
Modulation of EZH2 Activity Induces an Antitumoral Effect and Cell Redifferentiation in Anaplastic Thyroid Cancer

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
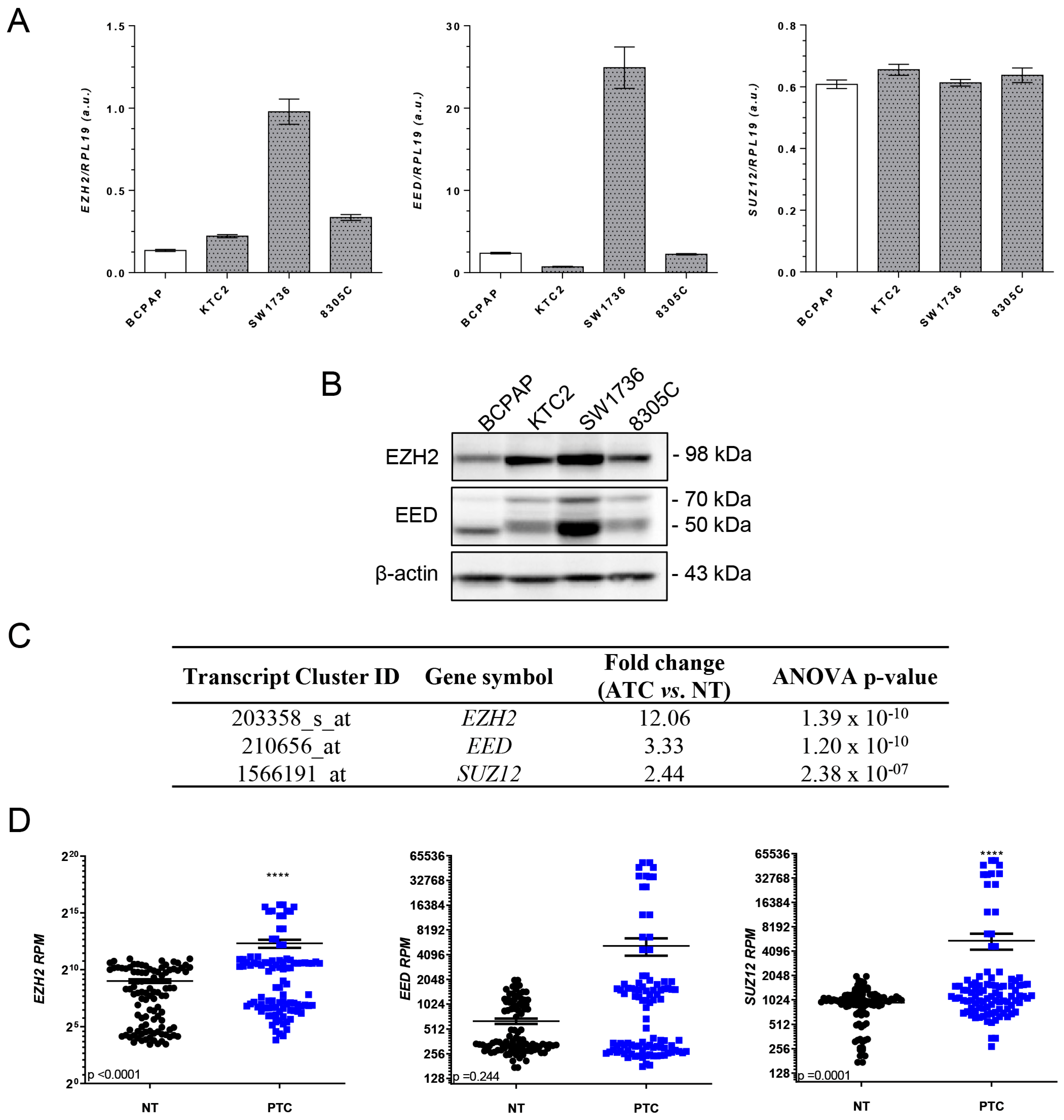
2.1. EZH2 Is Overexpressed in Thyroid Cancer
2.2. CRISPR/Cas9-Mediated EZH2 Gene Editing Improves Differentiation of ATC Cells
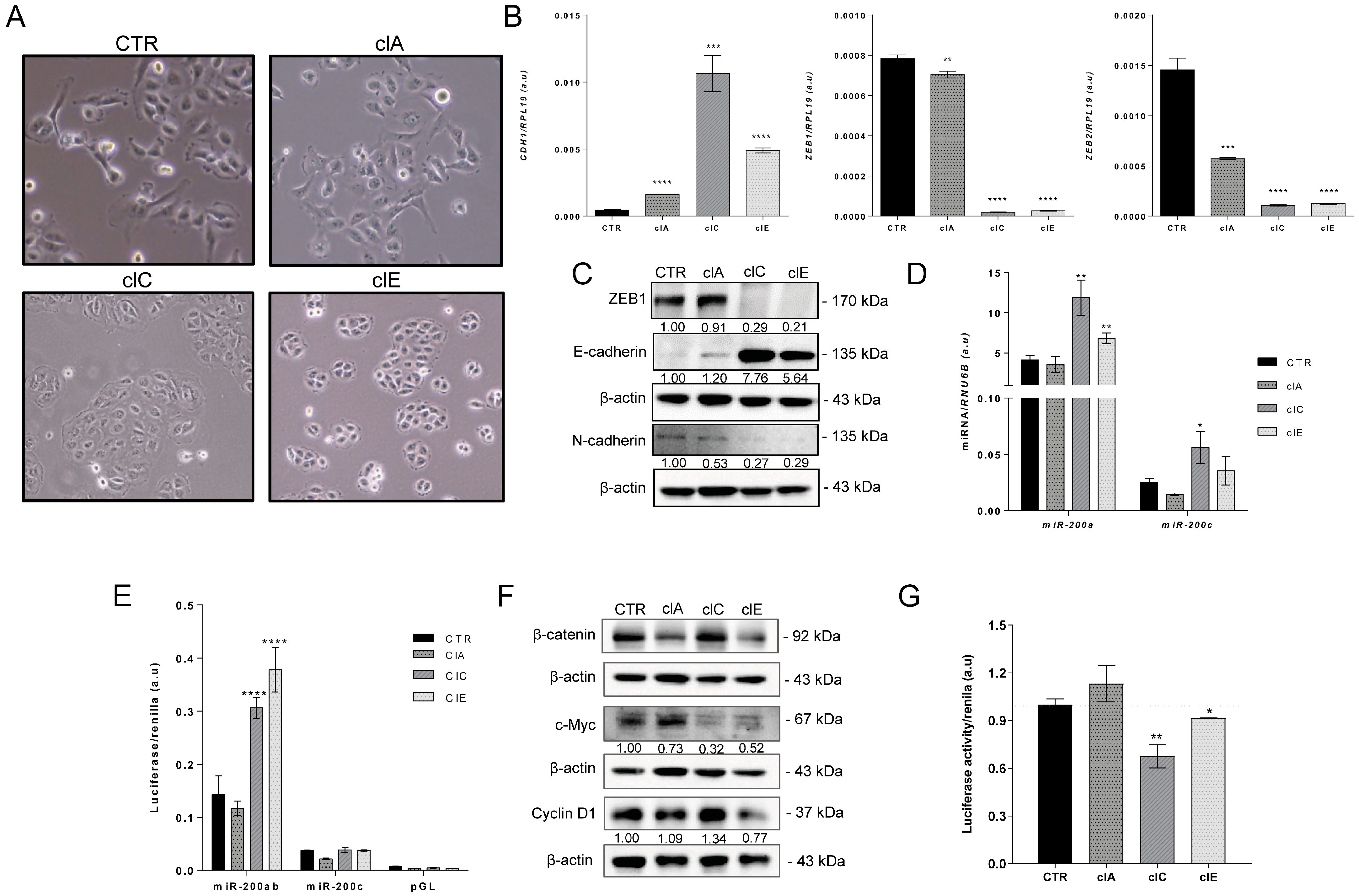
2.3. CRISPR/Cas9-Mediated EZH2 Gene Editing Induces Mesenchymal–Epithelial Transition (MET)
2.4. CRISPR/Cas9-Mediated EZH2 Gene Editing Has an Antitumoral Effect In Vitro
2.5. CRISPR/Cas9-Mediated EZH2 Gene Editing Has an Antitumoral Effect In Vivo
2.6. Pharmacological Blockage of EZH2 Methyltransferase Activity Induces MET and Differentiation of ATC Cells and Reduces Colony Formation
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.1.1. Cell Culture
4.1.2. Cell Treatments
EZH2 Pharmacological Inhibition
4.2. EZH2 Gene Editing with CRISPR/Cas9
Plasmid Cloning
4.3. Genomic DNA Sequencing for Gene Editing Validation
4.4. Cell Function Assays
4.4.1. Cell Count
4.4.2. Cell Invasion
4.4.3. Cell Migration
4.4.4. Colony Formation
4.5. Gene Expression Analysis
4.6. Western Blot
4.7. Luciferase Gene Reporter Assay
4.7.1. Wnt-β-Catenin Signaling
4.7.2. miR-200 Promoter Reporter
4.8. In Vivo Xenotransplant in Nude Mice
4.9. Analysis of Publicly Available Data from Thyroid Tumors
4.9.1. The Cancer Genome Atlas (TCGA) Data
4.9.2. Microarray Data
4.9.3. Cistrome Data Browser
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bible, K.C.; Kebebew, E.; Brierley, J.; Brito, J.P.; Cabanillas, M.E.; Clark, T.J., Jr.; Di Cristofano, A.; Foote, R.; Giordano, T.; Kasperbauer, J.; et al. 2021 American Thyroid Association Guidelines for Management of Patients with Anaplastic Thyroid Cancer. Thyroid 2021, 31, 337–386. [Google Scholar] [CrossRef]
- Saini, S.; Tulla, K.; Maker, A.V.; Burman, K.D.; Prabhakar, B.S. Therapeutic advances in anaplastic thyroid cancer: A current perspective. Mol. Cancer 2018, 17, 154. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.P.; Lopez-Marquez, A.; Santisteban, P. Thyroid transcription factors in development, differentiation and disease. Nat. Rev. Endocrinol. 2015, 11, 29–42. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Fuziwara, C.S.; Kimura, E.T. MicroRNAs in thyroid development, function and tumorigenesis. Mol. Cell. Endocrinol. 2017, 456, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Lin, Y.; Liang, J. Radioactive Iodine-Refractory Differentiated Thyroid Cancer and Redifferentiation Therapy. Endocrinol. Metab. 2019, 34, 215–225. [Google Scholar] [CrossRef]
- Alobuia, W.; Gillis, A.; Kebebew, E. Contemporary Management of Anaplastic Thyroid Cancer. Curr. Treat. Options Oncol. 2020, 21, 78. [Google Scholar] [CrossRef]
- Nagaiah, G.; Hossain, A.; Mooney, C.J.; Parmentier, J.; Remick, S.C. Anaplastic thyroid cancer: A review of epidemiology, pathogenesis, and treatment. J. Oncol. 2011, 2011, 542358. [Google Scholar] [CrossRef]
- Fagin, J.A.; Wells, S.A., Jr. Biologic and Clinical Perspectives on Thyroid Cancer. N. Engl. J. Med. 2016, 375, 1054–1067. [Google Scholar] [CrossRef]
- Zaballos, M.A.; Santisteban, P. Key signaling pathways in thyroid cancer. J. Endocrinol. 2017, 235, R43–R61. [Google Scholar] [CrossRef]
- Liu, R.; Xing, M. TERT promoter mutations in thyroid cancer. Endocr. Relat. Cancer 2016, 23, R143–R155. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Donghi, R.; Longoni, A.; Pilotti, S.; Michieli, P.; Della Porta, G.; Pierotti, M.A. Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. J. Clin. Investig. 1993, 91, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Melo, M.; da Rocha, A.G.; Vinagre, J.; Batista, R.; Peixoto, J.; Tavares, C.; Celestino, R.; Almeida, A.; Salgado, C.; Eloy, C.; et al. TERT promoter mutations are a major indicator of poor outcome in differentiated thyroid carcinomas. J. Clin. Endocrinol. Metab. 2014, 99, E754–E765. [Google Scholar] [CrossRef]
- Pita, J.M.; Figueiredo, I.F.; Moura, M.M.; Leite, V.; Cavaco, B.M. Cell cycle deregulation and TP53 and RAS mutations are major events in poorly differentiated and undifferentiated thyroid carcinomas. J. Clin. Endocrinol. Metab. 2014, 99, E497–E507. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Ferrarotto, R.; Garden, A.S.; Ahmed, S.; Busaidy, N.L.; Dadu, R.; Williams, M.D.; Skinner, H.; Gunn, G.B.; Grosu, H.; et al. Neoadjuvant BRAF- and Immune-Directed Therapy for Anaplastic Thyroid Carcinoma. Thyroid 2018, 28, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Ferrari, K.J.; Scelfo, A.; Jammula, S.; Cuomo, A.; Barozzi, I.; Stutzer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef]
- Hojfeldt, J.W.; Laugesen, A.; Willumsen, B.M.; Damhofer, H.; Hedehus, L.; Tvardovskiy, A.; Mohammad, F.; Jensen, O.N.; Helin, K. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat. Struct. Mol. Biol. 2018, 25, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wu, X. Polycomb-group proteins in the initiation and progression of cancer. J. Genet. Genomics 2021, 48, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Pang, B.; Wang, Q.; Yang, S.; Gao, T.; Ding, Q.; Liu, H.; Yang, Y.; Fan, H.; Zhang, R.; et al. EZH2 upregulation correlates with tumor invasiveness, proliferation, and angiogenesis in human pituitary adenomas. Hum. Pathol. 2017, 66, 101–107. [Google Scholar] [CrossRef]
- Raman, J.D.; Mongan, N.P.; Tickoo, S.K.; Boorjian, S.A.; Scherr, D.S.; Gudas, L.J. Increased expression of the polycomb group gene, EZH2, in transitional cell carcinoma of the bladder. Clin. Cancer Res. 2005, 11 Pt 1, 8570–8576. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Xia, L.; Zhu, X.; Zhang, L.; Xu, Y.; Chen, G.; Luo, J. EZH2 enhances expression of CCL5 to promote recruitment of macrophages and invasion in lung cancer. Biotechnol. Appl. Biochem. 2020, 67, 1011–1019. [Google Scholar] [CrossRef]
- Simeone, N.; Frezza, A.M.; Zaffaroni, N.; Stacchiotti, S. Tazemetostat for advanced epithelioid sarcoma: Current status and future perspectives. Future Oncol. 2021, 17, 1253–1263. [Google Scholar] [CrossRef]
- von Keudell, G.; Salles, G. The role of tazemetostat in relapsed/refractory follicular lymphoma. Ther. Adv. Hematol. 2021, 12, 20406207211015882. [Google Scholar] [CrossRef] [PubMed]
- Borbone, E.; Troncone, G.; Ferraro, A.; Jasencakova, Z.; Stojic, L.; Esposito, F.; Hornig, N.; Fusco, A.; Orlando, V. Enhancer of zeste homolog 2 overexpression has a role in the development of anaplastic thyroid carcinomas. J. Clin. Endocrinol. Metab. 2011, 96, 1029–1038. [Google Scholar] [CrossRef]
- Masudo, K.; Suganuma, N.; Nakayama, H.; Oshima, T.; Rino, Y.; Iwasaki, H.; Matsuzu, K.; Sugino, K.; Ito, K.; Kondo, T.; et al. EZH2 Overexpression as a Useful Prognostic Marker for Aggressive Behaviour in Thyroid Cancer. In Vivo 2018, 32, 25–31. [Google Scholar] [PubMed]
- Tsai, C.C.; Chien, M.N.; Chang, Y.C.; Lee, J.J.; Dai, S.H.; Cheng, S.P. Overexpression of Histone H3 Lysine 27 Trimethylation Is Associated with Aggressiveness and Dedifferentiation of Thyroid Cancer. Endocr. Pathol. 2019, 30, 305–311. [Google Scholar] [CrossRef]
- Fu, H.; Cheng, L.; Sa, R.; Jin, Y.; Chen, L. Combined tazemetostat and MAPKi enhances differentiation of papillary thyroid cancer cells harbouring BRAF(V600E) by synergistically decreasing global trimethylation of H3K27. J. Cell. Mol. Med. 2020, 24, 3336–3345. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Foukakis, T.; Hashemi, J.; Grimelius, L.; Heldin, N.E.; Wallin, G.; Rudduck, C.; Lui, W.O.; Hoog, A.; Larsson, C. Molecular cytogenetic profiles of novel and established human anaplastic thyroid carcinoma models. Thyroid 2007, 17, 289–301. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef]
- Fuziwara, C.S.; Kimura, E.T. MicroRNA Deregulation in Anaplastic Thyroid Cancer Biology. Int. J. Endocrinol. 2014, 2014, 743450. [Google Scholar] [CrossRef] [PubMed]
- Mongroo, P.S.; Rustgi, A.K. The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol. Ther. 2010, 10, 219–222. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, X.; Feng, X.; Fan, X.; Jin, Z. The crosstalk between microRNAs and the Wnt/beta-catenin signaling pathway in cancer. Oncotarget 2017, 8, 14089–14106. [Google Scholar] [CrossRef] [PubMed]
- Sastre-Perona, A.; Santisteban, P. Role of the wnt pathway in thyroid cancer. Front. Endocrinol. 2012, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef] [PubMed]
- Coelho, B.P.; Fernandes, C.F.L.; Boccacino, J.M.; Souza, M.; Melo-Escobar, M.I.; Alves, R.N.; Prado, M.B.; Iglesia, R.P.; Cangiano, G.; Mazzaro, G.R.; et al. Multifaceted WNT Signaling at the Crossroads between Epithelial-Mesenchymal Transition and Autophagy in Glioblastoma. Front. Oncol. 2020, 10, 597743. [Google Scholar] [CrossRef]
- Liao, Z.; Tan, Z.W.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts in tumor microenvironment—Accomplices in tumor malignancy. Cell. Immunol. 2019, 343, 103729. [Google Scholar] [CrossRef]
- Margueron, R.; Justin, N.; Ohno, K.; Sharpe, M.L.; Son, J.; Drury, W.J., 3rd; Voigt, P.; Martin, S.R.; Taylor, W.R.; De Marco, V.; et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 2009, 461, 762–767. [Google Scholar] [CrossRef]
- Kim, H.; Kang, K.; Kim, J. AEBP2 as a potential targeting protein for Polycomb Repression Complex PRC2. Nucleic Acids Res. 2009, 37, 2940–2950. [Google Scholar] [CrossRef]
- Li, G.; Margueron, R.; Ku, M.; Chambon, P.; Bernstein, B.E.; Reinberg, D. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 2010, 24, 368–380. [Google Scholar] [CrossRef]
- Maenner, S.; Blaud, M.; Fouillen, L.; Savoye, A.; Marchand, V.; Dubois, A.; Sanglier-Cianferani, S.; Van Dorsselaer, A.; Clerc, P.; Avner, P.; et al. 2-D structure of the A region of Xist RNA and its implication for PRC2 association. PLoS Biol. 2010, 8, e1000276. [Google Scholar] [CrossRef]
- Song, J.J.; Garlick, J.D.; Kingston, R.E. Structural basis of histone H4 recognition by p55. Genes Dev. 2008, 22, 1313–1318. [Google Scholar] [CrossRef]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef]
- Yu, Y.; Deng, P.; Yu, B.; Szymanski, J.M.; Aghaloo, T.; Hong, C.; Wang, C.Y. Inhibition of EZH2 Promotes Human Embryonic Stem Cell Differentiation into Mesoderm by Reducing H3K27me3. Stem Cell Rep. 2017, 9, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Caretti, G.; Di Padova, M.; Micales, B.; Lyons, G.E.; Sartorelli, V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004, 18, 2627–2638. [Google Scholar] [CrossRef] [PubMed]
- Ezhkova, E.; Pasolli, H.A.; Parker, J.S.; Stokes, N.; Su, I.H.; Hannon, G.; Tarakhovsky, A.; Fuchs, E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 2009, 136, 1122–1135. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gou, H.; Yao, J.; Yi, K.; Jin, Z.; Matsuoka, M.; Zhao, T. The noncanonical role of EZH2 in cancer. Cancer Sci. 2021, 112, 1376–1382. [Google Scholar] [CrossRef]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef]
- Lawrence, C.L.; Baldwin, A.S. Non-Canonical EZH2 Transcriptionally Activates RelB in Triple Negative Breast Cancer. PLoS ONE 2016, 11, e0165005. [Google Scholar] [CrossRef]
- Yuen, G.; Khan, F.J.; Gao, S.; Stommel, J.M.; Batchelor, E.; Wu, X.; Luo, J. CRISPR/Cas9-mediated gene knockout is insensitive to target copy number but is dependent on guide RNA potency and Cas9/sgRNA threshold expression level. Nucleic Acids Res. 2017, 45, 12039–12053. [Google Scholar] [CrossRef]
- Riesco-Eizaguirre, G.; Santisteban, P.; De la Vieja, A. The complex regulation of NIS expression and activity in thyroid and extrathyroidal tissues. Endocr. Relat. Cancer 2021, 28, T141–T165. [Google Scholar] [CrossRef]
- Choudhury, P.S.; Gupta, M. Differentiated thyroid cancer theranostics: Radioiodine and beyond. Br. J. Radiol. 2018, 91, 20180136. [Google Scholar] [CrossRef]
- Kang, H.S.; Kumar, D.; Liao, G.; Lichti-Kaiser, K.; Gerrish, K.; Liao, X.H.; Refetoff, S.; Jothi, R.; Jetten, A.M. GLIS3 is indispensable for TSH/TSHR-dependent thyroid hormone biosynthesis and follicular cell proliferation. J. Clin. Investig. 2017, 127, 4326–4337. [Google Scholar] [CrossRef] [PubMed]
- Ku, M.; Koche, R.P.; Rheinbay, E.; Mendenhall, E.M.; Endoh, M.; Mikkelsen, T.S.; Presser, A.; Nusbaum, C.; Xie, X.; Chi, A.S.; et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008, 4, e1000242. [Google Scholar] [CrossRef] [PubMed]
- Tanay, A.; O’Donnell, A.H.; Damelin, M.; Bestor, T.H. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc. Natl. Acad. Sci. USA 2007, 104, 5521–5526. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ortiz, J.A.; Taing, L.; Meyer, C.A.; Lee, B.; Zhang, Y.; Shin, H.; Wong, S.S.; Ma, J.; Lei, Y.; et al. Cistrome: An integrative platform for transcriptional regulation studies. Genome Biol. 2011, 12, R83. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, D.; Di Loreto, C.; Beltrami, C.A.; Belfiore, A.; Di Lauro, R.; Damante, G. Expression of thyroid-specific transcription factors TTF-1 and PAX-8 in human thyroid neoplasms. Cancer Res. 1994, 54, 4744–4749. [Google Scholar]
- Miccadei, S.; De Leo, R.; Zammarchi, E.; Natali, P.G.; Civitareale, D. The synergistic activity of thyroid transcription factor 1 and Pax 8 relies on the promoter/enhancer interplay. Mol. Endocrinol. 2002, 16, 837–846. [Google Scholar] [CrossRef]
- Pasca di Magliano, M.; Di Lauro, R.; Zannini, M. Pax8 has a key role in thyroid cell differentiation. Proc. Natl. Acad. Sci. USA 2000, 97, 13144–13149. [Google Scholar] [CrossRef]
- Schweppe, R.E.; Klopper, J.P.; Korch, C.; Pugazhenthi, U.; Benezra, M.; Knauf, J.A.; Fagin, J.A.; Marlow, L.A.; Copland, J.A.; Smallridge, R.C.; et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J. Clin. Endocrinol. Metab. 2008, 93, 4331–4341. [Google Scholar] [CrossRef]
- Lopez-Marquez, A.; Carrasco-Lopez, C.; Fernandez-Mendez, C.; Santisteban, P. Unraveling the Complex Interplay between Transcription Factors and Signaling Molecules in Thyroid Differentiation and Function, from Embryos to Adults. Front. Endocrinol. 2021, 12, 654569. [Google Scholar] [CrossRef]
- Cao, Q.; Mani, R.S.; Ateeq, B.; Dhanasekaran, S.M.; Asangani, I.A.; Prensner, J.R.; Kim, J.H.; Brenner, J.C.; Jing, X.; Cao, X.; et al. Coordinated regulation of polycomb group complexes through microRNAs in cancer. Cancer Cell 2011, 20, 187–199. [Google Scholar] [CrossRef]
- Cao, Q.; Yu, J.; Dhanasekaran, S.M.; Kim, J.H.; Mani, R.S.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Cao, X.; Yu, J.; et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008, 27, 7274–7284. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Ochiai, A. Enhancer of zeste homolog 2 downregulates E-cadherin by mediating histone H3 methylation in gastric cancer cells. Cancer Sci. 2008, 99, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.S.; Lau, S.S.; Chen, Y.; Kondo, Y.; Li, M.S.; Feng, H.; Ching, A.K.; Cheung, K.F.; Wong, H.K.; Tong, J.H.; et al. EZH2-mediated concordant repression of Wnt antagonists promotes beta-catenin-dependent hepatocarcinogenesis. Cancer Res. 2011, 71, 4028–4039. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Wang, J.; Yu, X.; Gong, W.; Liu, X.; Park, K.S.; Ma, A.; Tsai, Y.H.; Shen, Y.; Onikubo, T.; Pi, W.C.; et al. EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nat. Cell Biol. 2022, 24, 384–399. [Google Scholar] [CrossRef]
- Shi, B.; Liang, J.; Yang, X.; Wang, Y.; Zhao, Y.; Wu, H.; Sun, L.; Zhang, Y.; Chen, Y.; Li, R.; et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol. Cell. Biol. 2007, 27, 5105–5119. [Google Scholar] [CrossRef]
- Liu, J.; Ruan, B.; You, N.; Huang, Q.; Liu, W.; Dang, Z.; Xu, W.; Zhou, T.; Ji, R.; Cao, Y.; et al. Downregulation of miR-200a induces EMT phenotypes and CSC-like signatures through targeting the beta-catenin pathway in hepatic oval cells. PLoS ONE 2013, 8, e79409. [Google Scholar]
- Su, J.; Zhang, A.; Shi, Z.; Ma, F.; Pu, P.; Wang, T.; Zhang, J.; Kang, C.; Zhang, Q. MicroRNA-200a suppresses the Wnt/beta-catenin signaling pathway by interacting with beta-catenin. Int. J. Oncol. 2012, 40, 1162–1170. [Google Scholar]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Xue, B.; Chuang, C.H.; Prosser, H.M.; Fuziwara, C.S.; Chan, C.; Sahasrabudhe, N.; Kuhn, M.; Wu, Y.; Chen, J.; Biton, A.; et al. miR-200 deficiency promotes lung cancer metastasis by activating Notch signaling in cancer-associated fibroblasts. Genes Dev. 2021, 35, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Labun, K.; Montague, T.G.; Krause, M.; Torres Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef]
- Sastre-Perona, A.; Santisteban, P. Wnt-independent role of beta-catenin in thyroid cell proliferation and differentiation. Mol. Endocrinol. 2014, 28, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Simon, P. Q-Gene: Processing quantitative real-time RT-PCR data. Bioinformatics 2003, 19, 1439–1440. [Google Scholar] [CrossRef] [PubMed]
- Geraldo, M.V.; Nakaya, H.I.; Kimura, E.T. Down-regulation of 14q32-encoded miRNAs and tumor suppressor role for miR-654–3p in papillary thyroid cancer. Oncotarget 2017, 8, 9597–9607. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Mello, D.C.; Saito, K.C.; Cristovão, M.M.; Kimura, E.T.; Fuziwara, C.S. Modulation of EZH2 Activity Induces an Antitumoral Effect and Cell Redifferentiation in Anaplastic Thyroid Cancer. Int. J. Mol. Sci. 2023, 24, 7872. https://doi.org/10.3390/ijms24097872
de Mello DC, Saito KC, Cristovão MM, Kimura ET, Fuziwara CS. Modulation of EZH2 Activity Induces an Antitumoral Effect and Cell Redifferentiation in Anaplastic Thyroid Cancer. International Journal of Molecular Sciences. 2023; 24(9):7872. https://doi.org/10.3390/ijms24097872
Chicago/Turabian Stylede Mello, Diego Claro, Kelly Cristina Saito, Marcella Maringolo Cristovão, Edna Teruko Kimura, and Cesar Seigi Fuziwara. 2023. "Modulation of EZH2 Activity Induces an Antitumoral Effect and Cell Redifferentiation in Anaplastic Thyroid Cancer" International Journal of Molecular Sciences 24, no. 9: 7872. https://doi.org/10.3390/ijms24097872
APA Stylede Mello, D. C., Saito, K. C., Cristovão, M. M., Kimura, E. T., & Fuziwara, C. S. (2023). Modulation of EZH2 Activity Induces an Antitumoral Effect and Cell Redifferentiation in Anaplastic Thyroid Cancer. International Journal of Molecular Sciences, 24(9), 7872. https://doi.org/10.3390/ijms24097872