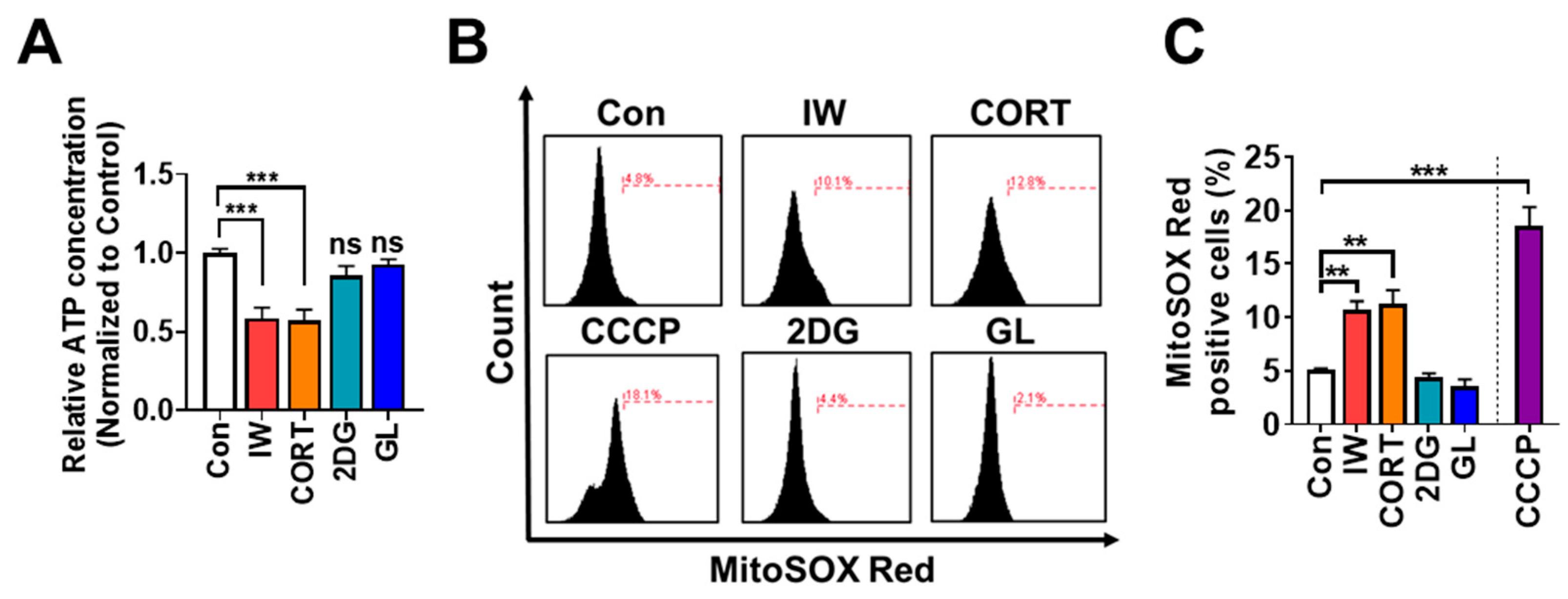
2.7. IW and CORT, but Not 2DG or GL, Reduce ATP Amounts and Increase Mitochondrial ROS Levels in HCN Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Cultures
4.3. Cell Death Assay
4.4. Generation of Stable Cell Lines
4.5. Transfection
4.6. Confocal Imaging
4.7. Western Blotting
4.8. MitoSOX Staining Using Fluorescence-Activated Cell Sorting (FACS) Analysis
4.9. ATP Measurement Assay
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Morishita, H.; Mizushima, N. Diverse Cellular Roles of Autophagy. Annu. Rev. Cell Dev. Biol. 2019, 35, 453–475. [Google Scholar] [CrossRef]
- Kawabata, T.; Yoshimori, T. Beyond starvation: An update on the autophagic machinery and its functions. J. Mol. Cell. Cardiol. 2016, 95, 2–10. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Chan, E.Y.; Tooze, S.A. Evolution of Atg1 function and regulation. Autophagy 2009, 5, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Burman, C.; Ktistakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Y.; Li, Y.; Jiang, W.Q.; Zhou, L.F. MAPK/JNK signalling: A potential autophagy regulation pathway. Biosci. Rep. 2015, 35, e00199. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell. Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Komatsu, M.; Finley, K.; Simonsen, A. Autophagy: More than a nonselective pathway. Int. J. Cell Biol. 2012, 2012, 219625. [Google Scholar] [CrossRef]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B.J.C. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Zou, Z.; Sun, Q.; Luby-Phelps, K.; Cheng, P.; Hogan, R.N.; Gilpin, C.; Levine, B.J.C. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007, 128, 931–946. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Kuma, A.; Mizushima, N. Physiological role of autophagy as an intracellular recycling system: With an emphasis on nutrient metabolism. Semin. Cell Dev. Biol. 2010, 21, 683–690. [Google Scholar] [CrossRef]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J.J.S. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004, 304, 1500–1502. [Google Scholar] [CrossRef]
- Lamy, L.; Vu, N.N.; Emre, N.C.T.; Shaffer, A.L.; Yang, Y.; Tian, E.; Nair, V.; Kruhlak, M.J.; Zingone, A.; Landgren, O.; et al. Control of Autophagic Cell Death by Caspase-10 in Multiple Myeloma. Cancer Cell 2013, 23, 435–449. [Google Scholar] [CrossRef]
- Dasari, S.K.; Bialik, S.; Levin-Zaidman, S.; Levin-Salomon, V.; Merrill, A.H.; Futerman, A.H.; Kimchi, A. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017, 24, 1288–1302. [Google Scholar] [CrossRef]
- Jiang, K.; Wang, W.; Jin, X.; Wang, Z.; Ji, Z.; Meng, G. Silibinin, a natural flavonoid, induces autophagy via ROS-dependent mitochondrial dysfunction and loss of ATP involving BNIP3 in human MCF7 breast cancer cells. Oncol. Rep. 2015, 33, 2711–2718. [Google Scholar] [CrossRef]
- Law, B.Y.K.; Michelangeli, F.; Qu, Y.Q.; Xu, S.-W.; Han, Y.; Mok, S.W.F.; Dias, I.R.d.S.R.; Javed, M.-u.-H.; Chan, W.-K.; Xue, W.-W.; et al. Neferine induces autophagy-dependent cell death in apoptosis-resistant cancers via ryanodine receptor and Ca2+-dependent mechanism. Sci. Rep. 2019, 9, 20034. [Google Scholar] [CrossRef]
- Yoshikawa, N.; Liu, W.; Nakamura, K.; Yoshida, K.; Ikeda, Y.; Tanaka, H.; Mizuno, M.; Toyokuni, S.; Hori, M.; Kikkawa, F.; et al. Plasma-activated medium promotes autophagic cell death along with alteration of the mTOR pathway. Sci. Rep. 2020, 10, 1614. [Google Scholar] [CrossRef]
- Kong, W.; Zhu, H.; Zheng, S.; Yin, G.; Yu, P.; Shan, Y.; Liu, X.; Ying, R.; Zhu, H.; Ma, S.J.J.o.C.; et al. Larotrectinib induces autophagic cell death through AMPK/mTOR signalling in colon cancer. J. Cell. Mol. Med. 2022, 26, 5539–5550. [Google Scholar] [CrossRef]
- Yu, J.E.; Yeo, I.J.; Lee, D.W.; Chang, J.Y.; Son, D.J.; Yun, J.; Han, S.-B.; Hong, J.T. Snake venom induces an autophagic cell death via activation of the JNK pathway in colorectal cancer cells. J. Cancer 2022, 13, 3333–3341. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.W.; Baek, S.H.; Brennan, R.T.; Bradley, C.J.; Park, S.K.; Lee, Y.S.; Jun, E.J.; Lookingland, K.J.; Kim, E.K.; Lee, H.; et al. Autophagic death of adult hippocampal neural stem cells following insulin withdrawal. Stem Cells 2008, 26, 2602–2610. [Google Scholar] [CrossRef]
- Baek, S.-H.; Kim, E.-K.; Goudreau, J.L.; Lookingland, K.J.; Kim, S.W.; Yu, S.-W. Insulin withdrawal-induced cell death in adult hippocampal neural stem cells as a model of autophagic cell death. Autophagy 2009, 5, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Choe, S.; Woo, H.; Jeong, H.; An, H.-K.; Moon, H.; Ryu, H.Y.; Yeo, B.K.; Lee, Y.W.; Choi, H.; et al. Autophagic death of neural stem cells mediates chronic stress-induced decline of adult hippocampal neurogenesis and cognitive deficits. Autophagy 2020, 16, 512–530. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Jeong, S.H.; Yi, K.; Chung, K.M.; Hong, C.J.; Kim, S.W.; Kim, E.K.; Yu, S.W. Phosphorylation of p62 by AMP-activated protein kinase mediates autophagic cell death in adult hippocampal neural stem cells. J. Biol. Chem. 2017, 292, 13795–13808. [Google Scholar] [CrossRef]
- Ha, S.; Ryu, H.Y.; Chung, K.M.; Baek, S.H.; Kim, E.K.; Yu, S.W. Regulation of autophagic cell death by glycogen synthase kinase-3beta in adult hippocampal neural stem cells following insulin withdrawal. Mol. Brain 2015, 8, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.M.; Park, H.; Jung, S.; Ha, S.; Yoo, S.J.; Woo, H.; Lee, H.J.; Kim, S.W.; Kim, E.K.; Moon, C.; et al. Calpain Determines the Propensity of Adult Hippocampal Neural Stem Cells to Autophagic Cell Death Following Insulin Withdrawal. Stem Cells 2015, 33, 3052–3064. [Google Scholar] [CrossRef]
- Yeo, B.K.; Hong, C.J.; Chung, K.M.; Woo, H.; Kim, K.; Jung, S.; Kim, E.-K.; Yu, S.-W. Valosin-containing protein is a key mediator between autophagic cell death and apoptosis in adult hippocampal neural stem cells following insulin withdrawal. Mol. Brain 2016, 9, 31. [Google Scholar] [CrossRef]
- Park, H.; Chung, K.M.; An, H.-K.; Gim, J.-E.; Hong, J.; Woo, H.; Cho, B.; Moon, C.; Yu, S.-W. Parkin Promotes Mitophagic Cell Death in Adult Hippocampal Neural Stem Cells Following Insulin Withdrawal. Front. Mol. Neurosci. 2019, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.M.; Jeong, E.-J.; Park, H.; An, H.-K.; Yu, S.-W. Mediation of Autophagic Cell Death by Type 3 Ryanodine Receptor (RyR3) in Adult Hippocampal Neural Stem Cells. Front. Cell. Neurosci. 2016, 10, 116. [Google Scholar] [CrossRef]
- Shen, H.-M.; Codogno, P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy 2011, 7, 457–465. [Google Scholar] [CrossRef]
- Tower, D.B. The effects of 2-deoxy-D-glucose on metabolism of slices of cerebral cortex incubated in vitro. J. Neurochem. 1958, 3, 185–205. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.T.; Hwang, E.S. 2-Deoxyglucose: An anticancer and antiviral therapeutic, but not any more a low glucose mimetic. Life Sci. 2006, 78, 1392–1399. [Google Scholar] [CrossRef]
- Xi, H.; Kurtoglu, M.; Liu, H.; Wangpaichitr, M.; You, M.; Liu, X.; Savaraj, N.; Lampidis, T.J. 2-Deoxy-D-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother. Pharmacol. 2011, 67, 899–910. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the Autophagosome Maturation Process by a Novel Reporter Protein, Tandem Fluorescent-Tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Jo, C.; Kim, S.; Cho, S.J.; Choi, K.J.; Yun, S.M.; Koh, Y.H.; Johnson, G.V.; Park, S.I. Sulforaphane induces autophagy through ERK activation in neuronal cells. FEBS Lett. 2014, 588, 3081–3088. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chang, F.; Li, F.; Fu, H.; Wang, J.; Zhang, S.; Zhao, J.; Yin, D. Palmitate promotes autophagy and apoptosis through ROS-dependent JNK and p38 MAPK. Biochem. Biophys. Res. Commun. 2015, 463, 262–267. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, X.; Ma, S. The Roles of Mitochondria in Autophagic Cell Death. Cancer Biother. Radiopharm. 2016, 31, 269–276. [Google Scholar] [CrossRef]
- Zamaraeva, M.V.; Sabirov, R.Z.; Maeno, E.; Ando-Akatsuka, Y.; Bessonova, S.V.; Okada, Y. Cells die with increased cytosolic ATP during apoptosis: A bioluminescence study with intracellular luciferase. Cell. Death Differ. 2005, 12, 1390–1397. [Google Scholar] [CrossRef]
- Tsujimoto, Y. Apoptosis and necrosis: Intracellular ATP level as a determinant for cell death modes. Cell. Death Differ. 1997, 4, 429–434. [Google Scholar] [CrossRef]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar] [PubMed]
- Zamzami, N.; Marchetti, P.; Castedo, M.; Decaudin, D.; Macho, A.; Hirsch, T.; Susin, S.A.; Petit, P.X.; Mignotte, B.; Kroemer, G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 1995, 182, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Warnsmann, V.; Marschall, L.M.; Osiewacz, H.D. Impaired F(1)F(o)-ATP-Synthase Dimerization Leads to the Induction of Cyclophilin D-Mediated Autophagy-Dependent Cell Death and Accelerated Aging. Cells 2021, 10, 757. [Google Scholar] [CrossRef]
- Tiscornia, G.; Singer, O.; Verma, I.M. Production and purification of lentiviral vectors. Nat. Protoc. 2006, 1, 241–245. [Google Scholar] [CrossRef]
- Ryu, H.Y.; Kim, L.E.; Jeong, H.; Yeo, B.K.; Lee, J.-W.; Nam, H.; Ha, S.; An, H.-K.; Park, H.; Jung, S.J.E.; et al. GSK3B induces autophagy by phosphorylating ULK1. Exp. Mol. Med. 2021, 53, 369–383. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, S.-H.; An, H.-K.; Ha, S.; Yu, S.-W. Distinct Signaling Pathways for Autophagy-Driven Cell Death and Survival in Adult Hippocampal Neural Stem Cells. Int. J. Mol. Sci. 2023, 24, 8289. https://doi.org/10.3390/ijms24098289
Jeong S-H, An H-K, Ha S, Yu S-W. Distinct Signaling Pathways for Autophagy-Driven Cell Death and Survival in Adult Hippocampal Neural Stem Cells. International Journal of Molecular Sciences. 2023; 24(9):8289. https://doi.org/10.3390/ijms24098289
Chicago/Turabian StyleJeong, Seol-Hwa, Hyun-Kyu An, Shinwon Ha, and Seong-Woon Yu. 2023. "Distinct Signaling Pathways for Autophagy-Driven Cell Death and Survival in Adult Hippocampal Neural Stem Cells" International Journal of Molecular Sciences 24, no. 9: 8289. https://doi.org/10.3390/ijms24098289
APA StyleJeong, S.-H., An, H.-K., Ha, S., & Yu, S.-W. (2023). Distinct Signaling Pathways for Autophagy-Driven Cell Death and Survival in Adult Hippocampal Neural Stem Cells. International Journal of Molecular Sciences, 24(9), 8289. https://doi.org/10.3390/ijms24098289