
Microdystrophin Gene Addition Significantly Improves Muscle Functionality and Diaphragm Muscle Histopathology in a Fibrotic Mouse Model of Duchenne Muscular Dystrophy

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
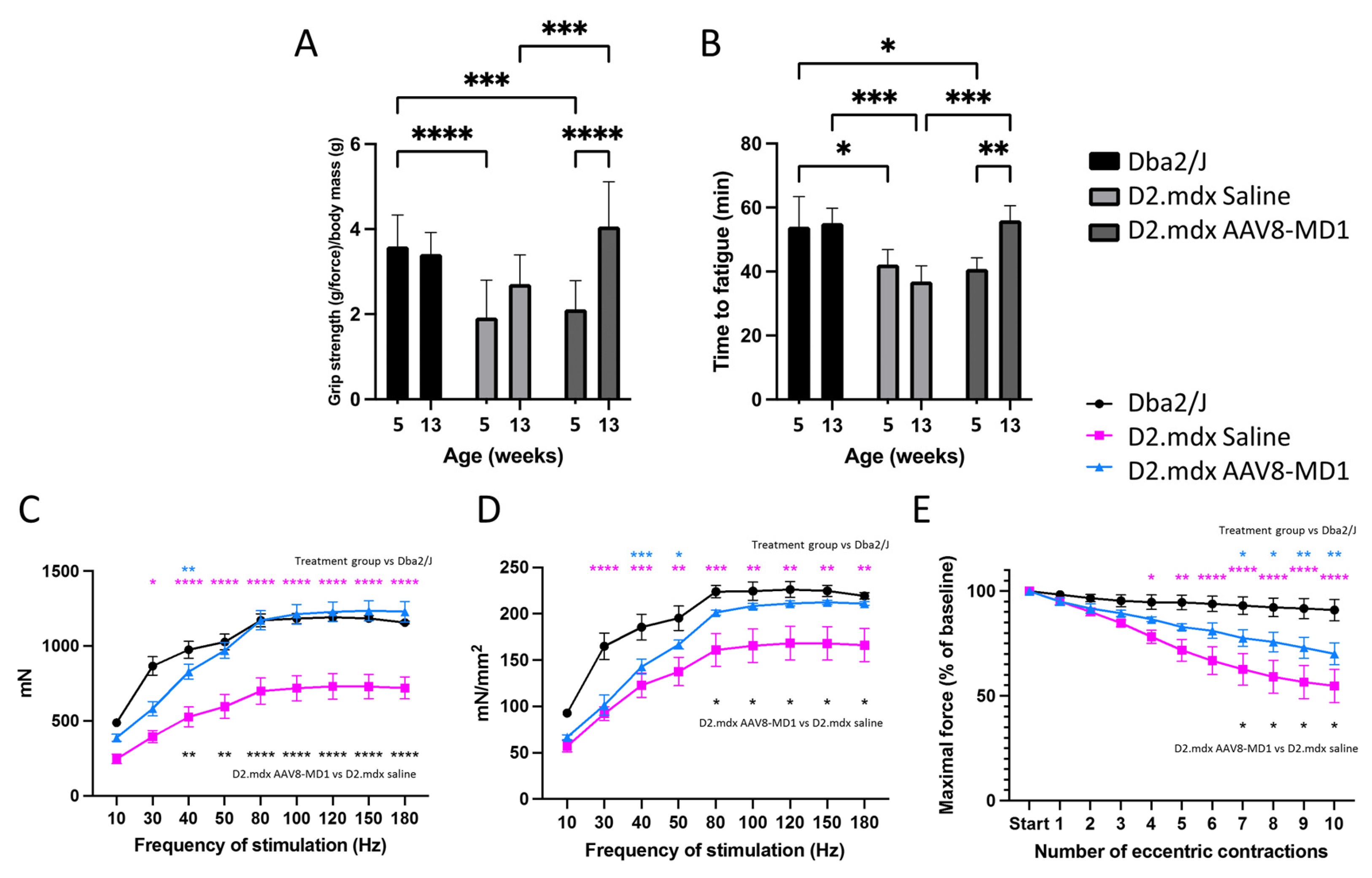
2.1. Administration of AAV8-MD1 Improves the Physical Abilities of D2.mdx Mice
2.2. Single Intravenous Administration of AAV8-MD1 Results in Significant Microdystrophin Protein Expression in the Diaphragm of D2.mdx Mice
2.3. Administration of AAV8-MD1 Improves Diaphragm Histopathology in D2.mdx Mice
2.4. Microdystrophin Gene Addition in D2.mdx Mice Prevents Fibrotic Tissue Deposition in Diaphragm Muscle
2.5. Administration of AAV8-MD1 Leads to Downregulation of Fibrosis and Inflammation Related Genes in the Diaphragm of D2.mdx Mice
3. Discussion
4. Materials and Methods
4.1. AAV8-MD1 Production
4.2. Animals and In Vivo Experimental Design
4.3. Forelimb Grip Strength Analysis
4.4. Fatigue Resistance Analysis
4.5. In Situ Muscle Electrophysiology
4.6. Sample Collection and Processing
4.7. Immunohistochemistry Staining
4.8. Histological Analysis
4.9. RNA Extraction from Tissues, cDNA Synthesis, and Quantitative Polymerase Chain Reaction
4.10. Protein Extraction from Tissue, Protein Assay, and Western Blot
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Monaco, A.P.; Kunkel, L.M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 1988, 53, 219–228. [Google Scholar] [CrossRef]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Aupy, P.; Goyenvalle, A. Exon-skipping advances for Duchenne muscular dystrophy. Hum. Mol. Genet. 2018, 27, R163–R172. [Google Scholar] [CrossRef] [PubMed]
- Duan, D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef]
- Malerba, A.; Sidoli, C.; Lu-Nguyen, N.B.; Herath, S.; Le Heron, A.; Abdul-Razak, H.; Jarmin, S.; Vandendriessche, T.; Chuah, M.K.L.; Dickson, G.; et al. Dose-dependent microdystrophin expression enhancement in cardiac muscle by a cardiac specific regulatory element. Hum. Gene Ther. 2021, 32, 1138–1146. [Google Scholar] [CrossRef]
- Sarcar, S.; Tulalamba, W.; Rincon, M.Y.; Tipanee, J.; Pham, H.Q.; Evens, H.; Boon, D.; Samara-Kuko, E.; Keyaerts, M.; Loperfido, M.; et al. Next-generation muscle-directed gene therapy by in silico vector design. Nat. Commun. 2019, 10, 492. [Google Scholar] [CrossRef]
- Li, D.; Long, C.; Yue, Y.; Duan, D. Sub-physiological sarcoglycan expression contributes to compensatory muscle protection in mdx mice. Hum. Mol. Genet. 2009, 18, 1209–1220. [Google Scholar] [CrossRef]
- Fukada, S.; Morikawa, D.; Yamamoto, Y.; Yoshida, T.; Sumie, N.; Yamaguchi, M.; Ito, T.; Miyagoe-Suzuki, Y.; Takeda, S.; Tsujikawa, K.; et al. Genetic background affects properties of satellite cells and mdx phenotypes. Am. J. Pathol. 2010, 176, 2414–2424. [Google Scholar] [CrossRef]
- Coley, W.D.; Bogdanik, L.; Vila, M.C.; Yu, Q.; Van Der Meulen, J.H.; Rayavarapu, S.; Novak, J.S.; Nearing, M.; Quinn, J.L.; Saunders, A.; et al. Effect of genetic background on the dystrophic phenotype in mdx mice. Hum. Mol. Genet. 2016, 25, 130–145. [Google Scholar] [CrossRef]
- van Putten, M.; Putker, K.; Overzier, M.; Adamzek, W.A.; Pasteuning-Vuhman, S.; Plomp, J.J.; Aartsma-Rus, A. Natural disease history of the D2-mdx mouse model for Duchenne muscular dystrophy. FASEB J. 2019, 33, 8110–8124. [Google Scholar] [CrossRef] [PubMed]
- Mazala, D.A.; Novak, J.S.; Hogarth, M.W.; Nearing, M.; Adusumalli, P.; Tully, C.B.; Habib, N.F.; Gordish-Dressman, H.; Chen, Y.W.; Jaiswal, J.K.; et al. TGF-beta-driven muscle degeneration and failed regeneration underlie disease onset in a DMD mouse model. JCI Insight 2020, 5, e135703. [Google Scholar] [CrossRef] [PubMed]
- Heydemann, A.; Ceco, E.; Lim, J.E.; Hadhazy, M.; Ryder, P.; Moran, J.L.; Beier, D.R.; Palmer, A.A.; McNally, E.M. Latent TGF-beta-binding protein 4 modifies muscular dystrophy in mice. J. Clin. Investig. 2009, 119, 3703–3712. [Google Scholar] [CrossRef]
- Todorovic, V.; Rifkin, D.B. LTBPs, more than just an escort service. J. Cell. Biochem. 2012, 113, 410–418. [Google Scholar] [CrossRef]
- Ceco, E.; Bogdanovich, S.; Gardner, B.; Miller, T.; DeJesus, A.; Earley, J.U.; Hadhazy, M.; Smith, L.R.; Barton, E.R.; Molkentin, J.D.; et al. Targeting latent TGFbeta release in muscular dystrophy. Sci. Transl. Med. 2014, 6, 259ra144. [Google Scholar] [CrossRef] [PubMed]
- Florini, J.R.; Roberts, A.B.; Ewton, D.Z.; Falen, S.L.; Flanders, K.C.; Sporn, M.B. Transforming growth factor-beta. A very potent inhibitor of myoblast differentiation, identical to the differentiation inhibitor secreted by Buffalo rat liver cells. J. Biol. Chem. 1986, 261, 16509–16513. [Google Scholar] [CrossRef]
- Massague, J.; Cheifetz, S.; Endo, T.; Nadal-Ginard, B. Type beta transforming growth factor is an inhibitor of myogenic differentiation. Proc. Natl. Acad. Sci. USA 1986, 83, 8206–8210. [Google Scholar] [CrossRef]
- Olson, E.N.; Sternberg, E.; Hu, J.S.; Spizz, G.; Wilcox, C. Regulation of myogenic differentiation by type beta transforming growth factor. J. Cell Biol. 1986, 103, 1799–1805. [Google Scholar] [CrossRef]
- Allen, R.E.; Boxhorn, L.K. Regulation of skeletal muscle satellite cell proliferation and differentiation by transforming growth factor-beta, insulin-like growth factor I, and fibroblast growth factor. J. Cell Physiol. 1989, 138, 311–315. [Google Scholar] [CrossRef]
- Hakim, C.H.; Wasala, N.B.; Pan, X.; Kodippili, K.; Yue, Y.; Zhang, K.; Yao, G.; Haffner, B.; Duan, S.X.; Ramos, J.; et al. A Five-Repeat Micro-Dystrophin Gene Ameliorated Dystrophic Phenotype in the Severe DBA/2J-mdx Model of Duchenne Muscular Dystrophy. Mol. Ther. Methods Clin. Dev. 2017, 6, 216–230. [Google Scholar] [CrossRef]
- Kennedy, T.L.; Guiraud, S.; Edwards, B.; Squire, S.; Moir, L.; Babbs, A.; Odom, G.; Golebiowski, D.; Schneider, J.; Chamberlain, J.S.; et al. Micro-utrophin Improves Cardiac and Skeletal Muscle Function of Severely Affected D2/mdx Mice. Mol. Ther. Methods Clin. Dev. 2018, 11, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Hammers, D.W.; Hart, C.C.; Matheny, M.K.; Wright, L.A.; Armellini, M.; Barton, E.R.; Sweeney, H.L. The D2.mdx mouse as a preclinical model of the skeletal muscle pathology associated with Duchenne muscular dystrophy. Sci. Rep. 2020, 10, 14070. [Google Scholar] [CrossRef]
- de Greef, J.C.; Hamlyn, R.; Jensen, B.S.; O’Campo Landa, R.; Levy, J.R.; Kobuke, K.; Campbell, K.P. Collagen VI deficiency reduces muscle pathology, but does not improve muscle function, in the gamma-sarcoglycan-null mouse. Hum. Mol. Genet. 2016, 25, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Boldrin, L.; Ross, J.A.; Whitmore, C.; Doreste, B.; Beaver, C.; Eddaoudi, A.; Pearce, D.J.; Morgan, J.E. The effect of calorie restriction on mouse skeletal muscle is sex, strain and time-dependent. Sci. Rep. 2017, 7, 5160. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Dowling, P.; Meleady, P.; Henry, M.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Label-free mass spectrometric analysis of the mdx-4cv diaphragm identifies the matricellular protein periostin as a potential factor involved in dystrophinopathy-related fibrosis. Proteomics 2015, 15, 2318–2331. [Google Scholar] [CrossRef] [PubMed]
- Lu-Nguyen, N.; Dickson, G.; Malerba, A.; Popplewell, L. Long-Term Systemic Treatment of a Mouse Model Displaying Chronic FSHD-like Pathology with Antisense Therapeutics That Inhibit DUX4 Expression. Biomedicines 2022, 10, 1623. [Google Scholar] [CrossRef] [PubMed]
- Foster, H.; Sharp, P.S.; Athanasopoulos, T.; Trollet, C.; Graham, I.R.; Foster, K.; Wells, D.J.; Dickson, G. Codon and mRNA sequence optimization of microdystrophin transgenes improves expression and physiological outcome in dystrophic mdx mice following AAV2/8 gene transfer. Mol. Ther. 2008, 16, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Le Guiner, C.; Servais, L.; Montus, M.; Larcher, T.; Fraysse, B.; Moullec, S.; Allais, M.; Francois, V.; Dutilleul, M.; Malerba, A.; et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 16105. [Google Scholar] [CrossRef]
- Koo, T.; Malerba, A.; Athanasopoulos, T.; Trollet, C.; Boldrin, L.; Ferry, A.; Popplewell, L.; Foster, H.; Foster, K.; Dickson, G. Delivery of AAV2/9-microdystrophin genes incorporating helix 1 of the coiled-coil motif in the C-terminal domain of dystrophin improves muscle pathology and restores the level of alpha1-syntrophin and alpha-dystrobrevin in skeletal muscles of mdx mice. Hum. Gene. Ther. 2011, 22, 1379–1388. [Google Scholar] [CrossRef]
- Vohra, R.; Batra, A.; Forbes, S.C.; Vandenborne, K.; Walter, G.A. Magnetic Resonance Monitoring of Disease Progression in mdx Mice on Different Genetic Backgrounds. Am. J. Pathol. 2017, 187, 2060–2070. [Google Scholar] [CrossRef]
- Mann, C.J.; Perdiguero, E.; Kharraz, Y.; Aguilar, S.; Pessina, P.; Serrano, A.L.; Munoz-Canoves, P. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 2011, 1, 21. [Google Scholar] [CrossRef]
- Ozdemir, C.; Akpulat, U.; Sharafi, P.; Yildiz, Y.; Onbasilar, I.; Kocaefe, C. Periostin is temporally expressed as an extracellular matrix component in skeletal muscle regeneration and differentiation. Gene 2014, 553, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Lorts, A.; Schwanekamp, J.A.; Baudino, T.A.; McNally, E.M.; Molkentin, J.D. Deletion of periostin reduces muscular dystrophy and fibrosis in mice by modulating the transforming growth factor-beta pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 10978–10983. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Thomas, G.D.; Yue, Y.; Yang, H.T.; Li, D.; Long, C.; Judge, L.; Bostick, B.; Chamberlain, J.S.; Terjung, R.L.; et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Investig. 2009, 119, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Zhao, J.; Yue, Y.; Duan, D. alpha2 and alpha3 helices of dystrophin R16 and R17 frame a microdomain in the alpha1 helix of dystrophin R17 for neuronal NOS binding. Proc. Natl. Acad. Sci. USA 2013, 110, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yue, Y.; Lai, Y.; Hakim, C.H.; Duan, D. Nitrosative stress elicited by nNOSmicro delocalization inhibits muscle force in dystrophin-null mice. J. Pathol. 2011, 223, 88–98. [Google Scholar] [CrossRef]
- Walsh, F.S.; Moore, S.E.; Dhut, S. Monoclonal antibody to human fibronectin: Production and characterization using human muscle cultures. Dev. Biol. 1981, 84, 121–132. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cernisova, V.; Lu-Nguyen, N.; Trundle, J.; Herath, S.; Malerba, A.; Popplewell, L. Microdystrophin Gene Addition Significantly Improves Muscle Functionality and Diaphragm Muscle Histopathology in a Fibrotic Mouse Model of Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2023, 24, 8174. https://doi.org/10.3390/ijms24098174
Cernisova V, Lu-Nguyen N, Trundle J, Herath S, Malerba A, Popplewell L. Microdystrophin Gene Addition Significantly Improves Muscle Functionality and Diaphragm Muscle Histopathology in a Fibrotic Mouse Model of Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2023; 24(9):8174. https://doi.org/10.3390/ijms24098174
Chicago/Turabian StyleCernisova, Viktorija, Ngoc Lu-Nguyen, Jessica Trundle, Shan Herath, Alberto Malerba, and Linda Popplewell. 2023. "Microdystrophin Gene Addition Significantly Improves Muscle Functionality and Diaphragm Muscle Histopathology in a Fibrotic Mouse Model of Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 24, no. 9: 8174. https://doi.org/10.3390/ijms24098174
APA StyleCernisova, V., Lu-Nguyen, N., Trundle, J., Herath, S., Malerba, A., & Popplewell, L. (2023). Microdystrophin Gene Addition Significantly Improves Muscle Functionality and Diaphragm Muscle Histopathology in a Fibrotic Mouse Model of Duchenne Muscular Dystrophy. International Journal of Molecular Sciences, 24(9), 8174. https://doi.org/10.3390/ijms24098174