
Cadmium Disrupted ER Ca2+ Homeostasis by Inhibiting SERCA2 Expression and Activity to Induce Apoptosis in Renal Proximal Tubular Cells

 ,
, 

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. NPS R-467 Protects against Cd2+-Induced Apoptosis Associated with Restoring Cytosolic Ca2+ Homeostasis
2.2. Cd2+-Induced Cytosolic Ca2+ Level Depends on ER Store
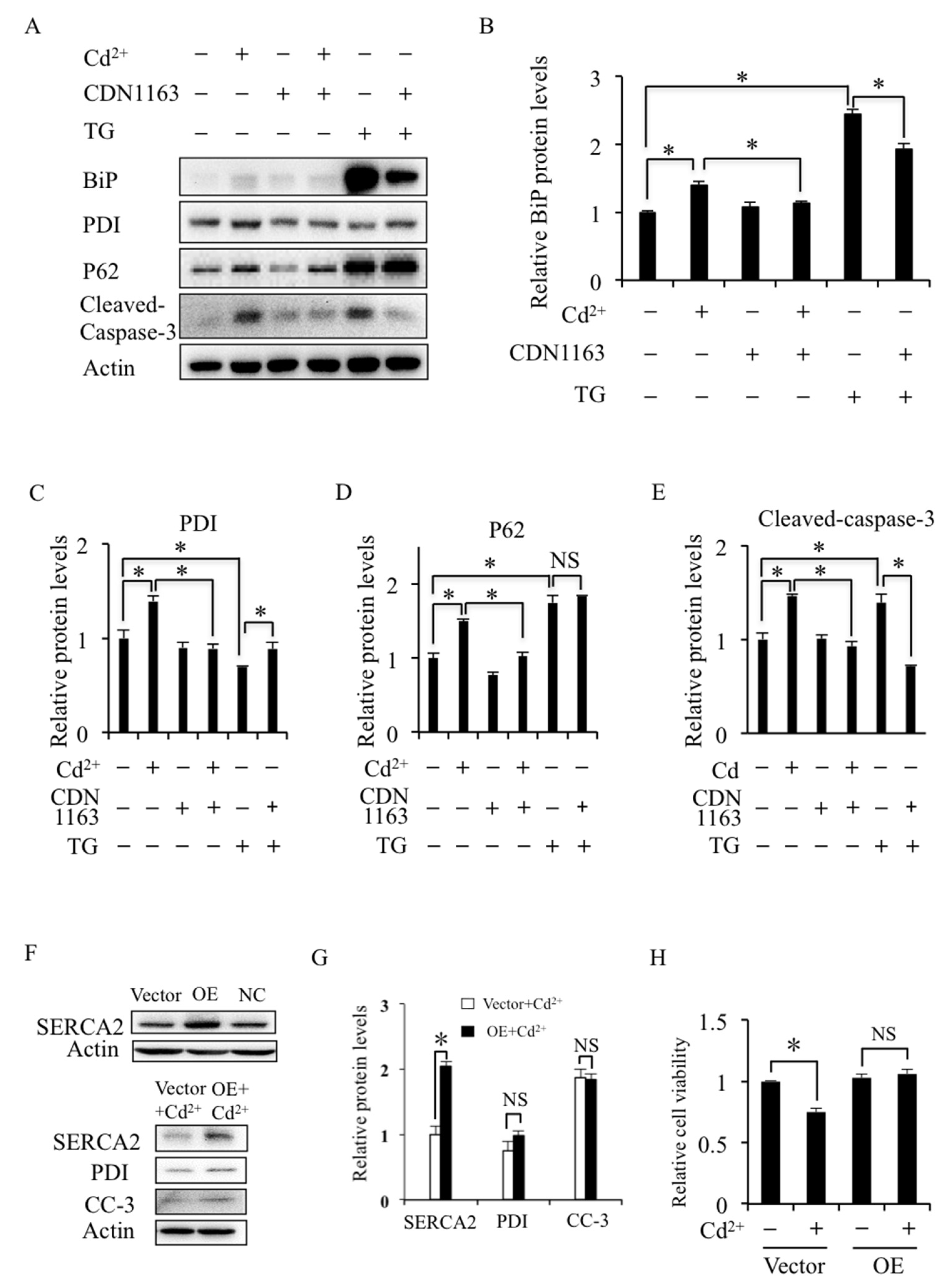
2.3. SERCA Inhibition Induces ER Stress and Apoptosis
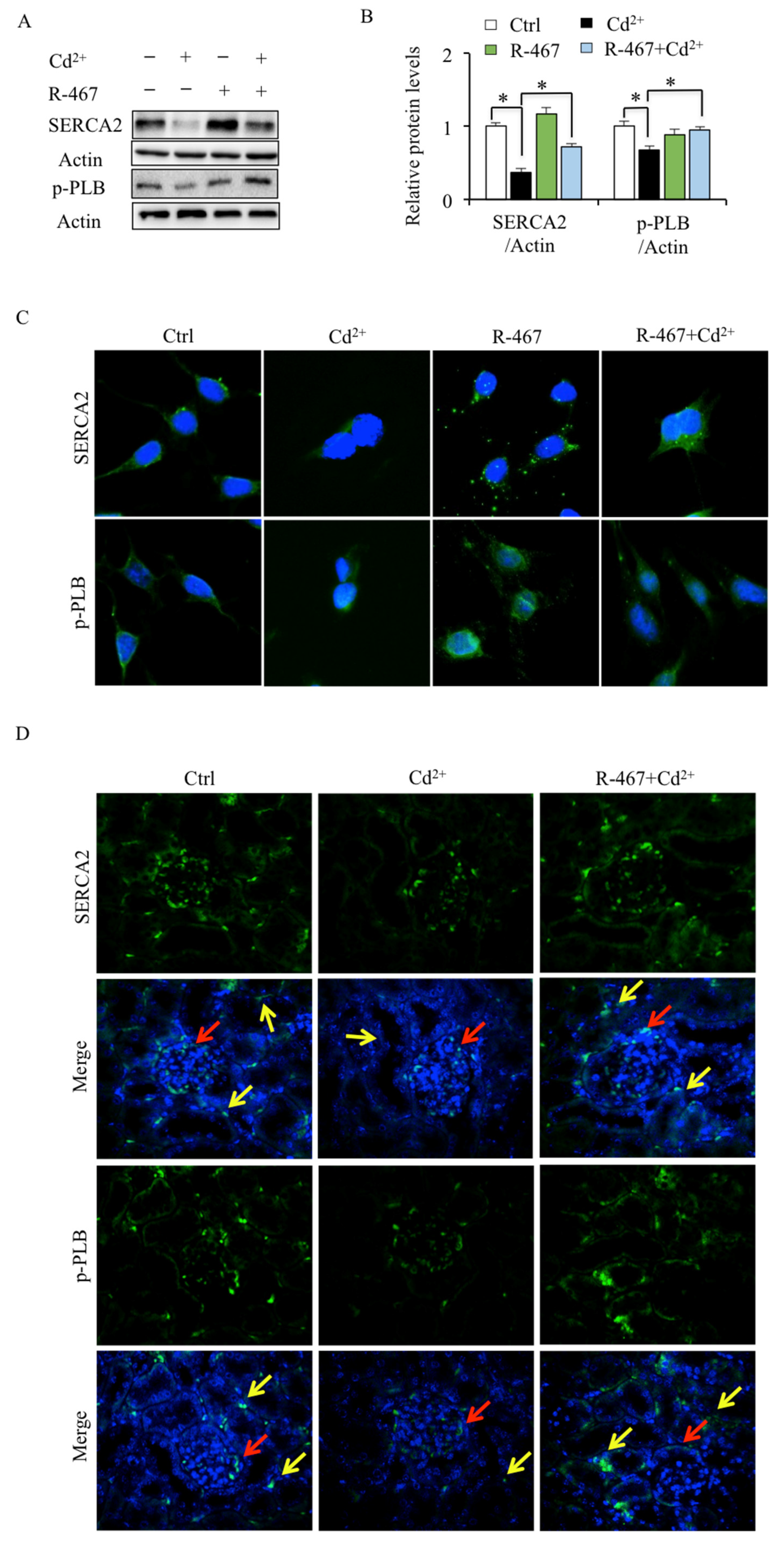
2.4. R-467 Reverses Cd2+-Inhibited SERCA2 and p-PLB Expression in Mice Kidney Cells and Tissues
2.5. Cd2+ Induces SERCA2 Protein Degradation through Proteasome Pathway
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animal Experiments and Cell Culture
4.3. Measurement of Cell Viability
4.4. Flow Cytometry
4.5. RNA Isolation and RT-PCR Analysis
4.6. Western Blotting Analysis
4.7. Plasmid Transfection
4.8. Protein Half-Life of SERCA2
4.9. Immunofluorescence
4.10. Measurement of Intracellular Ca2+ Level and ER Ca2+ Content
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jarup, L. Hazards of heavy metal contamination. Br. Med. Bull. 2003, 68, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Gerhardsson, L.; Englyst, V.; Lundström, N.; Sandberg, S.; Nordberg, G. Cadmium, copper and zinc in tissues of deceased copper smelter workers. J. Trace Elem. Med. Biol. 2002, 16, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Komoike, Y.; Inamura, H.; Matsuoka, M. Effects of salubrinal on cadmium-induced apoptosis in HK-2 human renal proximal tubular cells. Arch. Toxicol. 2012, 86, 37–44. [Google Scholar] [CrossRef]
- Fan, R.F.; Tang, K.K.; Wang, Z.Y.; Wang, L. Persistent activation of Nrf2 promotes a vicious cycle of oxidative stress and autophagy inhibition in cadmium-induced kidney injury. Toxicology 2021, 464, 152999. [Google Scholar] [CrossRef]
- Gu, J.; Wang, Y.; Liu, Y.; Shi, M.; Yin, L.; Hou, Y.; Zhou, Y.; Wong, C.K.C.; Chen, D.; Guo, Z.; et al. Inhibition of autophagy alleviates cadmium-induced mouse spleen and human B cells apoptosis. Toxicol. Sci. 2019, 170, 109–122. [Google Scholar] [CrossRef]
- Liu, F.; Wang, X.Y.; Zhou, X.P.; Liu, Z.P.; Song, X.B.; Wang, Z.Y.; Wang, L. Cadmium disrupts autophagic flux by inhibiting cytosolic Ca2+-dependent autophagosome-lysosome fusion in primary rat proximal tubular cells. Toxicology 2017, 383, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.; Guo, C.; Kong, A.; Li, K.; Xie, Y.; Shi, H.; Gu, J. Calcium signaling mediates cell death and crosstalk with autophagy in kidney disease. Cells 2021, 10, 3204. [Google Scholar] [CrossRef]
- Wang, X.Y.; Yang, H.; Wang, M.G.; Yang, D.B.; Wang, Z.Y.; Wang, L. Trehalose protects against cadmium-induced cytotoxicity in primary rat proximal tubular cells via inhibiting apoptosis and restoring autophagic flux. Cell Death Dis. 2017, 8, e3099. [Google Scholar] [CrossRef]
- Liu, F.; Li, Z.F.; Wang, Z.Y.; Wang, L. Role of subcellular calcium redistribution in regulating apoptosis and autophagy in cadmium-exposed primary rat proximal tubular cells. J. Inorg. Biochem. 2016, 164, 99–109. [Google Scholar] [CrossRef]
- Kosiba, A.A.; Wang, Y.; Chen, D.; Wong, C.K.C.; Gu, J.; Shi, H. The roles of calcium-sensing receptor (CaSR) in heavy metals-induced nephrotoxicity. Life Sci. 2020, 242, 117183. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.K.; Jiao, P.; Zhou, X.P.; Yang, D.B.; Wang, Z.Y.; Wang, L. Redistribution of subcellular calcium and its effect on apoptosis in primary cultures of rat proximal tubular cells exposed to lead. Toxicology 2015, 333, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Dai, S.; Liu, Y.; Liu, H.; Zhang, Y.; Ji, X.; Yu, F.; Zhou, Y.; Chen, L.; Tse, W.K.F.; et al. Activation of Ca2+-sensing receptor as a protective pathway to reduce Cadmium-induced cytotoxicity in renal proximal tubular cells. Sci. Rep. 2018, 8, 1092. [Google Scholar] [CrossRef] [PubMed]
- Biagioli, M.; Pifferi, S.; Ragghianti, M.; Bucci, S.; Rizzuto, R.; Pinton, P. Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium-induced apoptosis. Cell Calcium 2008, 43, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, M.; Tamma, G.D.M.A.; Vezzoli, G.; Soldati, L.; Svelto, M.; Valenti, G. Excessive signal transduction of gain-of-function variants of the calcium-sensing receptor (CaSR) are associated with increased ER to cytosol calcium gradient. PLoS ONE 2013, 8, e79113. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Ren, Z.; Zhao, J.; Peprah, F.A.; Xie, Y.; Cheng, D.; Wang, Y.; Liu, H.; Wong, C.K.C.; Zhou, Y.; et al. Calcimimetic compound NPS R-467 protects against chronic cadmium-induced mouse kidney injury by restoring autophagy process. Ecotoxicol. Environ. Saf. 2020, 189, 110052. [Google Scholar] [CrossRef] [PubMed]
- Orr, S.E.; Bridges, C.C. Chronic kidney disease and exposure to nephrotoxic metals. Int. J. Mol. Sci. 2017, 18, 1039. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Nie, S.; Ding, H.; Hou, F.F. Environmental pollution and kidney diseases. Nat. Rev. Nephrol. 2018, 14, 313–324. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17009. [Google Scholar] [CrossRef]
- Shi, H.; Sun, X.; Kong, A.; Ma, H.; Xie, Y.; Cheng, D.; Wong, C.K.C.; Zhou, Y.; Gu, J. Cadmium induces epithelial-mesenchymal transition and migration of renal cancer cells by increasing PGE2 through a cAMP/PKA-COX2 dependent mechanism. Ecotoxicol. Environ. Saf. 2021, 207, 111480. [Google Scholar] [CrossRef]
- Evenepoel, P.; Viaene, L.; Meijers, B. Calcium balance in chronic kidney disease: Walking the tightrope. Kidney Int. 2012, 81, 1057–1059. [Google Scholar] [CrossRef][Green Version]
- Luciani, D.S.; Gwiazda, K.S.; Yang, T.L.; Kalynyak, T.B.; Bychkivska, Y.; Frey, M.H.; Jeffrey, K.D.; Sampaio, A.V.; Underhill, T.M.; Johnson, J.D. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes 2009, 58, 422–432. [Google Scholar] [CrossRef]
- Seo, M.D.; Enomoto, M.; Ishiyama, N.; Stathopulos, P.B.; Ikura, M. Structural insights into endoplasmic reticulum stored calcium regulation by inositol 1,4,5-trisphosphate and ryanodine receptors. Biochim. Biophys. Acta 2015, 1853, 1980–1991. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Meng, Z.; Lu, J.; Chen, F.M.; Wong, W.T.; Tse, G.; Zheng, C.; Keung, W.; Tse, K.; Li, R.A.; et al. TRPV6 protects ER stress-induced apoptosis via ATF6α-TRPV6-JNK pathway in human embryonic stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 120, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Haustrate, A.; Prevarskaya, N.; Lehen’kyi, V. Role of the TRPV Channels in the Endoplasmic Reticulum Calcium Homeostasis. Cells 2020, 9, 317. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Danko, T.; Bergeron, M.J.; Balazs, B.; Suzuki, Y.; Zsembery, A.; Hediger, M.A. Heavy metal cations permeate the TRPV6 epithelial cation channel. Cell Calcium 2011, 49, 43–55. [Google Scholar] [CrossRef]
- Kovacs, G.; Montalbetti, N.; Franz, M.C.; Graeter, S.; Simonin, A.; Hediger, M.A. Human TRPV5 and TRPV6: Key players in cadmium and zinc toxicity. Cell Calcium 2013, 54, 276–286. [Google Scholar] [CrossRef]
- Yang, H.; Shu, Y. Cadmium transporters in the kidney and cadmium-induced nephrotoxicity. Int. J. Mol. Sci. 2015, 16, 1484–1494. [Google Scholar] [CrossRef]
- Aizman, O.; Uhlen, P.; Lal, M.; Brismar, H.; Aperia, A. Ouabain, a steroid hormone that signals with slow calcium oscillations. Proc. Natl. Acad. Sci. USA 2001, 98, 13420–13424. [Google Scholar] [CrossRef]
- Li, J.; Zelenin, S.; Aperia, A.; Aizman, O. Low doses of ouabain protect from serum deprivation-triggered apoptosis and stimulate kidney cell proliferation via activation of NF-kappaB. J. Am. Soc. Nephrol. 2006, 17, 1848–1857. [Google Scholar] [CrossRef]
- Yuan, Y.; Jiang, C.Y.; Xu, H.; Sun, Y.; Hu, F.F.; Bian, J.C.; Liu, X.Z.; Gu, J.H.; Liu, Z.P. Cadmium-induced apoptosis in primary rat cerebral cortical neurons culture is mediated by a calcium signaling pathway. PLoS ONE 2013, 8, e64330. [Google Scholar] [CrossRef]
- Kong, A.; Zhang, Y.; Ning, B.; Li, K.; Ren, Z.; Dai, S.; Chen, D.; Zhou, Y.; Gu, J.; Shi, H. Cadmium induces triglyceride levels via microsomal triglyceride transfer protein (MTTP) accumulation caused by lysosomal deacidification regulated by endoplasmic reticulum (ER) Ca2+ homeostasis. Chem. Biol. Interact. 2021, 348, 109649. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, Y.; Zhang, X.; Zang, Y.; Zhang, Y.; Wang, L.; Wang, H.; Wang, Y.; Cao, A.; Peng, W. Astragaloside IV protects against podocyte injury via SERCA2-dependent ER stress reduction and AMPKalpha-regulated autophagy induction in streptozotocin-induced diabetic nephropathy. Sci. Rep. 2017, 7, 6852. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Cao, A.; Chu, S.; Wang, Y.; Zang, Y.; Mao, X.; Wang, H.; Wang, Y.; Liu, C.; Zhang, X.; et al. Astragaloside IV attenuates podocyte apoptosis mediated by endoplasmic reticulum stress through upregulating sarco/endoplasmic reticulum Ca2+-ATPase 2 expression in diabetic nephropathy. Front. Pharmacol. 2016, 7, 500. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Contreras, C.; Saenz-Medina, J.; Munoz, M.; Corbacho, C.; Carballido, J.; Garcia-Sacristan, A.; Hernandez, M.; Lopez, M.; Rivera, L.; et al. Activation of the AMP-related kinase (AMPK) induces renal vasodilatation and downregulates Nox-derived reactive oxygen species (ROS) generation. Redox Biol. 2020, 34, 101575. [Google Scholar] [CrossRef]
- Lacave, J.M.; Bilbao, E.; Gilliland, D.; Mura, F.; Dini, L.; Cajaraville, M.P.; Orbea, A. Bioaccumulation, cellular and molecular effects in adult zebrafish after exposure to cadmium sulphide nanoparticles and to ionic cadmium. Chemosphere 2020, 238, 124588. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanjurjo, C.I.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Lysosomes shape Ins(1,4,5)P3-evoked Ca2+ signals by selectively sequestering Ca2+ released from the endoplasmic reticulum. J. Cell Sci. 2013, 126, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Xu, D.; Gu, J.; Xue, C.; Yang, B.; Fu, L.; Song, S.; Liu, D.; Zhou, W.; Lv, J.; et al. Saikosaponin-d inhibits proliferation by up-regulating autophagy via the CaMKKβ-AMPK-mTOR pathway in ADPKD cells. Mol. Cell Biochem. 2018, 449, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Zou, T.; Lin, Z. The roles of the ubiquitin-proteasome system in the endoplasmic reticulum stress pathway. Int. J. Mol. Sci. 2021, 22, 1526. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.S.; Shruthi, K.; Joy, D.; Reddy, G.B. 4-PBA prevents diabetic muscle atrophy in rats by modulating ER stress response and ubiquitin-proteasome system. Chem. Biol. Interact. 2019, 306, 70–77. [Google Scholar] [CrossRef]
- Shruthi, K.; Reddy, S.S.; Chitra, P.S.; Reddy, G.B. Ubiquitin-proteasome system and ER stress in the brain of diabetic rats. J. Cell. Biochem. 2019, 120, 5962–5973. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, K.; Guo, C.; Ruan, J.; Ning, B.; Wong, C.K.-C.; Shi, H.; Gu, J. Cadmium Disrupted ER Ca2+ Homeostasis by Inhibiting SERCA2 Expression and Activity to Induce Apoptosis in Renal Proximal Tubular Cells. Int. J. Mol. Sci. 2023, 24, 5979. https://doi.org/10.3390/ijms24065979
Li K, Guo C, Ruan J, Ning B, Wong CK-C, Shi H, Gu J. Cadmium Disrupted ER Ca2+ Homeostasis by Inhibiting SERCA2 Expression and Activity to Induce Apoptosis in Renal Proximal Tubular Cells. International Journal of Molecular Sciences. 2023; 24(6):5979. https://doi.org/10.3390/ijms24065979
Chicago/Turabian StyleLi, Kongdong, Chuanzhi Guo, Jiacheng Ruan, Bo Ning, Chris Kong-Chu Wong, Haifeng Shi, and Jie Gu. 2023. "Cadmium Disrupted ER Ca2+ Homeostasis by Inhibiting SERCA2 Expression and Activity to Induce Apoptosis in Renal Proximal Tubular Cells" International Journal of Molecular Sciences 24, no. 6: 5979. https://doi.org/10.3390/ijms24065979
APA StyleLi, K., Guo, C., Ruan, J., Ning, B., Wong, C. K.-C., Shi, H., & Gu, J. (2023). Cadmium Disrupted ER Ca2+ Homeostasis by Inhibiting SERCA2 Expression and Activity to Induce Apoptosis in Renal Proximal Tubular Cells. International Journal of Molecular Sciences, 24(6), 5979. https://doi.org/10.3390/ijms24065979