

Breaking the Mold: Epigenetics and Genomics Approaches Addressing Novel Treatments and Chemoresponse in TGCT Patients
 , , , , and
, , , , and
Abstract
1. Introduction
2. Summary of TGCT’s Major Features
2.1. Histological TGCT Subtypes
2.2. TGCT Genomic Hallmarks
2.2.1. Actionable Mutations
2.2.2. Chromosomal Aberrations
2.2.3. Potential Genomic Biomarkers for Diagnosis
3. Chemoresponse as a Clinically Unresolved Problem
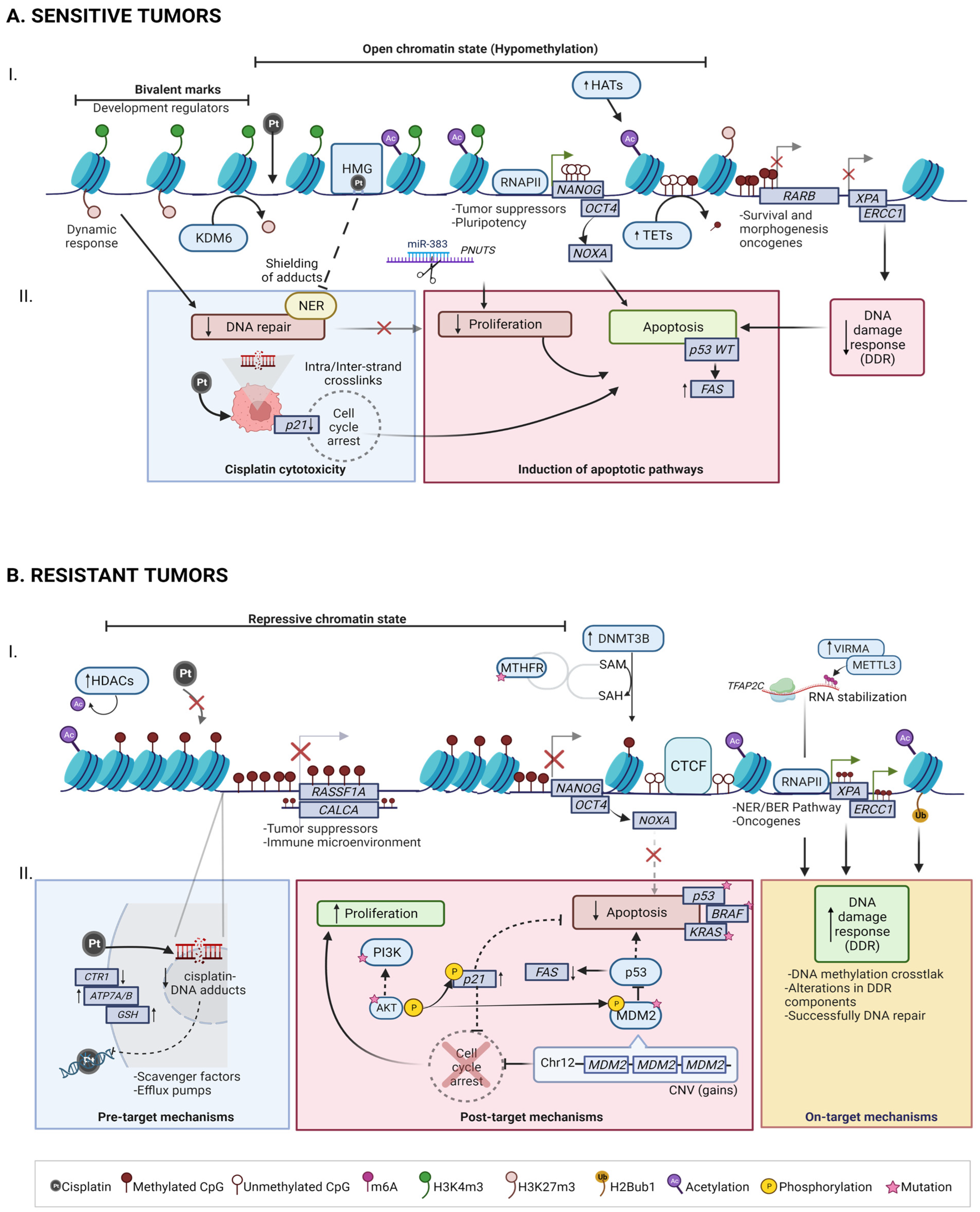
4. Molecular Basis of Chemoresponse

{kind=link}
{kind=link}
| Marker | Type | Phenotype Associated | Sample Type | Assays | Main Results and Marker Function | Reference | |
|---|---|---|---|---|---|---|---|
| Numeric variants | 2q11.1 | CNV (gains) | Sensitivity | Tumor tissue | WES | Amplifications were present in 100% of sensitive patients and not found in resistant tumors | [16] |
| 2q32.1 | CNV (gains) | Resistance | Tumor tissue | WES | Amplification was correlated with refractory and metastatic tumors | [18] | |
| 3p25.3 | CNV (gains) | Resistance | Cell lines | Genomic profiling/qPCR | Gains were detected at low frequencies in primary tumors but at higher frequencies in inducted cisplatin-resistant tumors | [51] | |
| Genes | ERCC1 | ↑exp | Resistance | Tumor tissue and cell lines | qPCR | Overexpression in both cell lines and tumor tissue is a finding in acquired resistant phenotypes | [52] |
| ERCC1 | ↓exp | Sensitivity | Tumor tissue and cell lines | qPCR | Downregulation in cell lines and tumor tissue is a finding in sensitive phenotypes | [52] | |
| HMGB4 | depletion | Resistance | Cell lines | HMGB4 Knockout | Plays a major role in sensitizing TGCTs to cisplatin knockout cause differences in cell cycle progression following cisplatin treatment | [53] | |
| HMGN5 | ↓exp | Resistance | Cell lines | Exp microarray | mRNA levels were remarkably upregulated in resistant subclones compared with the corresponding parental cells. Knockdown substantially reduced the viability of cisplatin-resistant TGCT cells in the presence of cisplatin | [54] | |
| REV7 | ↓exp | Sensitivity | Cell lines | qPCR | Depletion promoted chemosensitivity. In addition, inactivation in cisplatin-resistant TGCT cells meant they recovered chemosensitivity at almost equal levels to parental cells in vitro and in vivo | [55] | |
| CCND1 | ↑exp | Resistance | Tumor tissue | qPCR/IHC | Expression was significantly higher in resistant cases compared with sensitive samples | [56] | |
| OCT4 | ↓exp | Resistance | Tumor tissue and cell lines | qPCR/IHC | Decreased expression promotes higher differentiation, thus inducing a resistant phenotype | [39] | |
| CTR1 | depletion | Resistance | Cell lines | qPCR/WB | Increased protein expression was observed for the most cisplatin-sensitive cell lines, and depletion promotes a resistant phenotype | [57] | |
| MDM2 | CNV (gains) | Resistance | Cell lines | qPCR | CNV gains induced a resistant phenotype through inhibition of the p53 pathway | [58] | |
| MDM2 | ↑exp | Resistance | Tumor tissue | IHC | Overexpression at tissue level in TGCT correlates with more aggressive phenotypes that tend to acquire resistance | [59] | |
| KRAS | CNV (gains) | Resistance | Tumor tissue | qPCR | Amplifications are associated with poor prognosis in 80% of cases | [60] | |
| AKT1/PIK3CA | somatic mutations | Resistance | Tumor tissue | WES | Somatic mutations are present with a high frequency exclusively in resistant tumors | [61] | |
| TEX11 | ↑exp | Resistance | Cell lines | Exp microarray | Gene silencing in cisplatin-resistant TGCT cells increased the percentage of double-strand break marker γH2AX-positive cells. Overexpression promotes resistant phenotypes | [54] | |
| HIF-1α | ↓exp | Sensitivity | Tumor tissue | IHC | Low expression levels in TGCTs, specifically SE and mixed NS, promotes a sensitive phenotype | [62,63] | |
| TDRG1 | ↑exp | Resistance | Tumor tissue and cell lines | qPCR/IF | Overexpression regulates chemosensitivity to cisplatin in cell lines through PI3K/Akt/mTOR signaling and mitochondria-mediated apoptotic pathways both in vitro and in vivo | [64] | |
| ALDH1 | ↑exp | Resistance | Cell lines | qPCR | The ALDH inhibitor disulfiram restored sensitivity to cisplatin upon combinatorial treatment in both resistant cell lines and significantly inhibited tumor growth | [65] |
4.1. Mechanisms of Cisplatin Sensitivity in TGCT
4.1.1. Cisplatin cytotoxicity
4.1.2. Induction of Apoptotic Pathways
4.2. Mechanisms of Cisplatin Resistance in TGCT
4.2.1. Pre-Target: CTR1 Receptor Alterations Promotes Cisplatin Uptake Failure
4.2.2. On-Target: BRAF, ERCC1 and NER/BER Pathways
4.2.3. Post-Target: Pro-Apoptotic Pathway Dysfunction (P53, PI3K/AKT)
5. TGCT Epigenomic Hallmarks in Chemoresponse
5.1. DNA Methylation
5.2. RNA Methylation
| Marker | Role in Response | Mechanism of Resistance | Sample Type | Assays | Main Results and Marker Function | Reference |
|---|---|---|---|---|---|---|
| RASSF1A (↑5mC) | Resistance | Intrinsic | NS tissue (31Se; 39Re) | qMSP | (52% Re vs 28% Se) Negative regulator of cell growth | [98] |
| HIC1 (↑5mC) | Resistance | Intrinsic | (47% Re vs 24% Se) Transcription factor that acts as a tumor suppressor | |||
| RARB (↑5mC) | Sensitivity | Intrinsic | (0% Re vs 14% Se) Receptor involved in morphogenesis, cell growth and differentiation | |||
| MGMT (↑5mC) | Sensitivity | Intrinsic | (31% Re vs 13% Se) MGMT is a DNA repair enzyme | |||
| CALCA (↑5mC) | Resistance | Intrinsic | TGCT tissues (47Se; 15Re) | qMSP | 47.4% (9/19, p = 0.005) of samples with methylated loci presented refractory disease, also associated with NS tumors. Gene is involved in calcium regulation, acts as a vasodilator | [99] |
| MGMT (↑5mC) | Resistance | Intrinsic | 38.1% (08/21, p = 0.067) of tumors presenting MGMT methylation were refractory, which was also associated with NS histology | |||
| Global ↑5mC | Resistance | Acquired | NT2/D1, 833K, and 2102EP and cisplatin-resistant sublines | EPIC 850 K array and RNA-Seq | Acquired cisplatin resistance in TGCT triggers net ↑5mC. Hypermethylation in resistant cells is associated with repression of cancer suppressor genes and nuclear organization of repressive chromatin, while hypomethylation is associated with the polycomb pathway | [96] |
| Global ↑5mC | Resistance | Acquired | Matched primary and metastatic tissue from four patients | EPIC 850 K array | Hypermethylation in promoters of genes related to regulation of the immune microenvironment. Hypomethylation of promoters on pathways related to DNA/chromatin binding | [102] |
| VIRMA (↑exp) | Resistance | Acquired | TCam-2, NCCIT, 2102Ep, and NT2 and cisplatin-resistant sublines | RT-qPCR, ELISA and dot blot (m6A quantification). CRISPR/Cas9 (knockdown of VIRMA) followed by cell viability, proliferation, invasion, and CAM assays. | The component of the m6A writer complex VIRMA contributes to tumor aggressiveness and to cisplatin resistance, both in vitro and in vivo, by regulating DNA damage response | [115] |
| miR-371-373 cluster | Resistance | Acquired | NTERA-2, NCCIT, and 2102EP, and cisplatin-resistant sublines | RT-qPCR and LDA | Upregulated in NTERA-2 and NCCIT resistant cells; possibly promotes resistance by counteracting wild-type p53-induced senescence | [117] |
| hsa-miR-99a/-100/-145 | Resistance | Acquired | About 10-fold down-regulated in NTERA-2- and NCCIT resistant clones | |||
| miR-302a | Sensitivity | Acquired | NTERA-2 and its cisplatin-resistant subline | Overexpression via transfecting vector, RT-qPCR (expression). Cell proliferation and drug-sensitivity assay | Up-regulation of miR-302a significantly increased the sensitivity of NT2 cells to cisplatin by enhancing cisplatin-induced G2/M phase arrest and the subsequent progression to apoptosis | [118] |
| miR-302 cluster | Resistance | Acquired | NT2-D1, 833 K, and cisplatin-resistant sublines | Inhibitor-mediated transient transfection. RT-qPCr (expression). Cell survival, proliferation, and invasion assays | miR-302s act as TGCT oncogenes by inducing the expression of SPRY4 and activating the MAPK/ERK pathway while inhibiting apoptosis | [119] |
| miR-383 | Sensitivity | Acquired | NTERA-2 and its cisplatin-resistant subline | miR-383 mimics and miR-383 inhibitor transfection, RT-PCR/WB, cisplatin sensitivity assay | This miRNA ↓PNUTS levels; this blocks the phosphorylation of H2A and induces cell cycle arrest | [120] |
| Molecular signature: miR-218-5p, miR-31-5p, miR-375-5p, miR-517-3p, miR-20b-5p and miR-378a-3p | Resistance | Acquired | Discovery: Cisplatin-sensitive and -resistant TGCT cell linesValidation: TGCT tissue (n = 53) and control (n = 33) | miRNA microarray profiling (discovery), RT-qPCR (validation) | New panel of biomarkers for better prediction of chemoresistance and more aggressive phenotypes | [121] |
| ↓H3K27me3 (polycomb activity) | Resistance | Acquired | NT2/D1, 833K, and 2102EP and cisplatin-resistant sublines | RNA-Seq and GSEA. Drug Tx with GSK126 (EZH2 inhibitor) and GSKJ4 (JMJD3 inhibitor). Cell viability and proliferation assays | Resistant lines express genes normally repressed by polycomb. Repression of H3K27me3 conferred cisplatin resistance to parental cells while induction of the mark resulted in increased cisplatin sensitivity in resistant cells | [122] |
| Crosstalk: ↓DNMT3B → ↑H3K27me3 | Sensitivity | Acquired | 5-aza-resistant cell lines | Drug Tx 85-aza) and cell viability and proliferation assays. Lentiviral shRNA (DNMT3B knockdown) followed by RNA-Seq | DNMT3B knockdown alone in parental cells resulted in increased expression of H3K27me3, EZH2, and BMI1, and conferred 5-aza resistance and cisplatin sensitization. Patients resistant to cisplatin may have high levels of DNMT3B and KDM6B and low levels of H3K27me3 | [108] |
| ↑H2Bub1 in Lys120 | Resistance | Acquired | NCCIT and 2102EP and cisplatin-resistant sublines | WB for H2Bub1 levels with and w/o ATRA Tx. H2Bub1 knockdown followed by MTT and colony formation assay | ↑H2Bub1 levels in resistant cells; inhibition of H2Bub1 formation impaired DNA repair and decreased cellular survival (enhanced sensitivity) | [123] |
5.3. Non-Coding RNAs
5.4. Histone Post-Transcriptional Modifications
5.5. Integrative Landscape
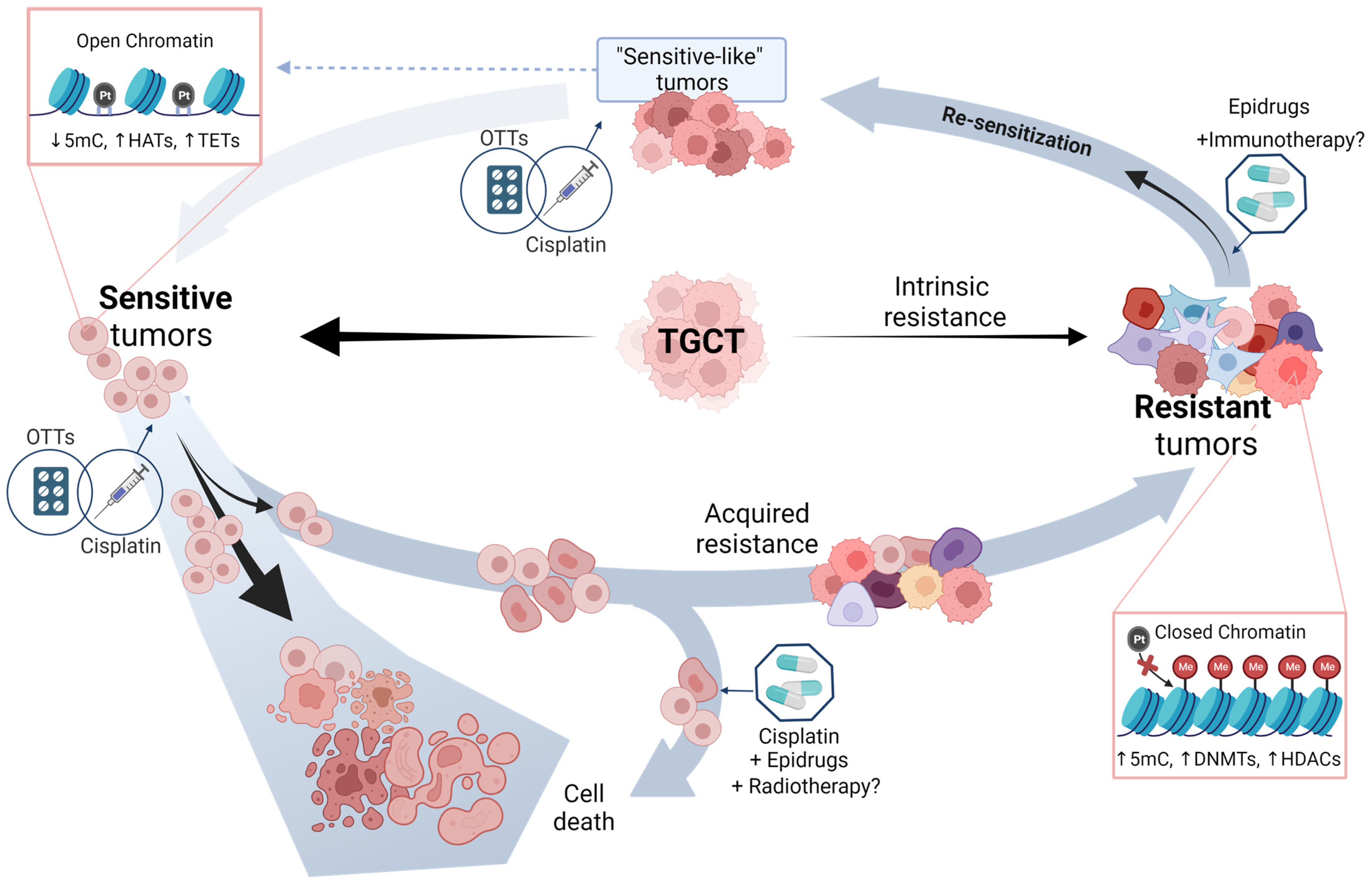
6. Potential Approaches of TGCT Therapy
6.1. Epridrugs
Therapy Enhancement by Epidrugs
6.2. Other Targeted Therapies (OTTs)
7. Driving the Future of TGCT Therapy
8. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Znaor, A.; Skakkebæk, N.E.; Rajpert-De Meyts, E.; Laversanne, M.; Kuliš, T.; Gurney, J.; Sarfati, D.; McGlynn, K.A.; Bray, F. Testicular Cancer Incidence Predictions in Europe 2010–2035: A Rising Burden despite Population Ageing. Int. J. Cancer 2020, 147, 820–828. [Google Scholar] [CrossRef]
- Gurney, J.K.; Florio, A.A.; Znaor, A.; Ferlay, J.; Laversanne, M.; Sarfati, D.; Bray, F.; McGlynn, K.A. International Trends in the Incidence of Testicular Cancer: Lessons from 35 Years and 41 Countries. Eur. Urol. 2019, 76, 615–623. [Google Scholar] [CrossRef]
- Buljubašić, R.; Buljubašić, M.; Bojanac, A.K.; Ulamec, M.; Vlahović, M.; Ježek, D.; Bulić-Jakuš, F.; Sinčić, N. Epigenetics and Testicular Germ Cell Tumors. Gene 2018, 661, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.H.; Einhorn, L.H. Testicular Cancer—Discoveries and Updates. N. Engl. J. Med. 2014, 371, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Chovanec, M.; Cheng, L. Advances in Diagnosis and Treatment of Testicular Cancer. BMJ 2022, 379, e070499. [Google Scholar] [CrossRef]
- Jacobsen, C.; Honecker, F. Cisplatin Resistance in Germ Cell Tumours: Models and Mechanisms. Andrology 2015, 3, 111–121. [Google Scholar] [CrossRef]
- Baroni, T.; Arato, I.; Mancuso, F.; Calafiore, R.; Luca, G. On the Origin of Testicular Germ Cell Tumors: From Gonocytes to Testicular Cancer. Front. Endocrinol. 2019, 10, 343. [Google Scholar] [CrossRef]
- Albers, P.; Albrecht, W.; Algaba, F.; Bokemeyer, C.; Cohn-Cedermark, G.; Fizazi, K.; Horwich, A.; Laguna, M.P.; Nicolai, N.; Oldenburg, J. Guidelines on Testicular Cancer: 2015 Update. Eur. Urol. 2015, 68, 1054–1068. [Google Scholar] [CrossRef]
- Medvedev, K.E.; Savelyeva, A.V.; Chen, K.S.; Bagrodia, A.; Jia, L.; Grishin, N.V. Integrated Molecular Analysis Reveals 2 Distinct Subtypes of Pure Seminoma of the Testis. Cancer Inform. 2022, 21, 11769351221132634. [Google Scholar] [CrossRef]
- Howitt, B.E.; Berney, D.M. Tumors of the Testis: Morphologic Features and Molecular Alterations. Surg. Pathol. Clin. 2015, 8, 687–716. [Google Scholar] [CrossRef] [PubMed]
- Rajpert-De Meyts, E.; McGlynn, K.A.; Okamoto, K.; Jewett, M.A.S.; Bokemeyer, C. Testicular Germ Cell Tumours. Lancet 2016, 387, 1762–1774. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Fazal, Z.; Freemantle, S.J.; Spinella, M.J. Mechanisms of Cisplatin Sensitivity and Resistance in Testicular Germ Cell Tumors. Cancer Drug. Resist. 2019, 2, 580–594. [Google Scholar] [CrossRef]
- Singh, R.; Fazal, Z.; Freemantle, S.J.; Spinella, M.J. Between a Rock and a Hard Place: An Epigenetic-Centric View of Testicular Germ Cell Tumors. Cancers 2021, 13, 1506. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, A.; Pfister, D.; Paffenholz, P. Salvage Management of Patients with Relapsing Testicular Germ Cell Tumors. Curr. Opin. Urol. 2021, 31, 206–213. [Google Scholar] [CrossRef] [PubMed]
- González-Barrios, R.; Alcaraz, N.; Montalvo-Casimiro, M.; Cervera, A.; Arriaga-Canon, C.; Munguia-Garza, P.; Hinojosa-Ugarte, D.; Sobrevilla-Moreno, N.; Torres-Arciga, K.; Mendoza-Perez, J.; et al. Genomic Profile in a Non-Seminoma Testicular Germ-Cell Tumor Cohort Reveals a Potential Biomarker of Sensitivity to Platinum-Based Therapy. Cancers 2022, 14, 2065. [Google Scholar] [CrossRef]
- Litchfield, K.; Levy, M.; Huddart, R.A.; Shipley, J.; Turnbull, C. The Genomic Landscape of Testicular Germ Cell Tumours: From Susceptibility to Treatment. Nat. Rev. Urol. 2016, 13, 409–419. [Google Scholar] [CrossRef]
- Loveday, C.; Litchfield, K.; Proszek, P.Z.; Cornish, A.J.; Santo, F.; Levy, M.; Macintyre, G.; Holryod, A.; Broderick, P.; Dudakia, D.; et al. Genomic Landscape of Platinum Resistant and Sensitive Testicular Cancers. Nat. Commun. 2020, 11, 2189. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; Puccini, A.; Grothey, A.; Raghavan, D.; Goldberg, R.M.; Xiu, J.; Korn, W.M.; Weinberg, B.A.; Hwang, J.J.; Shields, A.F.; et al. Landscape of Tumor Mutation Load, Mismatch Repair Deficiency, and PD-L1 Expression in a Large Patient Cohort of Gastrointestinal Cancers. Mol. Cancer Res. 2018, 16, 805–812. [Google Scholar] [CrossRef]
- Sheikh, E.; Tran, T.; Vranic, S.; Levy, A.; Bonfil, R.D. Role and Significance of C-KIT Receptor Tyrosine Kinase in Cancer: A Review. Bosn. J. Basic. Med. Sci. 2022, 22, 5. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, A.; Summersgill, B.; Grygalewicz, B.; Gillis, A.J.M.; Stoop, J.; van Gurp, R.J.H.L.M.; Dennis, N.; Fisher, C.; Huddart, R.; Cooper, C.; et al. Amplification and Overexpression of the KIT Gene Is Associated with Progression in the Seminoma Subtype of Testicular Germ Cell Tumors of Adolescents and Adults. Cancer Res. 2005, 65, 8085–8089. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Yao, X.; Zhou, H.; Duan, C.; Wu, X.; Li, B.; Liu, H.; Zhang, Y. Comprehensive Characteristics of Pathological Subtypes in Testicular Germ Cell Tumor: Gene Expression, Mutation and Alternative Splicing. Front. Immunol. 2023, 13, 1096494. [Google Scholar] [CrossRef] [PubMed]
- Kazemi-Sefat, G.E.; Keramatipour, M.; Talebi, S.; Kavousi, K.; Sajed, R.; Kazemi-Sefat, N.A.; Mousavizadeh, K. The Importance of CDC27 in Cancer: Molecular Pathology and Clinical Aspects. Cancer Cell Int. 2021, 21, 160. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Singla, N.; Lafin, J.T.; Ghandour, R.A.; Kaffenberger, S.; Amatruda, J.F.; Bagrodia, A. Genetics of Testicular Germ Cell Tumors. Curr. Opin. Urol. 2019, 29, 344–349. [Google Scholar] [CrossRef]
- Taylor-Weiner, A.; Zack, T.; O’Donnell, E.; Guerriero, J.L.; Bernard, B.; Reddy, A.; Han, G.C.; AlDubayan, S.; Amin-Mansour, A.; Schumacher, S.E.; et al. Genomic Evolution and Chemoresistance in Germ-Cell Tumours. Nature 2016, 540, 114–118. [Google Scholar] [CrossRef]
- Chovanec, M.; Albany, C.; Mego, M.; Montironi, R.; Cimadamore, A.; Cheng, L. Emerging Prognostic Biomarkers in Testicular Germ Cell Tumors: Looking Beyond Established Practice. Front. Oncol. 2018, 8, 571. [Google Scholar] [CrossRef]
- Litchfield, K.; Levy, M.; Orlando, G.; Loveday, C.; Law, P.J.; Migliorini, G.; Holroyd, A.; Broderick, P.; Karlsson, R.; Haugen, T.B.; et al. Identification of 19 New Risk Loci and Potential Regulatory Mechanisms Influencing Susceptibility to Testicular Germ Cell Tumor. Nat. Genet. 2017, 49, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Pluta, J.; Pyle, L.C.; Nead, K.T.; Wilf, R.; Li, M.; Mitra, N.; Weathers, B.; D’Andrea, K.; Almstrup, K.; Anson-Cartwright, L.; et al. Identification of 22 Susceptibility Loci Associated with Testicular Germ Cell Tumors. Nat. Commun. 2021, 12, 4487. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Summersgill, B.; Yost, S.; Sultana, R.; Labreche, K.; Dudakia, D.; Renwick, A.; Seal, S.; Al-Saadi, R.; Broderick, P.; et al. Whole-Exome Sequencing Reveals the Mutational Spectrum of Testicular Germ Cell Tumours. Nat. Commun. 2015, 6, 5973. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, A.M.; Skakkebaek, N.E.; Lundsteen, C.; Leffers, H.; Larsen, J.; Rajpert-De Meyts, E. High-Resolution Comparative Genomic Hybridization Detects Extra Chromosome Arm 12p Material in Most Cases of Carcinoma in Situ Adjacent to Overt Germ Cell Tumors, but Not before the Invasive Tumor Development: 12p Gain in Carcinoma In Situ of Germ Cell Tumors. Genes. Chromosom. Cancer 2003, 38, 117–125. [Google Scholar] [CrossRef]
- Rahimi, M.; Behjati, F.; Khorshid, H.R.K.; Karimlou, M.; Keyhani, E. The Relationship between KIT Copy Number Variation, Protein Expression, and Angiogenesis in Sporadic Breast Cancer. Rep. Biochem. Molecul. 2020, 9, 40–49. [Google Scholar] [CrossRef]
- Landero-Huerta, D.A.; Vigueras-Villaseñor, R.M.; Yokoyama-Rebollar, E.; García-Andrade, F.; Rojas-Castañeda, J.C.; Herrera-Montalvo, L.A.; Díaz-Chávez, J.; Pérez-Añorve, I.X.; Aréchaga-Ocampo, E.; Chávez-Saldaña, M.D. Cryptorchidism and Testicular Tumor: Comprehensive Analysis of Common Clinical Features and Search of SNVs in the KIT and AR Genes. Front. Cell Dev. Biol. 2020, 8, 762. [Google Scholar] [CrossRef]
- Califf, R.M. Biomarker Definitions and Their Applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, M.; Kollmannsberger, C.; Nappi, L. Emerging Role of Biomarkers in Testicular Germ Cell Tumors. Curr. Oncol. Rep. 2022, 24, 437–442. [Google Scholar] [CrossRef]
- Abada, P.B.; Howell, S.B. Cisplatin Induces Resistance by Triggering Differentiation of Testicular Embryonal Carcinoma Cells. PLoS ONE 2014, 9, e87444. [Google Scholar] [CrossRef]
- Mego, M.; Cierna, Z.; Svetlovska, D.; Macak, D.; Machalekova, K.; Miskovska, V.; Chovanec, M.; Usakova, V.; Obertova, J.; Babal, P.; et al. PARP Expression in Germ Cell Tumours. J. Clin. Pathol. 2013, 66, 607–612. [Google Scholar] [CrossRef]
- Országhová, Z.; Kalavska, K.; Mego, M.; Chovanec, M. Overcoming Chemotherapy Resistance in Germ Cell Tumors. Biomedicines 2022, 10, 972. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.C.; Kirschner, A.; Scarpato, K.R.; Morgans, A.K. Current Management of Refractory Germ Cell Tumors and Future Directions. Curr. Oncol. Rep. 2017, 19, 8. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Jerónimo, C.; Henrique, R. Cisplatin Resistance in Testicular Germ Cell Tumors: Current Challenges from Various Perspectives. Cancers 2020, 12, 1601. [Google Scholar] [CrossRef] [PubMed]
- Boer, H.; Proost, J.H.; Nuver, J.; Bunskoek, S.; Gietema, J.Q.; Geubels, B.M.; Altena, R.; Zwart, N.; Oosting, S.F.; Vonk, J.M.; et al. Long-Term Exposure to Circulating Platinum Is Associated with Late Effects of Treatment in Testicular Cancer Survivors. Ann. Oncol. 2015, 26, 2305–2310. [Google Scholar] [CrossRef]
- Chovanec, M.; Lauritsen, J.; Bandak, M.; Oing, C.; Kier, G.G.; Kreiberg, M.; Rosenvilde, J.; Wagner, T.; Bokemeyer, C.; Daugaard, G. Late Adverse Effects and Quality of Life in Survivors of Testicular Germ Cell Tumour. Nat. Rev. Urol. 2021, 18, 227–245. [Google Scholar] [CrossRef]
- Oing, C.; Seidel, C.; Bokemeyer, C. Therapeutic Approaches for Refractory Germ Cell Cancer. Expert. Rev. Anticancer Ther. 2018, 18, 389–397. [Google Scholar] [CrossRef]
- Fenske, A.E.; Glaesener, S.; Bokemeyer, C.; Thomale, J.; Dahm-Daphi, J.; Honecker, F.; Dartsch, D.C. Cisplatin Resistance Induced in Germ Cell Tumour Cells Is Due to Reduced Susceptibility towards Cell Death but Not to Altered DNA Damage Induction or Repair. Cancer Lett. 2012, 324, 171–178. [Google Scholar] [CrossRef]
- Lim, Z.-F.; Ma, P.C. Emerging Insights of Tumor Heterogeneity and Drug Resistance Mechanisms in Lung Cancer Targeted Therapy. J. Hematol. Oncol. 2019, 12, 134. [Google Scholar] [CrossRef]
- Amable, L. Cisplatin Resistance and Opportunities for Precision Medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef]
- LeBron, C.; Pal, P.; Brait, M.; Dasgupta, S.; Guerrero-Preston, R.; Looijenga, L.H.J.; Kowalski, J.; Netto, G.; Hoque, M.O. Genome-Wide Analysis of Genetic Alterations in Testicular Primary Seminoma Using High Resolution Single Nucleotide Polymorphism Arrays. Genomics 2011, 97, 341–349. [Google Scholar] [CrossRef]
- Timmerman, D.M.; Eleveld, T.F.; Sriram, S.; Dorssers, L.C.J.; Gillis, A.J.M.; Schmidtova, S.; Kalavska, K.; van de Werken, H.J.G.; Oing, C.; Honecker, F.; et al. Chromosome 3p25.3 Gain Is Associated With Cisplatin Resistance and Is an Independent Predictor of Poor Outcome in Male Malignant Germ Cell Tumors. JCO 2022, 40, 3077–3087. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, J.; Martínez, J.; Hernández, C.; Pérez-Montiel, D.; Castro, C.; Fabián-Morales, E.; Santibáñez, M.; González-Barrios, R.; Díaz-Chávez, J.; Andonegui, M.A.; et al. Association between ERCC1 and XPA Expression and Polymorphisms and the Response to Cisplatin in Testicular Germ Cell Tumours. Br. J. Cancer 2013, 109, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Awuah, S.G.; Riddell, I.A.; Lippard, S.J. Repair Shielding of Platinum-DNA Lesions in Testicular Germ Cell Tumors by High-Mobility Group Box Protein 4 Imparts Cisplatin Hypersensitivity. Proc. Natl. Acad. Sci. USA 2017, 114, 950–955. [Google Scholar] [CrossRef]
- Kitayama, S.; Ikeda, K.; Sato, W.; Takeshita, H.; Kawakami, S.; Inoue, S.; Horie, K. Testis-Expressed Gene 11 Inhibits Cisplatin-Induced DNA Damage and Contributes to Chemoresistance in Testicular Germ Cell Tumor. Sci. Rep. 2022, 12, 18423. [Google Scholar] [CrossRef]
- Sakurai, Y.; Ichinoe, M.; Yoshida, K.; Nakazato, Y.; Saito, S.; Satoh, M.; Nakada, N.; Sanoyama, I.; Umezawa, A.; Numata, Y.; et al. Inactivation of REV7 Enhances Chemosensitivity and Overcomes Acquired Chemoresistance in Testicular Germ Cell Tumors. Cancer Lett. 2020, 489, 100–110. [Google Scholar] [CrossRef]
- Noel, E.E.; Yeste-Velasco, M.; Mao, X.; Perry, J.; Kudahetti, S.C.; Li, N.F.; Sharp, S.; Chaplin, T.; Xue, L.; McIntyre, A.; et al. The Association of CCND1 Overexpression and Cisplatin Resistance in Testicular Germ Cell Tumors and Other Cancers. Am. J. Pathol. 2010, 176, 2607–2615. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; McCormick, F.; Smith-McCune, K.; Hanahan, D. Enhancing Tumor-Specific Uptake of the Anticancer Drug Cisplatin with a Copper Chelator. Cancer Cell 2010, 17, 574–583. [Google Scholar] [CrossRef]
- Bauer, S.; Mühlenberg, T.; Leahy, M.; Hoiczyk, M.; Gauler, T.; Schuler, M.; Looijenga, L. Therapeutic Potential of Mdm2 Inhibition in Malignant Germ Cell Tumours. Eur. Urol. 2010, 57, 679–687. [Google Scholar] [CrossRef]
- Lobo, J.; Alzamora, M.A.; Guimarães, R.; Cantante, M.; Lopes, P.; Braga, I.; Maurício, J.; Jerónimo, C.; Henrique, R. P53 and MDM2 Expression in Primary and Metastatic Testicular Germ Cell Tumors: Association with Clinical Outcome. Andrology 2020, 8, 1233–1242. [Google Scholar] [CrossRef]
- Cabral, E.R.M.; Pacanhella, M.F.; van Helvoort Lengert, A.; dos Reis, M.B.; Leal, L.F.; de Lima, M.A.; da Silva, A.L.V.; Pinto, I.A.; Reis, R.M.; Pinto, M.T.; et al. Somatic Mutation Detection and KRAS Amplification in Testicular Germ Cell Tumors. Front. Oncol. 2023, 13, 1133363. [Google Scholar] [CrossRef]
- Feldman, D.R.; Iyer, G.; Van Alstine, L.; Patil, S.; Al-Ahmadie, H.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S.; Solit, D.B. Presence of Somatic Mutations within PIK3CA, AKT, RAS, and FGFR3 but Not BRAF in Cisplatin-Resistant Germ Cell Tumors. Clin. Cancer Res. 2014, 20, 3712–3720. [Google Scholar] [CrossRef]
- Vranic, S.; Hes, O.; Grossmann, P.; Gatalica, Z. Low Frequency of HIF-1α Overexpression in Germ Cell Tumors of the Testis. Appl. Immunohistochem. Mol. Morphol. 2013, 21, 165–169. [Google Scholar] [CrossRef]
- Shenoy, N.; Dronca, R.; Quevedo, F.; Boorjian, S.A.; Cheville, J.; Costello, B.; Kohli, M.; Witzig, T.; Pagliaro, L. Low Hypoxia Inducible Factor-1α (HIF-1α) Expression in Testicular Germ Cell Tumors—A Major Reason for Enhanced Chemosensitivity? Chin. J. Cancer Res. 2017, 29, 374–378. [Google Scholar] [CrossRef]
- Gan, Y.; Wang, Y.; Tan, Z.; Zhou, J.; Kitazawa, R.; Jiang, X.; Tang, Y.; Yang, J. TDRG1 Regulates Chemosensitivity of Seminoma TCam-2 Cells to Cisplatin via PI3K/Akt/MTOR Signaling Pathway and Mitochondria-Mediated Apoptotic Pathway. Cancer Biol. Ther. 2016, 17, 741–750. [Google Scholar] [CrossRef]
- Schmidtova, S.; Kalavska, K.; Gercakova, K.; Cierna, Z.; Miklikova, S.; Smolkova, B.; Buocikova, V.; Miskovska, V.; Durinikova, E.; Burikova, M.; et al. Disulfiram Overcomes Cisplatin Resistance in Human Embryonal Carcinoma Cells. Cancers 2019, 11, 1224. [Google Scholar] [CrossRef]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the Anticancer Drug Cisplatin Mediated by the Copper Transporter Ctr1 in Yeast and Mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaidy, K.I.; Chovanec, M.; Cheng, L. Molecular Characteristics of Testicular Germ Cell Tumors: Pathogenesis and Mechanisms of Therapy Resistance. Expert. Rev. Anticancer Ther. 2020, 20, 75–79. [Google Scholar] [CrossRef]
- Cavallo, F.; Feldman, D.R.; Barchi, M. Revisiting DNA Damage Repair, P53-Mediated Apoptosis and Cisplatin Sensitivity in Germ Cell Tumors. Int. J. Dev. Biol. 2013, 57, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, C.; Freemantle, S.; Spinella, M. Testicular Germ Cell Tumors: A Paradigm for the Successful Treatment of Solid Tumor Stem Cells. CCTR 2006, 2, 255–270. [Google Scholar] [CrossRef]
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin−DNA Adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef] [PubMed]
- Boublikova, L.; Buchler, T.; Stary, J.; Abrahamova, J.; Trka, J. Molecular Biology of Testicular Germ Cell Tumors: Unique Features Awaiting Clinical Application. Crit. Rev. Oncol./Hematol. 2014, 89, 366–385. [Google Scholar] [CrossRef]
- Woldu, S.L.; Amatruda, J.F.; Bagrodia, A. Testicular Germ Cell Tumor Genomics. Curr. Opin. Urol. 2017, 27, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Guillou, L.; Estreicher, A.; Chaubert, P.; Hurlimann, J.; Kurt, A.M.; Metthez, G.; Iggo, R.; Gray, A.C.; Jichlinski, P.; Leisinger, H.J.; et al. Germ Cell Tumors of the Testis Overexpress Wild-Type P53. Am. J. Pathol. 1996, 149, 1221–1228. [Google Scholar] [PubMed]
- Heidenreich, A.; Schenkman, N.S.; Sesterhenn, I.A.; Mostofi, K.F.; Moul, J.W.; Srivastava, S.; Engelmann, U.H. Immunohistochemical and Mutational Analysis of the P53 Tumour Suppressor Gene and the Bcl-2 Oncogene in Primary Testicular Germ Cell Tumours. APMIS 1998, 106, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Koster, R.; Timmer-Bosscha, H.; Bischoff, R.; Gietema, J.A.; de Jong, S. Disruption of the MDM2–P53 Interaction Strongly Potentiates P53-Dependent Apoptosis in Cisplatin-Resistant Human Testicular Carcinoma Cells via the Fas/FasL Pathway. Cell Death Dis. 2011, 2, e148. [Google Scholar] [CrossRef]
- Spierings, D.C.J.; de Vries, E.G.E.; Stel, A.J.; te Rietstap, N.; Vellenga, E.; de Jong, S. Low P21Waf1/Cip1 Protein Level Sensitizes Testicular Germ Cell Tumor Cells to Fas-Mediated Apoptosis. Oncogene 2004, 23, 4862–4872. [Google Scholar] [CrossRef]
- Looijenga, L.H.J.; Stoop, H.; de Leeuw, H.P.J.C.; de Gouveia Brazao, C.A.; Gillis, A.J.M.; van Roozendaal, K.E.P.; van Zoelen, E.J.J.; Weber, R.F.A.; Wolffenbuttel, K.P.; van Dekken, H.; et al. POU5F1 (OCT3/4) Identifies Cells with Pluripotent Potential in Human Germ Cell Tumors. Cancer Res. 2003, 63, 2244–2250. [Google Scholar]
- Morsi, R.Z.; Hage-Sleiman, R.; Kobeissy, H.; Dbaibo, G. Noxa: Role in Cancer Pathogenesis and Treatment. Curr. Cancer Drug Targets 2018, 18, 914–928. [Google Scholar] [CrossRef]
- Gutekunst, M.; Mueller, T.; Weilbacher, A.; Dengler, M.A.; Bedke, J.; Kruck, S.; Oren, M.; Aulitzky, W.E.; van der Kuip, H. Cisplatin Hypersensitivity of Testicular Germ Cell Tumors Is Determined by High Constitutive Noxa Levels Mediated by Oct-4. Cancer Res. 2013, 73, 1460–1469. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular Mechanisms of Cisplatin Resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef]
- Chen, H.H.W.; Kuo, M.T. Role of Glutathione in the Regulation of Cisplatin Resistance in Cancer Chemotherapy. Met.-Based Drugs 2010, 2010, 430909. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Constâncio, V.; Guimarães-Teixeira, C.; Leite-Silva, P.; Miranda-Gonçalves, V.; Sequeira, J.P.; Pistoni, L.; Guimarães, R.; Cantante, M.; Braga, I.; et al. Promoter Methylation of DNA Homologous Recombination Genes Is Predictive of the Responsiveness to PARP Inhibitor Treatment in Testicular Germ Cell Tumors. Mol. Oncol. 2021, 15, 846–865. [Google Scholar] [CrossRef] [PubMed]
- Fichtner, A.; Bohnenberger, H.; Elakad, O.; Richter, A.; Lenz, C.; Oing, C.; Ströbel, P.; Kueffer, S.; Nettersheim, D.; Bremmer, F. Proteomic Profiling of Cisplatin-Resistant and Cisplatin-Sensitive Germ Cell Tumour Cell Lines Using Quantitative Mass Spectrometry. World J. Urol. 2022, 40, 373–383. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Mouw, K.W.; Kim, P.; Iyer, G.; Wagle, N.; Al-Ahmadie, H.; Zhu, C.; Ostrovnaya, I.; Kryukov, G.V.; O’Connor, K.W.; et al. Somatic ERCC2 Mutations Correlate with Cisplatin Sensitivity in Muscle-Invasive Urothelial Carcinoma. Cancer Discov. 2014, 4, 1140–1153. [Google Scholar] [CrossRef]
- Barrett, M.T.; Lenkiewicz, E.; Malasi, S.; Stanton, M.; Slack, J.; Andrews, P.; Pagliaro, L.; Bryce, A.H. Clonal Analyses of Refractory Testicular Germ Cell Tumors. PLoS ONE 2019, 14, e0213815. [Google Scholar] [CrossRef]
- Lin, W.-Y.; Camp, N.J.; Cannon-Albright, L.A.; Allen-Brady, K.; Balasubramanian, S.; Reed, M.W.R.; Hopper, J.L.; Apicella, C.; Giles, G.G.; Southey, M.C.; et al. A Role for XRCC2 Gene Polymorphisms in Breast Cancer Risk and Survival. J. Med. Genet. 2011, 48, 477–484. [Google Scholar] [CrossRef]
- Honecker, F.; Wermann, H.; Mayer, F.; Gillis, A.J.M.; Stoop, H.; van Gurp, R.J.L.M.; Oechsle, K.; Steyerberg, E.; Hartmann, J.T.; Dinjens, W.N.M.; et al. Microsatellite Instability, Mismatch Repair Deficiency, and BRAF Mutation in Treatment-Resistant Germ Cell Tumors. JCO 2009, 27, 2129–2136. [Google Scholar] [CrossRef] [PubMed]
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic Determinants of Cisplatin Resistance in Patients With Advanced Germ Cell Tumors. JCO 2016, 34, 4000–4007. [Google Scholar] [CrossRef] [PubMed]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.M.; Stoop, H.; Nagel, R.; Liu, Y.-P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A Genetic Screen Implicates MiRNA-372 and MiRNA-373 As Oncogenes in Testicular Germ Cell Tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef]
- Zhang, S.; Carlsen, L.; Hernandez Borrero, L.; Seyhan, A.A.; Tian, X.; El-Deiry, W.S. Advanced Strategies for Therapeutic Targeting of Wild-Type and Mutant P53 in Cancer. Biomolecules 2022, 12, 548. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Li, X. Targeting the Untargetable KRAS in Cancer Therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.R.; Burmeister, A.; Skowron, M.A.; Stephan, A.; Bremmer, F.; Wakileh, G.A.; Petzsch, P.; Köhrer, K.; Albers, P.; Nettersheim, D. Therapeutical Interference with the Epigenetic Landscape of Germ Cell Tumors: A Comparative Drug Study and New Mechanistical Insights. Clin. Epigenet. 2022, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Nicu, A.-T.; Medar, C.; Chifiriuc, M.C.; Gradisteanu Pircalabioru, G.; Burlibasa, L. Epigenetics and Testicular Cancer: Bridging the Gap Between Fundamental Biology and Patient Care. Front. Cell Dev. Biol. 2022, 10, 861995. [Google Scholar] [CrossRef]
- Meng, H.; Cao, Y.; Qin, J.; Song, X.; Zhang, Q.; Shi, Y.; Cao, L. DNA Methylation, Its Mediators and Genome Integrity. Int. J. Biol. Sci. 2015, 11, 604–617. [Google Scholar] [CrossRef]
- Wermann, H.; Stoop, H.; Gillis, A.J.M.; Honecker, F.; Van Gurp, R.J.H.L.M.; Ammerpohl, O.; Richter, J.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H.J. Global DNA Methylation in Fetal Human Germ Cells and Germ Cell Tumours: Association with Differentiation and Cisplatin Resistance. J. Pathol. 2010, 221, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Fazal, Z.; Singh, R.; Fang, F.; Bikorimana, E.; Baldwin, H.; Corbet, A.; Tomlin, M.; Yerby, C.; Adra, N.; Albany, C.; et al. Hypermethylation and Global Remodelling of DNA Methylation Is Associated with Acquired Cisplatin Resistance in Testicular Germ Cell Tumours. Epigenetics 2021, 16, 1071–1084. [Google Scholar] [CrossRef]
- Sonnenburg, D.; Spinella, M.J.; Albany, C. Epigenetic Targeting of Platinum Resistant Testicular Cancer. Curr. Cancer Drug Targets 2016, 16, 789–795. [Google Scholar] [CrossRef]
- Koul, S.; McKiernan, J.M.; Narayan, G.; Houldsworth, J.; Bacik, J.; Dobrzynski, D.L.; Assaad, A.M.; Mansukhani, M.; Reuter, V.E.; Bosl, G.J.; et al. Role of Promoter Hypermethylation in Cisplatin Treatment Response of Male Germ Cell Tumors. Mol. Cancer 2004, 3, 16. [Google Scholar] [CrossRef]
- da Silva Martinelli, C.M.; van Helvoort Lengert, A.; Cárcano, F.M.; Silva, E.C.A.; Brait, M.; Lopes, L.F.; Vidal, D.O. MGMT and CALCA Promoter Methylation Are Associated with Poor Prognosis in Testicular Germ Cell Tumor Patients. Oncotarget 2016, 8, 50608–50617. [Google Scholar] [CrossRef]
- Lobo, J.; van Zogchel, L.M.J.; Nuru, M.G.; Gillis, A.J.M.; van der Schoot, C.E.; Tytgat, G.A.M.; Looijenga, L.H.J. Combining Hypermethylated Rassf1a Detection Using Ddpcr with Mir-371a-3p Testing: An Improved Panel of Liquid Biopsy Biomarkers for Testicular Germ Cell Tumor Patients. Cancers 2021, 13, 5228. [Google Scholar] [CrossRef]
- Fitzpatrick, G.V.; Pugacheva, E.M.; Shin, J.-Y.; Abdullaev, Z.; Yang, Y.; Khatod, K.; Lobanenkov, V.V.; Higgins, M.J. Allele-Specific Binding of CTCF to the Multipartite Imprinting Control Region KvDMR1. Mol. Cell. Biol. 2007, 27, 2636–2647. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Constâncio, V.; Leite-Silva, P.; Guimarães, R.; Cantante, M.; Braga, I.; Maurício, J.; Looijenga, L.H.J.; Henrique, R.; Jerónimo, C. Differential Methylation EPIC Analysis Discloses Cisplatin-Resistance Related Hypermethylation and Tumor-Specific Heterogeneity within Matched Primary and Metastatic Testicular Germ Cell Tumor Patient Tissue Samples. Clin. Epigenet. 2021, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Oing, C.; Skowron, M.A.; Bokemeyer, C.; Nettersheim, D. Epigenetic Treatment Combinations to Effectively Target Cisplatin-Resistant Germ Cell Tumors: Past, Present, and Future Considerations. Andrology 2019, 7, 487–497. [Google Scholar] [CrossRef]
- Benešová, M.; Trejbalová, K.; Kučerová, D.; Vernerová, Z.; Hron, T.; Szabó, A.; Amouroux, R.; Klézl, P.; Hajkova, P.; Hejnar, J. Overexpression of TET Dioxygenases in Seminomas Associates with Low Levels of DNA Methylation and Hydroxymethylation. Mol. Carcinog. 2017, 56, 1837–1850. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Guimarães, R.; Miranda-Gonçalves, V.; Monteiro-Reis, S.; Cantante, M.; Antunes, L.; Braga, I.; Maurício, J.; Looijenga, L.H.; Jerónimo, C.; et al. Differential Expression of DNA Methyltransferases and Demethylases among the Various Testicular Germ Cell Tumor Subtypes. Epigenomics 2020, 12, 1579–1592. [Google Scholar] [CrossRef]
- Lobo, J.; Henrique, R.; Jerónimo, C. The Role of DNA/Histone Modifying Enzymes and Chromatin Remodeling Complexes in Testicular Germ Cell Tumors. Cancers 2019, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.R.; Lobo, J.; Miranda-Gonçalves, V.; Henrique, R.; Jerónimo, C. Epigenetic Alterations as Therapeutic Targets in Testicular Germ Cell Tumours: Current and Future Application of ‘Epidrugs’. Epigenetics 2021, 16, 353–372. [Google Scholar] [CrossRef]
- Singh, R.; Fazal, Z.; Bikorimana, E.; Boyd, R.I.; Yerby, C.; Tomlin, M.; Baldwin, H.; Shokry, D.; Corbet, A.K.; Shahid, K.; et al. Reciprocal Epigenetic Remodeling Controls Testicular Cancer Hypersensitivity to Hypomethylating Agents and Chemotherapy. Mol. Oncol. 2022, 16, 683–698. [Google Scholar] [CrossRef]
- Munari, E.; Chaux, A.; Vaghasia, A.M.; Taheri, D.; Karram, S.; Bezerra, S.M.; Gonzalez Roibon, N.; Nelson, W.G.; Yegnasubramanian, S.; Netto, G.J.; et al. Global 5-Hydroxymethylcytosine Levels Are Profoundly Reduced in Multiple Genitourinary Malignancies. PLoS ONE 2016, 11, e0146302. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of MRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-Methyladenosine and Its Role in Cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Hu, Y.; Zhou, B.; Bao, Y.; Li, Z.; Gong, C.; Yang, H.; Wang, S.; Xiao, Y. The Role of M6A Modification in Physiology and Disease. Cell Death Dis. 2020, 11, 960. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Barros-Silva, D.; Henrique, R.; Jerónimo, C. The Emerging Role of Epitranscriptomics in Cancer: Focus on Urological Tumors. Genes 2018, 9, 552. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Liu, D.; Dong, S.; Zeng, L.; Wu, Z.; Zhao, P.; Zhang, L.; Chen, Z.S.; Zou, C. Epitranscriptomics and Epiproteomics in Cancer Drug Resistance: Therapeutic Implications. Signal Transduct. Target. Ther. 2020, 5, 193. [Google Scholar] [CrossRef]
- Miranda-Gonçalves, V.; Lobo, J.; Guimarães-Teixeira, C.; Barros-Silva, D.; Guimarães, R.; Cantante, M.; Braga, I.; Maurício, J.; Oing, C.; Honecker, F.; et al. The Component of the M6A Writer Complex VIRMA Is Implicated in Aggressive Tumor Phenotype, DNA Damage Response and Cisplatin Resistance in Germ Cell Tumors. J. Exp. Clin. Cancer Res. 2021, 40, 268. [Google Scholar] [CrossRef]
- Wei, J.; Yin, Y.; Zhou, J.; Chen, H.; Peng, J.; Yang, J.; Tang, Y. METTL3 Potentiates Resistance to Cisplatin through M6A Modification of TFAP2C in Seminoma. J. Cell. Mol. Med. 2020, 24, 11366–11380. [Google Scholar] [CrossRef]
- Port, M.; Glaesener, S.; Ruf, C.; Riecke, A.; Bokemeyer, C.; Meineke, V.; Honecker, F.; Abend, M. Micro-RNA Expression in Cisplatin Resistant Germ Cell Tumor Cell Lines. Mol. Cancer 2011, 10, 52. [Google Scholar] [CrossRef]
- Liu, L.; Lian, J.; Zhang, H.; Tian, H.; Liang, M.; Yin, M.; Sun, F. MicroRNA-302a Sensitizes Testicular Embryonal Carcinoma Cells to Cisplatin-Induced Cell Death. J. Cell. Physiol. 2013, 228, 2294–2304. [Google Scholar] [CrossRef]
- Das, M.K.; Evensen, H.S.F.; Furu, K.; Haugen, T.B. MiRNA-302s May Act as Oncogenes in Human Testicular Germ Cell Tumours. Sci. Rep. 2019, 9, 9189. [Google Scholar] [CrossRef]
- Huang, H.; Tian, H.; Duan, Z.; Cao, Y.; Zhang, X.-S.; Sun, F. MicroRNA-383 Impairs Phosphorylation of H2AX by Targeting PNUTS and Inducing Cell Cycle Arrest in Testicular Embryonal Carcinoma Cells. Cell. Signal. 2014, 26, 903–911. [Google Scholar] [CrossRef]
- Roška, J.; Lobo, J.; Ivovič, D.; Wachsmannová, L.; Mueller, T.; Henrique, R.; Jerónimo, C.; Chovanec, M.; Jurkovičová, D. Integrated Microarray-Based Data Analysis of MiRNA Expression Profiles: Identification of Novel Biomarkers of Cisplatin-Resistance in Testicular Germ Cell Tumours. IJMS 2023, 24, 2495. [Google Scholar] [CrossRef]
- Singh, R.; Fazal, Z.; Corbet, A.K.; Bikorimana, E.; Rodriguez, J.C.; Khan, E.M.; Shahid, K.; Freemantle, S.J.; Spinella, M.J. Epigenetic Remodeling through Downregulation of Polycomb Repressive Complex 2 Mediates Chemotherapy Resistance in Testicular Germ Cell Tumors. Cancers 2019, 11, 796. [Google Scholar] [CrossRef]
- Oing, C.; Dyshlovoy, S.; Honecker, F.; Nordquist, L.; Rothkamm, K.; Bokemeyer, C.; Petersen, C.; Mansour, W.Y. EP-1618: Monoubiquitinylated Histone H2B as a Potential Target in Treatment Resistant Germ Cell Tumors. Radiother. Oncol. 2018, 127, S871. [Google Scholar] [CrossRef]
- Huang, W.; Chen, T.Q.; Fang, K.; Zeng, Z.C.; Ye, H.; Chen, Y.Q. N6-Methyladenosine Methyltransferases: Functions, Regulation, and Clinical Potential. J. Hematol. Oncol. 2021, 14, 117. [Google Scholar] [CrossRef]
- Luo, Y.; Sun, Y.; Li, L.; Mao, Y. METTL3 May Regulate Testicular Germ Cell Tumors Through EMT and Immune Pathways. Cell Transplant. 2020, 29, 096368972094665. [Google Scholar] [CrossRef]
- Lobo, J.; Costa, A.L.; Cantante, M.; Guimarães, R.; Lopes, P.; Antunes, L.; Braga, I.; Oliveira, J.; Pelizzola, M.; Henrique, R.; et al. M6A RNA Modification and Its Writer/Reader VIRMA/YTHDF3 in Testicular Germ Cell Tumors: A Role in Seminoma Phenotype Maintenance. J. Transl. Med. 2019, 17, 79. [Google Scholar] [CrossRef]
- Cong, R.; Ji, C.; Zhang, J.; Zhang, Q.; Zhou, X.; Yao, L.; Luan, J.; Meng, X.; Song, N. M6A RNA Methylation Regulators Play an Important Role in the Prognosis of Patients with Testicular Germ Cell Tumor. Transl. Androl. Urol. 2021, 10, 662–679. [Google Scholar] [CrossRef]
- Tang, W.; Qian, J.; Qian, S. Biological Functions of RNA Modification Patterns That Define Tumor Microenvironment and Survival Outcomes in Testicular Germ Cell Tumors. Am. J. Transl. Res. 2022, 14, 6484–6503. [Google Scholar]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An Overview of MicroRNAs: Biology, Functions, Therapeutics, and Analysis Methods. J. Cell. Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene Regulation by Long Non-Coding RNAs and Its Biological Functions. Nat. Rev. Mol. Cell. Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Das, M.K.; Haugen, Ø.P.; Haugen, T.B. Diverse Roles and Targets of MiRNA in the Pathogenesis of Testicular Germ Cell Tumour. Cancers 2022, 14, 1190. [Google Scholar] [CrossRef]
- Bresesti, C.; Vezzoli, V.; Cangiano, B.; Bonomi, M. Long Non-Coding RNAs: Role in Testicular Cancers. Front. Oncol. 2021, 11, 605606. [Google Scholar] [CrossRef]
- Constâncio, V.; Tavares, N.T.; Henrique, R.; Jerónimo, C.; Lobo, J. MiRNA Biomarkers in Cancers of the Male Reproductive System: Are We Approaching Clinical Application? Andrology 2022, 11, 651–667. [Google Scholar] [CrossRef]
- Regouc, M.; Belge, G.; Lorch, A.; Dieckmann, K.-P.; Pichler, M. Non-Coding MicroRNAs as Novel Potential Tumor Markers in Testicular Cancer. Cancers 2020, 12, 749. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, J.; Hu, S.; Guo, Q.; Guo, X.; Ren, W.; Zhou, Q.; Duan, Y. Exosomal MiR-193b-3p Contributes to Cisplatin Sensitivity in Seminoma by Targeting ZBTB7A. Tohoku J. Exp. Med. 2022, 258, 309–317. [Google Scholar] [CrossRef]
- Wei, J.; Gan, Y.; Peng, D.; Jiang, X.; Kitazawa, R.; Xiang, Y.; Dai, Y.; Tang, Y.; Yang, J. Long Non-Coding RNA H19 Promotes TDRG1 Expression and Cisplatin Resistance by Sequestering MiRNA-106b-5p in Seminoma. Cancer Med. 2018, 7, 6247–6257. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhu, Q.-N.; Deng, J.-L.; Li, Z.-X.; Wang, G.; Zhu, Y.-S. Emerging Role of Long Non-Coding RNAs in Cisplatin Resistance. OTT 2018, 11, 3185–3194. [Google Scholar] [CrossRef]
- Liu, J.; Shi, H.; Li, X.; Chen, G.; Larsson, C.; Lui, W.-O. MiR-223-3p Regulates Cell Growth and Apoptosis via FBXW7 Suggesting an Oncogenic Role in Human Testicular Germ Cell Tumors. Int. J. Oncol. 2017, 50, 356–364. [Google Scholar] [CrossRef]
- Zhou, X.; Jin, W.; Jia, H.; Yan, J.; Zhang, G. MiR-223 Promotes the Cisplatin Resistance of Human Gastric Cancer Cells via Regulating Cell Cycle by Targeting FBXW7. J. Exp. Clin. Cancer Res. 2015, 34, 28. [Google Scholar] [CrossRef]
- Lobo, J.; Gillis, A.J.M.; van den Berg, A.; Dorssers, L.C.J.; Belge, G.; Dieckmann, K.-P.; Roest, H.P.; van der Laan, L.J.W.; Gietema, J.; Hamilton, R.J.; et al. Identification and Validation Model for Informative Liquid Biopsy-Based MicroRNA Biomarkers: Insights from Germ Cell Tumor In Vitro, In Vivo and Patient-Derived Data. Cells 2019, 8, 1637. [Google Scholar] [CrossRef]
- Peterson, C.L.; Laniel, M.-A. Histones and Histone Modifications. Curr. Biol. 2004, 14, R546–R551. [Google Scholar] [CrossRef]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the Roles of Histone-Modifying Enzymes and Their Associated Chromatin Modifications in Transcriptional Regulation. Nat. Genet. 2020, 52, 1271–1281. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic Modifications and Human Disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Samaržija, I.; Tomljanović, M.; Novak Kujundžić, R.; Trošelj, K.G. EZH2 Inhibition and Cisplatin as a Combination Anticancer Therapy: An Overview of Preclinical Studies. Cancers 2022, 14, 4761. [Google Scholar] [CrossRef]
- Chernikova, S.B.; Razorenova, O.V.; Higgins, J.P.; Sishc, B.J.; Nicolau, M.; Dorth, J.A.; Chernikova, D.A.; Kwok, S.; Brooks, J.D.; Bailey, S.M.; et al. Deficiency in Mammalian Histone H2B Ubiquitin Ligase Bre1 (Rnf20/Rnf40) Leads to Replication Stress and Chromosomal Instability. Cancer Res. 2012, 72, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Eckert, D.; Biermann, K.; Nettersheim, D.; Gillis, A.J.; Steger, K.; Jäck, H.-M.; Müller, A.M.; Looijenga, L.H.; Schorle, H. Expression of BLIMP1/PRMT5and Concurrent Histone H2A/H4 Arginine 3 Dimethylation in Fetal Germ Cells, CIS/IGCNU and Germ Cell Tumors. BMC Dev. Biol. 2008, 8, 106. [Google Scholar] [CrossRef]
- Grasso, C.; Popovic, M.; Isaevska, E.; Lazzarato, F.; Fiano, V.; Zugna, D.; Pluta, J.; Weathers, B.; D’Andrea, K.; Almstrup, K.; et al. Association Study between Polymorphisms in DNA Methylation-Related Genes and Testicular Germ Cell Tumor Risk. Cancer Epidemiol. Biomark. Prev. 2022, 31, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Jhuang, Y.L.; Yang, C.W.; Tseng, Y.F.; Hsu, C.L.; Li, H.Y.; Yuan, R.H.; Jeng, Y.M. SIN3-HDAC Complex-Associated Factor, a Chromatin Remodelling Gene Located in the 12p Amplicon, Is a Potential Germ Cell Tumour-Specific Oncogene. J. Pathol. 2022, 258, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; Berney, D.M.; Bokemeyer, C.; Climent, M.A.; Daugaard, G.; Gietema, J.A.; Giorgi, U.D.; Haugnes, H.S.; Huddart, R.A.; Leão, R.; et al. Testicular Seminoma and Non-Seminoma: ESMO-EURACAN Clinical Practice Guideline for Diagnosis, Treatment and Follow-Up☆. Ann. Oncol. 2022, 33, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Oing, C.; Hentrich, M.; Lorch, A.; Gläser, D.; Rumpold, H.; Ochsenreither, S.; Richter, S.; Dieing, A.; Zschäbitz, S.; Pereira, R.R.; et al. Treatment of Refractory Germ-Cell Tumours with Single-Agent Cabazitaxel: A German Testicular Cancer Study Group Case Series. J. Cancer Res. Clin. Oncol. 2020, 146, 449–455. [Google Scholar] [CrossRef]
- Albany, C.; Fazal, Z.; Singh, R.; Bikorimana, E.; Adra, N.; Hanna, N.H.; Einhorn, L.H.; Perkins, S.M.; Sandusky, G.E.; Christensen, B.C.; et al. A Phase 1 Study of Combined Guadecitabine and Cisplatin in Platinum Refractory Germ Cell Cancer. Cancer Med. 2021, 10, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Oing, C.; Verem, I.; Mansour, W.Y.; Bokemeyer, C.; Dyshlovoy, S.; Honecker, F. 5-Azacitidine Exerts Prolonged Pro-Apoptotic Effects and Overcomes Cisplatin-Resistance in Non-Seminomatous Germ Cell Tumor Cells. Int. J. Mol. Sci. 2018, 20, 21. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Cardoso, A.R.; Miranda-Gonçalves, V.; Looijenga, L.H.J.; Lopez, M.; Arimondo, P.B.; Henrique, R.; Jerónimo, C. Targeting Germ Cell Tumors with the Newly Synthesized Flavanone-Derived Compound MLo1302 Efficiently Reduces Tumor Cell Viability and Induces Apoptosis and Cell Cycle Arrest. Pharmaceutics 2021, 13, 73. [Google Scholar] [CrossRef] [PubMed]
- Steinemann, G.; Dittmer, A.; Schmidt, J.; Josuttis, D.; Fähling, M.; Biersack, B.; Beindorff, N.; Jolante Koziolek, E.; Schobert, R.; Brenner, W.; et al. Antitumor and Antiangiogenic Activity of the Novel Chimeric Inhibitor Animacroxam in Testicular Germ Cell Cancer. Mol. Oncol. 2019, 13, 2679–2696. [Google Scholar] [CrossRef]
- Lobo, J.; Guimarães-Teixeira, C.; Barros-Silva, D.; Miranda-Gonçalves, V.; Camilo, V.; Guimarães, R.; Cantante, M.; Braga, I.; Maurício, J.; Oing, C.; et al. Efficacy of HDAC Inhibitors Belinostat and Panobinostat against Cisplatin-Sensitive and Cisplatin-Resistant Testicular Germ Cell Tumors. Cancers 2020, 12, 2903. [Google Scholar] [CrossRef]
- Kurz, L.; Miklyaeva, A.; Skowron, M.A.; Overbeck, N.; Poschmann, G.; Becker, T.; Eul, K.; Kurz, T.; Schönberger, S.; Calaminus, G.; et al. ARID1A Regulates Transcription and the Epigenetic Landscape via POLE and DMAP1 While ARID1A Deficiency or Pharmacological Inhibition Sensitizes Germ Cell Tumor Cells to ATR Inhibition. Cancers 2020, 12, 905. [Google Scholar] [CrossRef]
- Jostes, S.; Nettersheim, D.; Fellermeyer, M.; Schneider, S.; Hafezi, F.; Honecker, F.; Schumacher, V.; Geyer, M.; Kristiansen, G.; Schorle, H. The Bromodomain Inhibitor JQ1 Triggers Growth Arrest and Apoptosis in Testicular Germ Cell Tumours in Vitro and in Vivo. J. Cell. Mol. Med. 2017, 21, 1300–1314. [Google Scholar] [CrossRef]
- Burmeister, A.; Stephan, A.; Alves Avelar, L.A.; Müller, M.R.; Seiwert, A.; Höfmann, S.; Fischer, F.; Torres-Gomez, H.; Hoffmann, M.J.; Niegisch, G.; et al. Establishment and Evaluation of Dual HDAC/BET Inhibitors as Therapeutic Options for Germ Cell Tumors and Other Urological Malignancies. Mol. Cancer 2022, 21, 1674–1688. [Google Scholar] [CrossRef]
- van Helvoort Lengert, A.; do Nascimento Braga Pereira, L.; Cabral, E.R.M.; Gomes, I.N.F.; de Jesus, L.M.; Gonçalves, M.F.S.; da Rocha, A.O.; Tassinari, T.A.; da Silva, L.S.; Laus, A.C.; et al. Potential New Therapeutic Approaches for Cisplatin-Resistant Testicular Germ Cell Tumors. FBL 2022, 27, 245. [Google Scholar] [CrossRef]
- Schepisi, G.; Gianni, C.; Cursano, M.C.; Gallà, V.; Menna, C.; Casadei, C.; Bleve, S.; Lolli, C.; Martinelli, G.; Rosti, G.; et al. Immune Checkpoint Inhibitors and Chimeric Antigen Receptor (CAR)-T Cell Therapy: Potential Treatment Options against Testicular Germ Cell Tumors. Front. Immunol. 2023, 14, 1118610. [Google Scholar] [CrossRef]
- Lobo, J.; Jerónimo, C.; Henrique, R. Targeting the Immune System and Epigenetic Landscape of Urological Tumors. IJMS 2020, 21, 829. [Google Scholar] [CrossRef]
- Semaan, A.; Haddad, F.G.; Eid, R.; Kourie, H.R.; Nemr, E. Immunotherapy:Last Bullet in Platinum Refractory Germ Cell Testicular Cancer. Future Oncol. 2019, 15, 533–541. [Google Scholar] [CrossRef]
- Jonska-Gmyrek, J.; Peczkowski, P.; Michalski, W.; Poniatowska, G.; Zolciak-Siwinska, A.; Kotowicz, B.; Wiechno, P.; Golawska, M.; Kowalska, M.; Demkow, T. Radiotherapy in Testicular Germ Cell Tumours—A Literature Review. Contemp. Oncol. 2017, 3, 203–208. [Google Scholar] [CrossRef]
- Oronsky, B.; Scicinski, J.; Kim, M.; Cabrales, P.; Salacz, M.; Carter, C.; Oronsky, N.; Lybeck, H.; Lybeck, M.; Larson, C.; et al. Turning on the Radio: Epigenetic Inhibitors as Potential Radiopriming Agents. Biomolecules 2016, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Avila, W.; Piulats, J.M.; Garcia Del Muro, X.; Vidal, A.; Condom, E.; Casanovas, O.; Mora, J.; Germà, J.R.; Capellà, G.; Villanueva, A.; et al. Sunitinib Inhibits Tumor Growth and Synergizes with Cisplatin in Orthotopic Models of Cisplatin-Sensitive and Cisplatin-Resistant Human Testicular Germ Cell Tumors. Clin. Cancer Res. 2009, 15, 3384–3395. [Google Scholar] [CrossRef]
- Juliachs, M.; Vidal, A.; del Muro, X.G.; Piulats, J.M.; Condom, E.; Casanovas, O.; Graupera, M.; Germà, J.R.; Villanueva, A.; Viñals, F. Effectivity of Pazopanib Treatment in Orthotopic Models of Human Testicular Germ Cell Tumors. BMC Cancer 2013, 13, 382. [Google Scholar] [CrossRef]
- Rossini, E.; Bosatta, V.; Abate, A.; Fragni, M.; Salvi, V.; Basnet, R.M.; Zizioli, D.; Bosisio, D.; Piovani, G.; Valcamonico, F.; et al. Cisplatin Cytotoxicity in Human Testicular Germ Cell Tumor Cell Lines Is Enhanced by the CDK4/6 Inhibitor Palbociclib. Clin. Genitourin. Cancer 2021, 19, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Schmidtova, S.; Kalavska, K.; Liskova, V.; Plava, J.; Miklikova, S.; Kucerova, L.; Matuskova, M.; Rojikova, L.; Cierna, Z.; Rogozea, A.; et al. Targeting of Deregulated Wnt/β-Catenin Signaling by PRI-724 and LGK974 Inhibitors in Germ Cell Tumor Cell Lines. Int. J. Mol. Sci. 2021, 22, 4263. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Li, Z.; Ye, K.; Zhou, Y.; Yan, M.; Qi, H.; Hu, H.; Dai, Y.; Tang, Y. Novel Sequential Therapy with Metformin Enhances the Effects of Cisplatin in Testicular Germ Cell Tumours via YAP1 Signalling. Cancer Cell Int. 2022, 22, 113. [Google Scholar] [CrossRef]
- Salatino, A.; Mirabelli, M.; Chiefari, E.; Greco, M.; Di Vito, A.; Bonapace, G.; Brunetti, F.S.; Crocerossa, F.; Epstein, A.L.; Foti, D.P.; et al. The Anticancer Effects of Metformin in the Male Germ Tumor SEM-1 Cell Line Are Mediated by HMGA1. Front. Endocrinol. 2022, 13, 1051988. [Google Scholar] [CrossRef] [PubMed]
- Beyrouthy, M.J.; Garner, K.M.; Hever, M.P.; Freemantle, S.J.; Eastman, A.; Dmitrovsky, E.; Spinella, M.J. High DNA Methyltransferase 3B Expression Mediates 5-Aza-Deoxycytidine Hypersensitivity in Testicular Germ Cell Tumors. Cancer Res. 2009, 69, 9360–9366. [Google Scholar] [CrossRef] [PubMed]
- Biswal, B.K.; Beyrouthy, M.J.; Hever-Jardine, M.P.; Armstrong, D.; Tomlinson, C.R.; Christensen, B.C.; Marsit, C.J.; Spinella, M.J. Acute Hypersensitivity of Pluripotent Testicular Cancer-Derived Embryonal Carcinoma to Low-Dose 5-Aza Deoxycytidine Is Associated with Global DNA Damage-Associated P53 Activation, Anti-Pluripotency and DNA Demethylation. PLoS ONE 2012, 7, e53003. [Google Scholar] [CrossRef]
- Nettersheim, D.; Jostes, S.; Fabry, M.; Honecker, F.; Schumacher, V.; Kirfel, J.; Kristiansen, G.; Schorle, H. A Signaling Cascade Including ARID1A, GADD45B and DUSP1 Induces Apoptosis and Affects the Cell Cycle of Germ Cell Cancers after Romidepsin Treatment. Oncotarget 2016, 7, 74931–74946. [Google Scholar] [CrossRef] [PubMed]
- Schmidtova, S.; Udvorkova, N.; Cierna, Z.; Horak, S.; Kalavska, K.; Chovanec, M.; Rojikova, L.; Vulevova, M.; Kucerova, L.; Mego, M. Effect of the PARP Inhibitor Veliparib on Germ Cell Tumor Cell Lines. Oncol. Lett. 2022, 24, 392. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, F.; Graziani, G.; Antinozzi, C.; Feldman, D.R.; Houldsworth, J.; Bosl, G.J.; Chaganti, R.S.K.; Moynahan, M.E.; Jasin, M.; Barchi, M. Reduced Proficiency in Homologous Recombination Underlies the High Sensitivity of Embryonal Carcinoma Testicular Germ Cell Tumors to Cisplatin and Poly (ADP-Ribose) Polymerase Inhibition. PLoS ONE 2012, 7, e51563. [Google Scholar] [CrossRef]


| Drug Class | Therapeutic Agent | Monotherapy | Therapeutic Target/Mechanism | Study Type | Produce Relevant Response to Cisplatin-Resistant Germ Cell Tumors | Cisplatin Sensitivity-Restoring/ Improvement | Main Results | Reference |
|---|---|---|---|---|---|---|---|---|
| Epidrugs | Guadecitabine | Yes | HMAS | Pre-clinical | Yes | Yes | Three responses in 14 patients, with two complete responses. Inhibited progression and regressed cisplatin-resistant testicular cancer cells | [151] |
| 5-azacytidine | Both, combined with cisplatin | HMAS/DNMT | Pre-clinical | Yes | No | Induced apoptosis at low nanomolar doses in both cisplatin-sensitive and resistant cells | [152] | |
| MLo1302 | Yes | HMAS/DNMT | Pre-clinical | Yes | No | Decreased cell viability by lowering the protein expression of pluripotency markers | [153] | |
| Decitabine | Yes | HMAS | Pre-clinical | Yes | Yes | Induced expression of tumor suppressor genes and p53 activation, encouraging a proapoptotic response and resensitizing GCT cells to cisplatin | [171,172] | |
| Trichostatin A/Romidepsin | Yes | HDACi | Pre-clinical | Yes | No | Antitumor activity in vitro and in vivo; induces apoptosis, reduces tumor size, and inhibits proliferation and angiogenesis | [41,173] | |
| Animacroxam | Yes | HDACi | Pre-clinical | Yes | No | Reduced tumor growth and angiogenesis | [154] | |
| Belinostat / Panobinostat | Yes | HDACis | Pre-clinical | Yes | No | Reduced acetylation, caused cell cycle arrest, decreased proliferation, lowered Ki67 index, and elevated p21, while enhancing apoptosis | [155] | |
| LAK-FFK11, LAK129; LAK-HGK7 | Yes | Dual inhibitor (HDACi/BETi) | Pre-clinical | Yes | No | Decreased cell viability, caused apoptosis, and changed the cell cycle in cisplatin-resistant TGCT | [158] | |
| JQ1 | Both, combined with romidepsin | BET inhibitor (BRD4) | Pre-clinical | Yes | No | Induced apoptosis, with a pronounced effect in resistant clones; reduced tumor size, proliferation rate, and angiogenesis | [157] | |
| C63 and BRD-K98645985 | Combined with romidepsin | ARID1A (chromatin remodeler) inhibitor | Pre-clinical | Yes | Yes | Enhanced the effectiveness of romidepsin and sensitized TGCT cells to ATR inhibition | [156] | |
| LP99, PRT4165, GSK343, Quisinostat, JIB-04, Chaetocin and MZ-1 | Yes | Epigenetic inhibitors | Pre-clinical | Yes | No | Cytotoxicity, ranging from nanomolar to micromolar. Most caused apoptosis or cell cycle arrest in GCT cell lines | [92] | |
| MG-132 | Yes | Proteasome inhibitor | Pre-clinical | Yes | Yes | Cytotoxic in the nanomolar range for TGCT cell lines; increased sensitivity to CDDP | [159] | |
| Inmunotherapy | BNT211 | CAR T-Cell therapy | Chimeric antigen receptor | Clinical study Phase I | Yes | No | Overall response rate of 57% in a TGCT patient cohort (N=13) | [160] |
| Other targeted therapies | Palbociclib | Combined with cisplatin | PARP inhibitor | Pre-clinical | Yes | No | Decreased cell viability; positive effect with regard to delaying cell recovery after the insult | [167] |
| Veliparib | Both, combined with cisplatin | PARP inhibitor | Pre-clinical | Yes | No | Synergistic effects when combined with cisplatin in vitro | [174] | |
| Olaparib | Yes | PARP inhibitor | Pre-clinical | Yes | Yes | DNA repair; sensitization to cisplatin and antitumor action | [175] | |
| Pazopanib | Combined with lapatinib | RTK inhibitor | Pre-clinical | Yes | No | Anti-angiogenesis properties | [41,166] | |
| Sunitinib | Yes | RTK inhibitor | Pre-clinical | Yes | No | In vivo antitumor action, including decreased vasculature and tumor growth inhibition | [41,165] | |
| Dissulfiram | Combined with cisplatin | ALDH inhibitor | Pre-clinical | Yes | Yes | An in vivo model with a synergistic antitumor effect with cisplatin | [65] | |
| PRI-724 | Yes | Wnt/β-catenin signaling Inhibitor | Pre-clinical | Yes | No | Pro-apoptotic effects | [168] | |
| Metformin | Combined with cisplatin | Biguanide (antihyperglycemic agent) | Pre-clinical | Yes | Yes | Inhibited cells in the G1 phase and decreased the levels of cyclin D1, CDK6, CDK4, and RB; induced apoptosis | [169,170] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuevas-Estrada, B.; Montalvo-Casimiro, M.; Munguia-Garza, P.; Ríos-Rodríguez, J.A.; González-Barrios, R.; Herrera, L.A. Breaking the Mold: Epigenetics and Genomics Approaches Addressing Novel Treatments and Chemoresponse in TGCT Patients. Int. J. Mol. Sci. 2023, 24, 7873. https://doi.org/10.3390/ijms24097873
Cuevas-Estrada B, Montalvo-Casimiro M, Munguia-Garza P, Ríos-Rodríguez JA, González-Barrios R, Herrera LA. Breaking the Mold: Epigenetics and Genomics Approaches Addressing Novel Treatments and Chemoresponse in TGCT Patients. International Journal of Molecular Sciences. 2023; 24(9):7873. https://doi.org/10.3390/ijms24097873
Chicago/Turabian StyleCuevas-Estrada, Berenice, Michel Montalvo-Casimiro, Paulina Munguia-Garza, Juan Alberto Ríos-Rodríguez, Rodrigo González-Barrios, and Luis A. Herrera. 2023. "Breaking the Mold: Epigenetics and Genomics Approaches Addressing Novel Treatments and Chemoresponse in TGCT Patients" International Journal of Molecular Sciences 24, no. 9: 7873. https://doi.org/10.3390/ijms24097873
APA StyleCuevas-Estrada, B., Montalvo-Casimiro, M., Munguia-Garza, P., Ríos-Rodríguez, J. A., González-Barrios, R., & Herrera, L. A. (2023). Breaking the Mold: Epigenetics and Genomics Approaches Addressing Novel Treatments and Chemoresponse in TGCT Patients. International Journal of Molecular Sciences, 24(9), 7873. https://doi.org/10.3390/ijms24097873