
The Histone Deacetylases Hst1 and Rpd3 Integrate De Novo NAD+ Metabolism with Phosphate Sensing in Saccharomyces cerevisiae
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
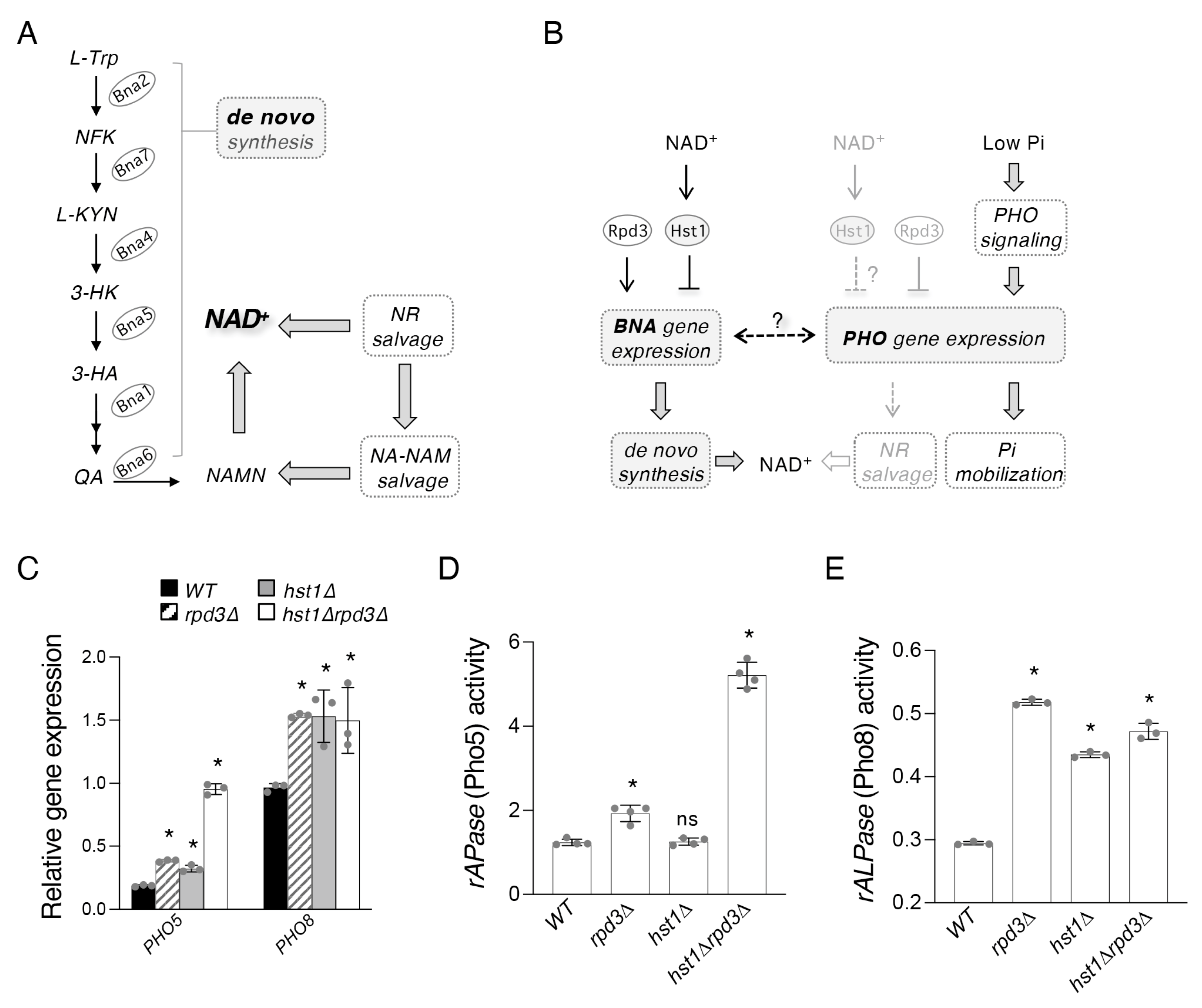
2.1. Hst1 and Rpd3 Regulate Targets of the PHO Pathway
2.2. The Bas1–Pho2 Complex Plays a Role in Hst1- and Rpd3-Mediated Regulation of De Novo NAD+ Metabolism
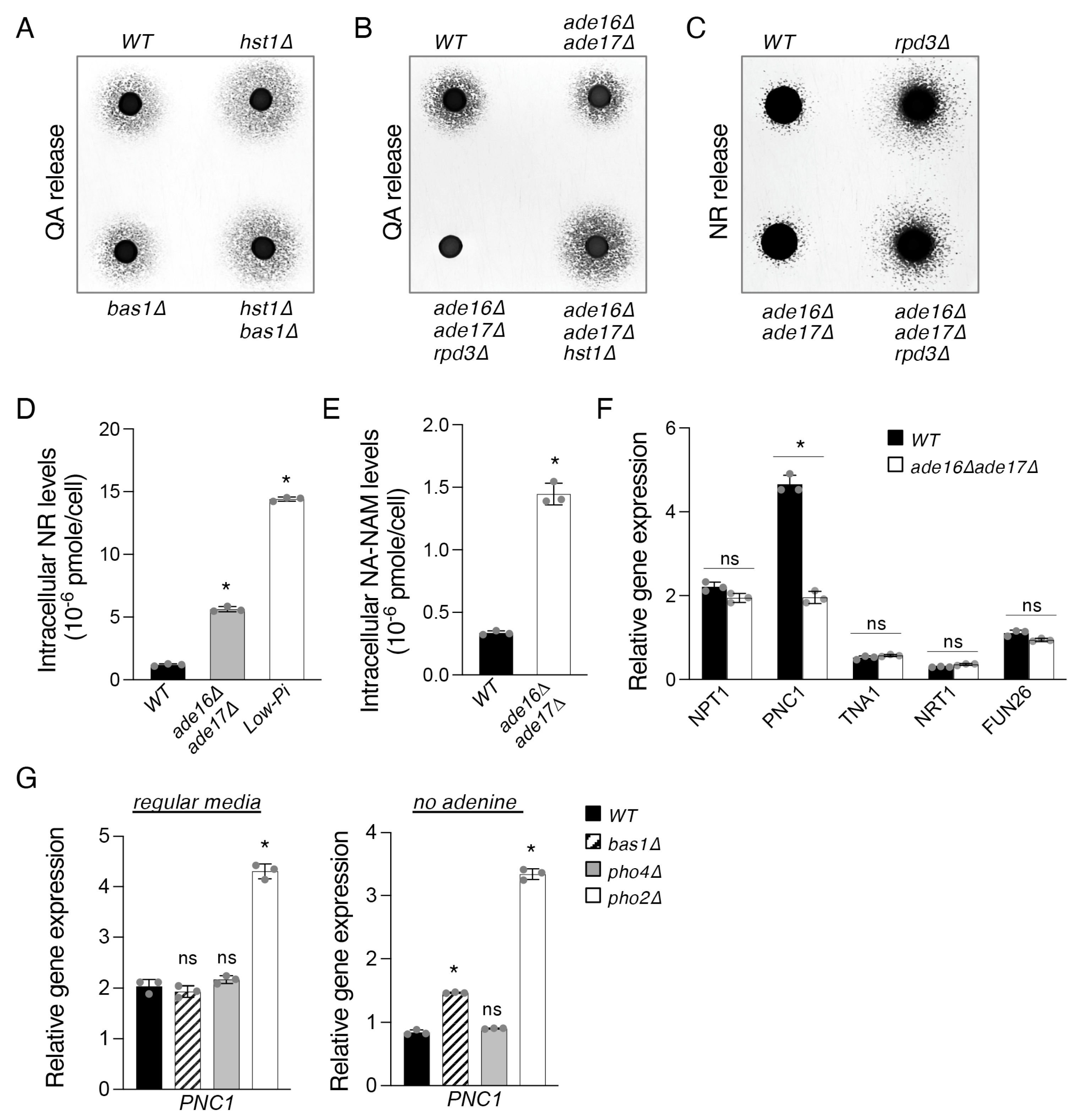
2.3. The ade16Δade17Δ Mutant Exhibits Altered Cellular NR and NA-NAM Metabolism
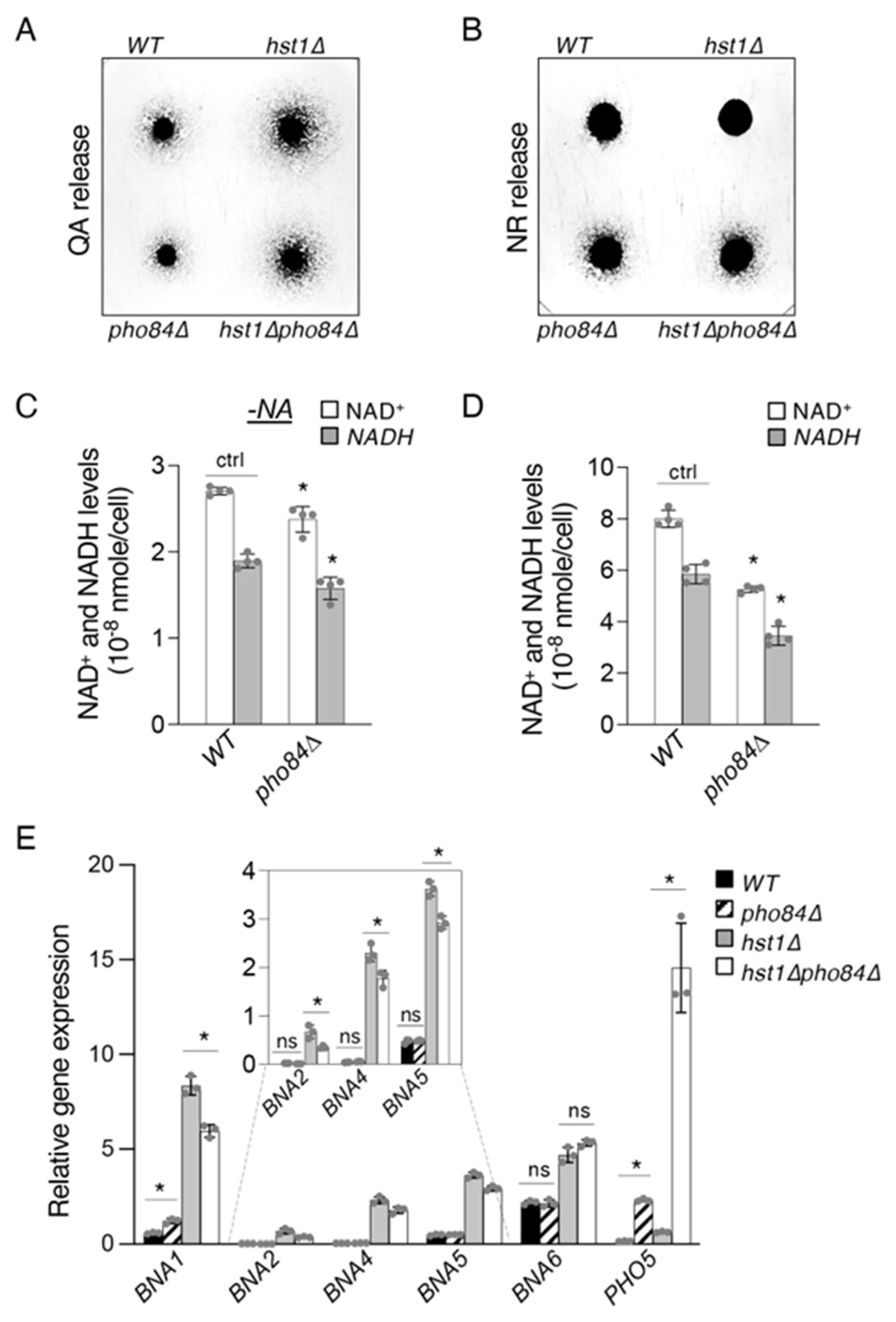
2.4. Pho84-Mediated Phosphate Transport Affects Homeostasis of NAD+ Precursors
2.5. Phosphate Depletion Increases Pho2 Binding Activity at the PHO5 Promoter but Not at the BNA2 Promoter
3. Discussion
4. Materials and Methods
4.1. Yeast Strains, Growth Media, and Plasmids
4.2. Quantitative PCR (qPCR) Quantitation of Gene Expression
4.3. Repressible Acid Phosphatase (rAPase) Activity and Alkaline Phosphatase (rALPase) Assays
4.4. QA and NR Cross-Feeding Plate Assays
4.5. Measurement(s) of NAD+, NADH, QA, NR, and NA-NAM
4.6. Protein Extraction and Western Blot Analysis
4.7. Chromatin Immunoprecipitation (ChIP) Assay
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD(+) metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Croft, T.; Venkatakrishnan, P.; Lin, S.J. NAD(+) Metabolism and Regulation: Lessons From Yeast. Biomolecules 2020, 10, 330. [Google Scholar] [CrossRef] [PubMed]
- Landry, J.; Sutton, A.; Tafrov, S.T.; Heller, R.C.; Stebbins, J.; Pillus, L.; Sternglanz, R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 5807–5811. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Smith, J.S.; Brachmann, C.B.; Celic, I.; Kenna, M.A.; Muhammad, S.; Starai, V.J.; Avalos, J.L.; Escalante-Semerena, J.C.; Grubmeyer, C.; Wolberger, C.; et al. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. USA 2000, 97, 6658–6663. [Google Scholar] [CrossRef]
- Kraus, W.L. PARPs and ADP-Ribosylation: 50 Years… and Counting. Mol. Cell 2015, 58, 902–910. [Google Scholar] [CrossRef]
- Okabe, K.; Yaku, K.; Tobe, K.; Nakagawa, T. Implications of altered NAD metabolism in metabolic disorders. J. Biomed. Sci. 2019, 26, 34. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD(+) in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Lin, Q.; Zuo, W.; Liu, Y.; Wu, K.; Liu, Q. NAD(+) and cardiovascular diseases. Clin. Chim. Acta Int. J. Clin. Chem. 2021, 515, 104–110. [Google Scholar] [CrossRef]
- Tummala, K.S.; Gomes, A.L.; Yilmaz, M.; Graña, O.; Bakiri, L.; Ruppen, I.; Ximénez-Embún, P.; Sheshappanavar, V.; Rodriguez-Justo, M.; Pisano, D.G.; et al. Inhibition of De Novo NAD+ Synthesis by Oncogenic URI Causes Liver Tumorigenesis through DNA Damage. Cancer Cell 2014, 26, 826–839. [Google Scholar] [CrossRef]
- Hong, S.M.; Hwang, S.W.; Wang, T.; Park, C.W.; Ryu, Y.M.; Jung, J.H.; Shin, J.H.; Kim, S.Y.; Lee, J.L.; Kim, C.W.; et al. Increased nicotinamide adenine dinucleotide pool promotes colon cancer progression by suppressing reactive oxygen species level. Cancer Sci. 2019, 110, 629–638. [Google Scholar] [CrossRef]
- Gujar, A.D.; Le, S.; Mao, D.D.; Dadey, D.Y.A.; Turski, A.; Sasaki, Y.; Aum, D.; Luo, J.; Dahiya, S.; Yuan, L.; et al. An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proc. Natl. Acad. Sci. USA 2016, 113, E8247–E8256. [Google Scholar] [CrossRef]
- Jahan, F.; Bagchi, R.A. Enhancing NAD(+) Metabolome in Cardiovascular Diseases: Promises and Considerations. Front. Cardiovasc. Med. 2021, 8, 716989. [Google Scholar] [CrossRef]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef]
- Zhen, X.; Zhang, S.; Xie, F.; Zhou, M.; Hu, Z.; Zhu, F.; Nie, J. Nicotinamide Supplementation Attenuates Renal Interstitial Fibrosis via Boosting the Activity of Sirtuins. Kidney Dis. 2021, 7, 186–199. [Google Scholar] [CrossRef]
- Lu, S.P.; Lin, S.J. Phosphate-responsive signaling pathway is a novel component of NAD+ metabolism in Saccharomyces cerevisiae. J. Biol. Chem. 2011, 286, 14271–14281. [Google Scholar] [CrossRef]
- Tsang, F.; James, C.; Kato, M.; Myers, V.; Ilyas, I.; Tsang, M.; Lin, S.J. Reduced Ssy1-Ptr3-Ssy5 (SPS) signaling extends replicative life span by enhancing NAD+ homeostasis in Saccharomyces cerevisiae. J. Biol. Chem. 2015, 290, 12753–12764. [Google Scholar] [CrossRef]
- Pinson, B.; Ceschin, J.; Saint-Marc, C.; Daignan-Fornier, B. Dual control of NAD+ synthesis by purine metabolites in yeast. eLife 2019, 8, e43808. [Google Scholar] [CrossRef]
- James Theoga Raj, C.; Croft, T.; Venkatakrishnan, P.; Groth, B.; Dhugga, G.; Cater, T.; Lin, S.J. The copper-sensing transcription factor Mac1, the histone deacetylase Hst1, and nicotinic acid regulate de novo NAD(+) biosynthesis in budding yeast. J. Biol. Chem. 2019, 294, 5562–5575. [Google Scholar] [CrossRef]
- Groth, B.; Venkatakrishnan, P.; Lin, S.J. NAD(+) Metabolism, Metabolic Stress, and Infection. Front. Mol. Biosci. 2021, 8, 686412. [Google Scholar] [CrossRef]
- Tsang, F.; Lin, S.-J. Less is more: Nutrient limitation induces cross-talk of nutrient sensing pathways with NAD+ homeostasis and contributes to longevity. Front. Biol. 2015, 10, 333–357. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ghislain, M.; Talla, E.; Francois, J.M. Identification and functional analysis of the Saccharomyces cerevisiae nicotinamidase gene, PNC1. Yeast 2002, 19, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Emanuelli, M.; Carnevali, F.; Lorenzi, M.; Raffaelli, N.; Amici, A.; Ruggieri, S.; Magni, G. Identification and characterization of YLR328W, the Saccharomyces cerevisiae structural gene encoding NMN adenylyltransferase. Expression and characterization of the recombinant enzyme. FEBS Lett. 1999, 455, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Bitterman, K.J.; Wood, J.G.; Medvedik, O.; Cohen, H.; Lin, S.S.; Manchester, J.K.; Gordon, J.I.; Sinclair, D.A. Manipulation of a nuclear NAD+ salvage pathway delays aging without altering steady-state NAD+ levels. J. Biol. Chem. 2002, 277, 18881–18890. [Google Scholar] [CrossRef]
- Bieganowski, P.; Pace, H.C.; Brenner, C. Eukaryotic NAD+ synthetase Qns1 contains an essential, obligate intramolecular thiol glutamine amidotransferase domain related to nitrilase. J. Biol. Chem. 2003, 278, 33049–33055. [Google Scholar] [CrossRef]
- Belenky, P.; Racette, F.G.; Bogan, K.L.; McClure, J.M.; Smith, J.S.; Brenner, C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 2007, 129, 473–484. [Google Scholar] [CrossRef]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef]
- Kato, M.; Lin, S.J. YCL047C/POF1 Is a Novel Nicotinamide Mononucleotide Adenylyltransferase (NMNAT) in Saccharomyces cerevisiae. J. Biol. Chem. 2014, 289, 15577–15587. [Google Scholar] [CrossRef]
- Bedalov, A.; Hirao, M.; Posakony, J.; Nelson, M.; Simon, J.A. NAD+-dependent deacetylase Hst1p controls biosynthesis and cellular NAD+ levels in Saccharomyces cerevisiae. Mol. Cell. Biol. 2003, 23, 7044–7054. [Google Scholar] [CrossRef]
- Groth, B.; Huang, C.C.; Lin, S.J. The histone deacetylases Rpd3 and Hst1 antagonistically regulate de novo NAD(+) metabolism in the budding yeast Saccharomyces cerevisiae. J. Biol. Chem. 2022, 298, 102410. [Google Scholar] [CrossRef]
- O’Neill, E.M.; Kaffman, A.; Jolly, E.R.; O’Shea, E.K. Regulation of PHO4 nuclear localization by the PHO80-PHO85 cyclin-CDK complex. Science 1996, 271, 209–212. [Google Scholar] [CrossRef]
- Kaffman, A.; Rank, N.M.; O’Shea, E.K. Phosphorylation regulates association of the transcription factor Pho4 with its import receptor Pse1/Kap121. Genes Dev. 1998, 12, 2673–2683. [Google Scholar] [CrossRef]
- Pinson, B.; Vaur, S.; Sagot, I.; Coulpier, F.; Lemoine, S.; Daignan-Fornier, B. Metabolic intermediates selectively stimulate transcription factor interaction and modulate phosphate and purine pathways. Genes Dev. 2009, 23, 1399–1407. [Google Scholar] [CrossRef]
- Vidal, M.; Gaber, R.F. RPD3 encodes a second factor required to achieve maximum positive and negative transcriptional states in Saccharomyces cerevisiae. Mol. Cell. Biol. 1991, 11, 6317–6327. [Google Scholar] [CrossRef]
- Wongwisansri, S.; Laybourn, P.J. Disruption of histone deacetylase gene RPD3 accelerates PHO5 activation kinetics through inappropriate Pho84p recycling. Eukaryot. Cell 2005, 4, 1387–1395. [Google Scholar] [CrossRef]
- Korber, P.; Barbaric, S. The yeast PHO5 promoter: From single locus to systems biology of a paradigm for gene regulation through chromatin. Nucleic Acids Res. 2014, 42, 10888–10902. [Google Scholar] [CrossRef]
- Lu, S.P.; Kato, M.; Lin, S.J. Assimilation of endogenous nicotinamide riboside is essential for calorie restriction-mediated life span extension in Saccharomyces cerevisiae. J. Biol. Chem. 2009, 284, 17110–17119. [Google Scholar] [CrossRef]
- Reinke, H.; Gregory, P.D.; Hörz, W. A transient histone hyperacetylation signal marks nucleosomes for remodeling at the PHO8 promoter in vivo. Mol. Cell 2001, 7, 529–538. [Google Scholar] [CrossRef]
- Boer, V.M.; Crutchfield, C.A.; Bradley, P.H.; Botstein, D.; Rabinowitz, J.D. Growth-limiting intracellular metabolites in yeast growing under diverse nutrient limitations. Mol. Biol. Cell 2010, 21, 198–211. [Google Scholar] [CrossRef]
- Gauthier, S.; Coulpier, F.; Jourdren, L.; Merle, M.; Beck, S.; Konrad, M.; Daignan-Fornier, B.; Pinson, B. Co-regulation of yeast purine and phosphate pathways in response to adenylic nucleotide variations. Mol. Microbiol. 2008, 68, 1583–1594. [Google Scholar] [CrossRef]
- Takaine, M.; Imamura, H.; Yoshida, S. High and stable ATP levels prevent aberrant intracellular protein aggregation in yeast. eLife 2022, 11, e67659. [Google Scholar] [CrossRef] [PubMed]
- Vogel, K.; Horz, W.; Hinnen, A. The two positively acting regulatory proteins PHO2 and PHO4 physically interact with PHO5 upstream activation regions. Mol. Cell. Biol. 1989, 9, 2050–2057. [Google Scholar] [CrossRef] [PubMed]
- Munsterkotter, M.; Barbaric, S.; Horz, W. Transcriptional regulation of the yeast PHO8 promoter in comparison to the coregulated PHO5 promoter. J. Biol. Chem. 2000, 275, 22678–22685. [Google Scholar] [CrossRef] [PubMed]
- Bun-Ya, M.; Nishimura, M.; Harashima, S.; Oshima, Y. The PHO84 gene of Saccharomyces cerevisiae encodes an inorganic phosphate transporter. Mol. Cell. Biol. 1991, 11, 3229–3238. [Google Scholar] [CrossRef]
- Bitterman, K.J.; Anderson, R.M.; Cohen, H.Y.; Latorre-Esteves, M.; Sinclair, D.A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002, 277, 45099–45107. [Google Scholar] [CrossRef]
- Jackson, M.D.; Schmidt, M.T.; Oppenheimer, N.J.; Denu, J.M. Mechanism of Nicotinamide Inhibition and Transglycosidation by Sir2 Histone/Protein Deacetylases. J. Biol. Chem. 2003, 278, 50985–50998. [Google Scholar] [CrossRef]
- Auesukaree, C.; Homma, T.; Tochio, H.; Shirakawa, M.; Kaneko, Y.; Harashima, S. Intracellular phosphate serves as a signal for the regulation of the PHO pathway in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 17289–17294. [Google Scholar] [CrossRef]
- Rudolph, H.; Hinnen, A. The yeast PHO5 promoter: Phosphate-control elements and sequences mediating mRNA start-site selection. Proc. Natl. Acad. Sci. USA 1987, 84, 1340–1344. [Google Scholar] [CrossRef]
- Barbarić, S.; Münsterkötter, M.; Svaren, J.; Hörz, W. The homeodomain protein Pho2 and the basic-helix-loop-helix protein Pho4 bind DNA cooperatively at the yeast PHO5 promoter. Nucleic Acids Res. 1996, 24, 4479–4486. [Google Scholar] [CrossRef][Green Version]
- Barbaric, S.; Münsterkötter, M.; Goding, C.; Hörz, W. Cooperative Pho2-Pho4 interactions at the PHO5 promoter are critical for binding of Pho4 to UASp1 and for efficient transactivation by Pho4 at UASp2. Mol. Cell. Biol. 1998, 18, 2629–2639. [Google Scholar] [CrossRef]
- Rundlett, S.E.; Carmen, A.A.; Kobayashi, R.; Bavykin, S.; Turner, B.M.; Grunstein, M. HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc. Natl. Acad. Sci. USA 1996, 93, 14503–14508. [Google Scholar] [CrossRef]
- Ceschin, J.; Saint-Marc, C.; Laporte, J.; Labriet, A.; Philippe, C.; Moenner, M.; Daignan-Fornier, B.; Pinson, B. Identification of yeast and human 5-aminoimidazole-4-carboxamide-1-beta-d-ribofuranoside (AICAr) transporters. J. Biol. Chem. 2014, 289, 16844–16854. [Google Scholar] [CrossRef]
- Ueda, Y.; Oshima, Y. A constitutive mutation, phoT, of the repressible acid phosphatase synthesis with inability to transport inorganic phosphate in Saccharomyces cerevisiae. Mol. Gen. Genet. 1975, 136, 255–259. [Google Scholar] [CrossRef]
- Som, I.; Mitsch, R.N.; Urbanowski, J.L.; Rolfes, R.J. DNA-bound Bas1 recruits Pho2 to activate ADE genes in Saccharomyces cerevisiae. Eukaryot. Cell 2005, 4, 1725–1735. [Google Scholar] [CrossRef]
- Brachmann, C.B.; Davies, A.; Cost, G.J.; Caputo, E.; Li, J.; Hieter, P.; Boeke, J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 1998, 14, 115–132. [Google Scholar] [CrossRef]
- Burke, D.; Dawson, D.; Sterns, T. Methods in Yeast Genetics; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2000; pp. 171–174. [Google Scholar]
- Goldstein, A.L.; McCusker, J.H. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 1999, 15, 1541–1553. [Google Scholar] [CrossRef]
- Guldener, U.; Heck, S.; Fielder, T.; Beinhauer, J.; Hegemann, J.H. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 1996, 24, 2519–2524. [Google Scholar] [CrossRef]
- Longtine, M.S.; McKenzie, A., 3rd; Demarini, D.J.; Shah, N.G.; Wach, A.; Brachat, A.; Philippsen, P.; Pringle, J.R. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 1998, 14, 953–961. [Google Scholar] [CrossRef]
- Toh-e, A.; Ueda, Y.; Kakimoto, S.I.; Oshima, Y. Isolation and characterization of acid phosphatase mutants in Saccharomyces cerevisiae. J. Bacteriol. 1973, 113, 727–738. [Google Scholar] [CrossRef]
- Noda, T.; Klionsky, D.J. The quantitative Pho8Δ60 assay of nonspecific autophagy. Methods Enzym. 2008, 451, 33–42. [Google Scholar] [CrossRef]
- Easlon, E.; Tsang, F.; Skinner, C.; Wang, C.; Lin, S.J. The malate-aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction-mediated life span extension in yeast. Genes Dev. 2008, 22, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Croft, T.; Raj, C.J.T.; Salemi, M.; Phinney, B.S.; Lin, S.-J. A functional link between NAD+ homeostasis and N-terminal protein acetylation in Saccharomyces cerevisiae. J. Biol. Chem. 2018, 293, 2927–2938. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Petteys, B.J.; McClure, J.M.; Valsakumar, V.; Bekiranov, S.; Frank, E.L.; Smith, J.S. Thiamine biosynthesis in Saccharomyces cerevisiae is regulated by the NAD+-dependent histone deacetylase Hst1. Mol. Cell. Biol. 2010, 30, 3329–3341. [Google Scholar] [CrossRef] [PubMed][Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groth, B.; Lee, Y.-C.; Huang, C.-C.; McDaniel, M.; Huang, K.; Lee, L.-H.; Lin, S.-J. The Histone Deacetylases Hst1 and Rpd3 Integrate De Novo NAD+ Metabolism with Phosphate Sensing in Saccharomyces cerevisiae. Int. J. Mol. Sci. 2023, 24, 8047. https://doi.org/10.3390/ijms24098047
Groth B, Lee Y-C, Huang C-C, McDaniel M, Huang K, Lee L-H, Lin S-J. The Histone Deacetylases Hst1 and Rpd3 Integrate De Novo NAD+ Metabolism with Phosphate Sensing in Saccharomyces cerevisiae. International Journal of Molecular Sciences. 2023; 24(9):8047. https://doi.org/10.3390/ijms24098047
Chicago/Turabian StyleGroth, Benjamin, Yi-Ching Lee, Chi-Chun Huang, Matilda McDaniel, Katie Huang, Lan-Hsuan Lee, and Su-Ju Lin. 2023. "The Histone Deacetylases Hst1 and Rpd3 Integrate De Novo NAD+ Metabolism with Phosphate Sensing in Saccharomyces cerevisiae" International Journal of Molecular Sciences 24, no. 9: 8047. https://doi.org/10.3390/ijms24098047
APA StyleGroth, B., Lee, Y.-C., Huang, C.-C., McDaniel, M., Huang, K., Lee, L.-H., & Lin, S.-J. (2023). The Histone Deacetylases Hst1 and Rpd3 Integrate De Novo NAD+ Metabolism with Phosphate Sensing in Saccharomyces cerevisiae. International Journal of Molecular Sciences, 24(9), 8047. https://doi.org/10.3390/ijms24098047