


Molecular Basic of Pharmacotherapy of Cytokine Imbalance as a Component of Intervertebral Disc Degeneration Treatment

 ,
,  , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. General Approaches to Intervertebral Disc Degeneration Therapy
3. Medication Methods for Correcting Cytokine Imbalance
3.1. Nonsteroidal Anti-Inflammatory Drugs
3.1.1. Salicylates
3.1.2. Pyrazolones
3.1.3. Derivatives of Organic Acids
3.1.4. Coxibs
3.1.5. Derivatives of Indazole
3.2. Hormones
3.2.1. Corticosteroids
3.2.2. Melatonin
3.2.3. Estrogens
3.3. Growth Factors
3.3.1. Bone Morphogenetic Proteins
3.3.2. Transforming Growth Factor Beta
3.3.3. Insulin-like Growth Factors
3.3.4. Osteogenic Protein 1
3.3.5. Human Recombinant Growth Factors/Differentiation
3.4. Chondroprotectors
3.4.1. Chondroitin Sulfate
3.4.2. Glucosamine Sulfate
3.5. Traps of Interleukin-1
3.6. Interleukins Regulating Pro-Inflammatory Cytokines
3.6.1. Interleukin 4 and Interleukin 10
3.6.2. Interleukin 12
3.7. Monoclonal Antibodies
3.7.1. Antibodies against Tumor Necrosis Factor Alpha
3.7.2. Antibodies against Interleukin 1 Alpha and 1 Beta
3.7.3. Trap Receptors for Pro-Inflammatory Cytokines
3.7.4. Antibodies against Interleukin 6
3.7.5. Janus Kinase Inhibitors
3.7.6. Antibodies against Interleukin 17
3.8. Cytostatics
3.9. Phosphodiesterase Inhibitors
3.10. Anticonvulsants and Antidepressants
3.11. Drugs of Other Groups
3.11.1. Suppressive Cytokine Anti-Inflammatory Agents
3.11.2. Renin Inhibitors
3.11.3. Acetylcholinesterase Inhibitors
3.11.4. Xanthines
3.11.5. Nitrates
3.11.6. Direct Oral Anticoagulants
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Byvaltsev, V.A.; Belykh, E.G.; Stepanov, I.A.; Giers, M.; Preul, M.C. Cytokine mechanisms of degenetation of intervertebral disc. Sib. Med. J. (Irkutsk) 2015, 6. Available online: https://cyberleninka.ru/article/n/tsitokinovye-mehanizmy-degeneratsii-mezhpozvonkovogo-diska (accessed on 10 April 2023).
- Li, Y.; Samartzis, D.; Campbell, D.D.; Cherny, S.S.; Cheung, K.M.; Luk, K.D.; Karppinen, J.; Song, Y.; Cheah, K.S.; Chan, D.; et al. Two subtypes of intervertebral disc degeneration distinguished by large-scale population-based study. Spine J. 2016, 16, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Rodrigues, S.; Sharp, C.; Wade, K.; Broom, N.; McCall, I.W.; Roberts, S. Staying connected: Structural integra-tion at the intervertebral disc-vertebra interface of human lumbar spines. Eur. Spine J. 2017, 26, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Grignon, B.; Grignon, Y.; Mainard, D.; Braun, M.; Netter, P.; Roland, J. The structure of the cartilaginous end-plates in elder people. Surg. Radiol. Anat. 2000, 22, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Kos, N.; Gradisnik, L.; Velnar, T. A brief review of the degenerative intervertebral disc disease. Med. Arch. 2019, 73, 421–424. [Google Scholar] [CrossRef]
- Shnayder, N.A.; Ashhotov, A.V.; Trefilova, V.V.; Nurgaliev, Z.A.; Novitsky, M.A.; Vaiman, E.E.; Petrova, M.M.; Nasyrova, R.F. Cytokine imbalance as a biomarker of intervertebral disk degeneration. Int. J. Mol. Sci. 2023, 24, 2360. [Google Scholar] [CrossRef]
- Kim, H.; Ham, C.H.; Kwon, W.K. Current knowledge and future therapeutic trospects in symptomatic intervertebral disc degeneration. Yonsei Med. J. 2022, 63, 199–210. [Google Scholar] [CrossRef]
- Kloppenburg, M.; Berenbaum, F. Osteoarthritis year in review 2019: Epidemiology and therapy. Osteoarthr. Cartil. 2020, 28, 242–248. [Google Scholar] [CrossRef]
- Okada, E.; Daimon, K.; Fujiwara, H.; Nishiwaki, Y.; Nojiri, K.; Watanabe, M.; Katoh, H.; Ishihama, H.; Fujita, N.; Tsuji, T.; et al. Ten-year longitudinal follow-up MRI study of age-related changes in thoracic intervertebral discs in asymptomatic subjects. Spine 2019, 44, E1317–E1324. [Google Scholar] [CrossRef]
- Molinos, M.; Almeida, C.R.; Caldeira, J.; Cunha, C.; Gonçalves, R.M.; Barbosa, M.A. Inflammation in intervertebral disc degeneration and regeneration. J. R. Soc. Interface 2015, 12, 20141191. [Google Scholar] [CrossRef]
- Weber, K.T.; Jacobsen, T.D.; Maidhof, R.; Virojanapa, J.; Overby, C.; Bloom, O.; Quraishi, S.; Levine, M.; Chahine, N.O. Developments in intervertebral disc disease research: Pathophysiology, mechanobiology, and therapeutics. Curr. Rev. Musculoskelet. Med. 2015, 8, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Purmessur, D.; Walter, B.; Roughley, P.; Laudier, D.; Hecht, A.; Iatridis, J. A role for TNFα in intervertebral disc degeneration: A non-recoverable catabolic shift. Biochem. Biophys. Res. Commun. 2013, 433, 151–156. [Google Scholar] [CrossRef]
- Hall, M.N.; Campos, H.; Li, H.; Sesso, H.D.; Stampfer, M.J.; Willett, W.C.; Ma, J. Blood levels of long-chain polyunsaturated fatty acids, aspirin, and the risk of colorectal cancer. Cancer Epidemiol. Biomark. Prev. 2007, 16, 314–321. [Google Scholar] [CrossRef]
- Drugbank. Drugs. Available online: https://go.drugbank.com/drugs/ (accessed on 25 February 2023).
- Yang, Z.; Kahn, B.B.; Shi, H.; Xue, B. Macrophage α1 AMP-activated protein kinase (α1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J. Biol. Chem. 2010, 285, 19051–19059. [Google Scholar] [CrossRef]
- Trefilova, V.V.; Shnayder, N.A.; Popova, T.E.; Balberova, O.V.; Nasyrova, R.F. The role of NO system in low back pain chronicity. Pers. Psychiatry Neurol. 2021, 1, 37–45. [Google Scholar] [CrossRef]
- Barreto-Torres, G.; Hernandez, J.S.; Jang, S.; Rodríguez-Muñoz, A.R.; Torres-Ramos, C.A.; Basnakian, A.G.; Javadov, S. The beneficial effects of AMP kinase activation against oxidative stress are associated with prevention of PPARα-cyclophilin D interaction in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H749–H758. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, J.; Wu, X.; Guo, X.; Sun, H.; Yu, B.; Shen, J.; Bai, J.; Chen, Z.; Yang, H.; et al. Aspirin-mediated attenuation of intervertebral disc degeneration by ameliorating reactive oxygen species in vivo and in vitro. Oxidative Med. Cell. Longev. 2019, 2019, 7189854. [Google Scholar] [CrossRef]
- Pelletier, J.P.; Jovanovic, D.V.; Lascau-Coman, V.; Fernandes, J.C.; Manning, P.T.; Connor, J.R.; Currie, M.G.; Martel-Pelletier, J. Selective inhibition of inducible nitric oxide synthase reduces progression of experimental osteoarthritis in vivo: Possible link with the reduction in chondrocyte apoptosis and caspase 3 level. Arthritis Rheum. 2000, 43, 1290–1299. [Google Scholar] [CrossRef]
- Tsoyi, K.; Jang, H.J.; Nizamutdinova, I.T.; Kim, Y.M.; Lee, Y.S.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Chang, K.C. Metformin inhibits HMGB1 release in LPS-treated RAW 264.7 cells and increases survival rate of endotoxaemic mice. Br. J. Pharmacol. 2011, 162, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xia, D.; Pan, Z.; Xu, D.; Zhou, Y.; Wu, Y.; Cai, N.; Tang, Q.; Wang, C.; Yan, M.; et al. Metformin protects against apoptosis and senescence in nucleus pulposus cells and ameliorates disc degeneration in vivo. Cell Death Dis. 2016, 7, e2441. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, D.Y.; Mariappan, M.M.; Feliers, D.; Ghosh-Choudhury, G.; Abboud, H.E.; Gorin, Y.; Kasinath, B.S. Hydrogen sulfide inhibits high glucose-induced NADPH oxidase 4 expression and matrix increase by recruiting inducible nitric oxide synthase in kidney proximal tubular epithelial cells. J. Biol. Chem. 2017, 292, 5665–5675. [Google Scholar] [CrossRef]
- Rousseaux, C.; Lefebvre, B.; Dubuquoy, L.; Lefebvre, P.; Romano, O.; Auwerx, J.; Metzger, D.; Wahli, W.; Desvergne, B.; Naccari, G.C.; et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J. Exp. Med. 2005, 201, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Linard, C.; Gremy, O.; Benderitter, M. Reduction of peroxisome proliferation-activated receptor gamma expression by gamma-irradiation as a mechanism contributing to inflammatory response in rat colon: Modulation by the 5-aminosalicylic acid agonist. J. Pharmacol. Exp. Ther. 2008, 324, 911–920. [Google Scholar] [CrossRef]
- Product Monograph: SALOFALK (Mesalamine) Delayed Release Tablets, for Oral Use. Available online: https://pdf.hres.ca/dpd_pm/00028807.PDF (accessed on 22 December 2022).
- Allgayer, H. Review article: Mechanisms of action of mesalazine in preventing colorectal carcinoma in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2003, 18 (Suppl. S2), 10–14. [Google Scholar] [CrossRef]
- Pruzanski, W.; Stefanski, E.; Vadas, P.; Ramamurthy, N.S. Inhibition of extracellular release of proinflammatory secretory phospholipase A2 (sPLA2) by sulfasalazine: A novel mechanism of anti-inflammatory activity. Biochem. Pharmacol. 1997, 53, 1901–1907. [Google Scholar] [CrossRef] [PubMed]
- Cevallos, S.A.; Lee, J.Y.; Velazquez, E.M.; Foegeding, N.J.; Shelton, C.D.; Tiffany, C.R.; Parry, B.H.; Stull-Lane, A.R.; Olsan, E.E.; Savage, H.P.; et al. 5-aminosalicylic acid ameliorates colitis and checks dysbiotic escherichia coli expansion by activating PPAR-gamma signaling in the intestinal epithelium. Mbio 2021, 12, e03227-20. [Google Scholar] [CrossRef]
- Watanabe, T.; Tahara, M.; Todo, S. The novel antioxidant edaravone: From bench to bedside. Cardiovasc. Ther. 2008, 26, 101–114. [Google Scholar] [CrossRef]
- Lucas, S. The pharmacology of indomethacin. Headache 2016, 56, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Brunelli, C.; Amici, C.; Angelini, M.; Fracassi, C.; Belardo, G.; Santoro, M.G. The non-steroidal anti-inflammatory drug indomethacin activates the eIF2α kinase PKR, causing a translational block in human colorectal cancer cells. Biochem. J. 2012, 443, 379–386. [Google Scholar] [CrossRef]
- Amici, C.; La Frazia, S.; Brunelli, C.; Balsamo, M.; Angelini, M.; Santoro, M.G. Inhibition of viral protein translation by indomethacin in vesicular stomatitis virus infection: Role of eIF2α kinase PKR. Cell Microbiol. 2015, 17, 1391–1404. [Google Scholar] [CrossRef] [PubMed]
- Rainsford, K.D. Discovery, mechanisms of action and safety of ibuprofen. Int. J. Clin. Pract. Suppl. 2003, 135, 3–8. [Google Scholar]
- Rao, P.; Knaus, E.E. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): Cyclooxygenase (COX) inhibition and beyond. J. Pharm. Pharm. Sci. 2008, 11, 81s–110s. [Google Scholar] [CrossRef] [PubMed]
- Palayoor, S.T.; J-Aryankalayil, M.; Makinde, A.Y.; Cerna, D.; Falduto, M.T.; Magnuson, S.R.; Coleman, C.N. Gene expression profile of coronary artery cells treated with nonsteroidal anti-inflammatory drugs reveals off-target effects. J. Cardiovasc. Pharmacol. 2012, 59, 487–499. [Google Scholar] [CrossRef]
- Bizzarri, C.; Pagliei, S.; Brandolini, L.; Mascagni, P.; Caselli, G.; Transidico, P.; Sozzani, S.; Bertini, R. Selective inhibition of interleukin-8-induced neutrophil chemotaxis by ketoprofen isomers. Biochem. Pharmacol. 2001, 61, 1429–1437. [Google Scholar] [CrossRef]
- Wang, L.M.; Toyoshima, A.; Mineshita, S.; Wang, X.X.; Yamamoto, T.; Nomura, Y.; Yang, L.; Koikei, Y.; Shiba, K.; Honda, Y. The anti-inflammatory effects of ketoprofen in animal experiments. Drugs Exp. Clin. Res. 1997, 23, 1–6. [Google Scholar] [PubMed]
- FDA. Approved Drug Products. Available online: https://www.fda.gov/drugs (accessed on 21 February 2023).
- Gong, L.; Thorn, C.F.; Bertagnolli, M.M.; Grosser, T.; Altman, R.B.; Klein, T.E. Celecoxib pathways: Pharmacokinetics and pharmacodynamics. Pharmacogenet. Genom. 2012, 22, 310–318. [Google Scholar] [CrossRef]
- Hawkey, C.J. COX-1 and COX-2 inhibitors. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 801–820. [Google Scholar] [CrossRef]
- Li, J.; Zhu, J.; Melvin, W.S.; Bekaii-Saab, T.S.; Chen, C.S.; Muscarella, P. A structurally optimized celecoxib derivative inhibits human pancreatic cancer cell growth. J. Gastrointest. Surg. 2006, 10, 207–214. [Google Scholar] [CrossRef]
- Tseng, P.H.; Wang, Y.C.; Weng, S.C.; Weng, J.R.; Chen, C.S.; Brueggemeier, R.W.; Shapiro, C.L.; Chen, C.Y.; Dunn, S.E.; Pollak, M.; et al. Overcoming trastuzumab resistance in HER2-overexpressing breast cancer cells by using a novel celecoxib-derived phosphoinositide-dependent kinase-1 inhibitor. Mol. Pharmacol. 2006, 70, 1534–1541. [Google Scholar] [CrossRef]
- Quane, P.A.; Graham, G.G.; Ziegler, J.B. Pharmacology of benzydamine. Inflammopharmacology 1998, 6, 95–107. [Google Scholar] [CrossRef]
- Sironi, M.; Massimiliano, L.; Transidico, P.; Pinza, M.; Sozzani, S.; Mantovani, A.; Vecchi, A. Differential effect of benzydamine on pro- versus anti-inflammatory cytokine production: Lack of inhibition of interleukin-10 and interleukin-1 receptor antagonist. Int. J. Clin. Lab. Res. 2000, 30, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Milanese, C.; Vecchi, A.; Polenzani, L.; Guglielmotti, A.; Coletta, I.; Landolfi, C.; Soldo, L.; Mantovani, A.; Pinza, M. Benzydamine inhibits the release of tumor necrosis factor-α and monocyte chemotactic protein-1 byCandida albicans-stimulated human peripheral blood cells. Int. J. Clin. Lab. Res. 1997, 27, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Riboldi, E.; Frascaroli, G.; Transidico, P.; Luini, W.; Bernasconi, S.; Mancini, F.; Guglielmotti, A.; Milanese, C.; Pinza, M.; Sozzani, S.; et al. Benzydamine inhibits monocyte migration and MAPK activation induced by chemotactic agonists. Br. J. Pharmacol. 2003, 140, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Wikipedia. Available online: https://ru.wikipedia.org/wiki/ (accessed on 24 February 2023).
- Berthelot, J.M.; Le Goff, B.; Maugars, Y. Side effects of corticosteroid injections: What’s new? Jt. Bone Spine 2013, 80, 363–367. [Google Scholar] [CrossRef]
- Buttermann, G.R. The effect of spinal steroid injections for degenerative disc disease. Spine J. 2004, 4, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Khot, A.; Bowditch, M.; Powell, J.; Sharp, D. The use of intradiscal steroid therapy for lumbar spinal discogenic pain: A randomized controlled trial. Spine J. 2004, 29, 833–837. [Google Scholar] [CrossRef]
- Teixeira, G.Q.; Leite Pereira, C.; Castro, F.; Ferreira, J.R.; Gomez-Lazaro, M.; Aguiar, P.; Barbosa, M.A.; Neidlinger-Wilke, C.; Goncalves, R.M. Anti-inflammatory chitosan/poly-γ-glutamic acid nanoparticles control inflammation while remodeling extracellular matrix in degenerated intervertebral disc. Acta Biomater. 2016, 42, 168–179. [Google Scholar] [CrossRef]
- Yasir, M.; Goyal, A.; Sonthalia, S. Corticosteroid Adverse Effects; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Cope, A.P.; Gibbons, D.L.; Aderka, D.; Foxwell, B.M.; Wallach, D.; Maini, R.N.; Feldmann, M.; Brennan, F.M. Differential regulation of tumour necrosis factor receptors (TNF-R) by IL-4; upregulation of P55 and P75 TNF-R on synovial joint mononuclear cells. Cytokine 1993, 5, 205–212. [Google Scholar] [CrossRef]
- Auphan, N.; Didonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: Inhibition of NF-κ B activity through induction of IκB synthesis. Science 1995, 270, 286–290. [Google Scholar] [CrossRef]
- Joosten, L.A.; Lubberts, E.; Helsen, M.M.; Saxne, T.; Coenen-de Roo, C.J.; Heinegård, D.; van den Berg, W.B. Protection against cartilage and bone destruction by systemic interleukin-4 treatment in established murine type II collagen-induced arthritis. Arthritis Res. 1999, 1, 81–91. [Google Scholar] [CrossRef]
- Pickup, M.E. Clinical pharmacokinetics of prednisone and prednisolone. Clin. Pharmacokinet. 1979, 4, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, F.; Scheffold, A. Rapid glucocorticoid effects on immune cells. Steroids 2002, 67, 529–534. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef]
- De Bosscher, K.; Haegeman, G. Minireview: Latest perspectives on antiinflammatory actions of glucocorticoids. Mol. Endocrinol. 2009, 23, 281–291. [Google Scholar] [CrossRef]
- Ratman, D.; Vanden Berghe, W.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.M.; De Bosscher, K. How glucocorticoid receptors modulate the activity of other transcription factors: A scope beyond tethering. Mol. Cell Endocrinol. 2013, 380, 41–54. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Wing, K.; Yamaguchi, T. Dynamics of peripheral tolerance and immune regulation mediated by Treg. Eur. J. Immunol. 2009, 39, 2331–2336. [Google Scholar] [CrossRef]
- Ugor, E.; Prenek, L.; Pap, R.; Berta, G.; Ernszt, D.; Najbauer, J.; Németh, P.; Boldizsár, F.; Berki, T. Glucocorticoid hormone treatment enhances the cytokine production of regulatory T cells by upregulation of Foxp3 expression. Immunobiology 2018, 223, 422–431. [Google Scholar] [CrossRef]
- Almawi, W.Y.; Melemedjian, O.K. Molecular mechanisms of glucocorticoid antiproliferative effects: Antagonism of transcription factor activity by glucocorticoid receptor. J. Leukoc. Biol. 2002, 71, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Corticosteroid effects on cell signalling. Eur. Respir. J. 2006, 27, 413–426. [Google Scholar] [CrossRef]
- Rebollo, A.; Pitton, C.; García, A.; Gómez, J.; Silva, A. A role for the intermediate affinity IL-2R in the protection against glucocorticoid-induced apoptosis. Immunology 1995, 84, 388–395. [Google Scholar] [PubMed]
- Nesbitt, A.; Fossati, G.; Bergin, M.; Stephens, P.; Stephens, S.; Foulkes, R.; Brown, D.; Robinson, M.; Bourne, T. Mechanism of action of certolizumab pegol (CDP870): In vitro comparison with other anti-tumor necrosis factor α agents. Inflamm. Bowel Dis. 2007, 13, 1323–1332. [Google Scholar] [CrossRef]
- Cheng, Z.; Xiang, Q.; Wang, J.; Zhang, Y. The potential role of melatonin in retarding intervertebral disc ageing and degeneration: A systematic review. Ageing Res. Rev. 2021, 70, 101394. [Google Scholar] [CrossRef]
- Zhang, Y.; He, F.; Chen, Z.; Su, Q.; Yan, M.; Zhang, Q.; Tan, J.; Qian, L.; Han, Y. Melatonin modulates IL-1β-induced extracellular matrix remodeling in human nucleus pulposus cells and attenuates rat intervertebral disc degeneration and inflammation. Aging 2019, 11, 10499–10512. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Jiang, G.; Liu, H.; Li, Z.; Pei, Y.; Wang, H.; Pan, H.; Cui, H.; Long, J.; Wang, J.; et al. Melatonin alleviates intervertebral disc degeneration by disrupting the IL-1β/NF-κB-NLRP3 inflammasome positive feedback loop. Bone Res. 2020, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Chao-Yang, G.; Peng, C.; Hai-Hong, Z. Roles of NLRP3 inflammasome in intervertebral disc degeneration. Osteoarthr. Cartil. 2021, 29, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Romacho, T.; Valencia, I.; Ramos-González, M.; Vallejo, S.; López-Esteban, M.; Lorenzo, O.; Cannata, P.; Romero, A.; San Hipólito-Luengo, A.; Gómez-Cerezo, J.F.; et al. Visfatin/eNampt induces endothelial dysfunction in vivo: A role for Toll-Like Receptor 4 and NLRP3 inflammasome. Sci. Rep. 2020, 10, 5386. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Peng, Y.; Sun, J.; Li, S.; Hong, J.; Zhou, J.; Chen, J.; Yan, J.; Huang, Z.; Wang, X.; et al. Nicotinamide phosphoribosyl transferase controls NLRP3 inflammasome activity through MAPK and NF-κB S\signaling in nucleus pulposus cells, as suppressed by melatonin. Inflammation 2020, 43, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Matta, A.; Karim, M.Z.; Gerami, H.; Jun, P.; Funabashi, M.; Kawchuk, G.; Goldstein, A.; Foltz, W.; Sussman, M.; Eek, B.C.; et al. NTG-101: A novel molecular therapy that halts the progression of degenerative disc disease. Sci. Rep. 2018, 8, 16809. [Google Scholar] [CrossRef]
- Turgut, M.; Oktem, G.; Uslu, S.; Yurtseven, M.E.; Aktuğ, H.; Uysal, A. The effect of exogenous melatonin administration on trabecular width, ligament thickness and TGF-β1 expression in degenerated intervertebral disk tissue in the rat. J. Clin. Neurosci. 2006, 13, 357–363. [Google Scholar] [CrossRef]
- Pettersson, K.; Gustafsson, J.A. Role of estrogen receptor beta in estrogen action. Annu. Rev. Physiol. 2001, 63, 165–192. [Google Scholar] [CrossRef]
- Cutolo, M.; Straub, R.H.; Bijlsma, J.W. Neuroendocrine-immune interactions in synovitis. Nat. Clin. Pract. Rheumatol. 2007, 3, 627–634. [Google Scholar] [CrossRef]
- Oestergaard, S.; Sondergaard, B.C.; Hoegh-Andersen, P.; Henriksen, K.; Qvist, P.; Christiansen, C.; Tankó, L.B.; Karsdal, M.A. Effects of ovariectomy and estrogen therapy on type II collagen degradation and structural integrity of articular cartilage in rats: Implications of the time of initiation. Arthritis Rheum. 2006, 54, 2441–2451. [Google Scholar] [CrossRef]
- Claassen, H.; Schunke, M.; Kurz, B. Estradiol protects cultured articular chondrocytes from oxygen-radical-induced damage. Cell Tissue Res. 2005, 319, 439–445. [Google Scholar] [CrossRef]
- Wang, Y.X.J. Postmenopausal chinese women show accelerated lumbar disc degeneration compared with chinese men. J. Orthop. Translat. 2015, 3, 205–211. [Google Scholar] [CrossRef]
- Baron, Y.M.; Brincat, M.P.; Galea, R.; Calleja, N. Intervertebral disc height in treated and untreated overweight post-menopausal women. Hum. Reprod. 2005, 20, 3566–3570. [Google Scholar] [CrossRef]
- Cauley, J.A. Estrogen and bone health in men and women. Steroids 2015, 99 Pt A, 11–15. [Google Scholar] [CrossRef]
- Liu, H.; Yang, S.D.; Xu, Y.; Ning, S.H.; Wang, T.; Yang, D.L.; Ding, W.Y. Protective role of 17β-estradiol on tumor necrosis factor-α-induced apoptosis in human nucleus pulposus cells. Mol. Med. Rep. 2017, 16, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhu, D.; Zhu, S.; Feng, F.; Gong, C.; Chen, C.; Chen, L. 17β-Estradiol/extrogen receptor β alleviates apoptosis and enhances matrix biosynthesis of nucleus pulposus cells through regulating oxidative damage under a high glucose condition. Biomed. Pharmacother. 2018, 107, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Ao, P.; Huang, W.; Li, J.; Wu, T.; Xu, L.; Deng, Z.; Chen, W.; Yin, C.; Cheng, X. 17β-estradiol protects nucleus pulposus cells from serum deprivation-induced apoptosis and regulates expression of MMP-3 and MMP-13 through promotion of autophagy. Biochem. Biophys. Res. Commun. 2018, 503, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.Y.; Song, X.X.; Li, X.F. The role of estrogen in intervertebral disc degeneration. Steroids 2020, 154, 108549. [Google Scholar] [CrossRef]
- Tinti, L.; Niccolini, S.; Lamboglia, A.; Pascarelli, N.A.; Cervone, R.; Fioravanti, A. Raloxifene protects cultured human chondrocytes from IL-1β induced damage: A biochemical and morphological study. Eur. J. Pharmacol. 2011, 670, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.L.; Liu, T.K. Estradiol-induced knee osteoarthrosis in ovariectomized rabbits. Clin. Orthop. Relat. Res. 1993, 291, 295–302. [Google Scholar] [CrossRef]
- Rosner, I.A.; Goldberg, V.M.; Getzy, L.; Moskowitz, R.W. Effects of estrogen on cartilage and experimentally induced osteoarthritis. Arthritis Rheum. 1979, 22, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Richette, P.; Dumontier, M.F.; François, M.; Tsagris, L.; Korwin-Zmijowska, C.; Rannou, F.; Corvol, M.T. Dual effects of 17β-oestradiol on interleukin 1β-induced proteoglycan degradation in chondrocytes. Ann. Rheum. Dis. 2004, 63, 191–199. [Google Scholar] [CrossRef]
- Riancho, J.A.; García-Ibarbia, C.; Gravani, A.; Raine, E.V.; Rodríguez-Fontenla, C.; Soto-Hermida, A.; Rego-Perez, I.; Dodd, A.W.; Gómez-Reino, J.J.; Zarrabeitia, M.T.; et al. Common variations in estrogen-related genes are associated with severe large-joint osteoarthritis: A multicenter genetic and functional study. Osteoarthr. Cartil. 2010, 18, 927–933. [Google Scholar] [CrossRef]
- Cirillo, D.J.; Wallace, R.B.; Wu, L.; Yood, R.A. Effect of hormone therapy on risk of hip and knee joint replacement in the Women’s Health Initiative. Arthritis Rheum. 2006, 54, 3194–3204. [Google Scholar] [CrossRef]
- Kou, X.X.; Wu, Y.W.; Ding, Y.; Hao, T.; Bi, R.Y.; Gan, Y.H.; Ma, X. 17β-estradiol aggravates temporomandibular joint inflammation through the NF-κB pathway in ovariectomized rats. Arthritis Rheum. 2011, 63, 1888–1897. [Google Scholar] [CrossRef]
- Martín-Millán, M.; Castaneda, S. Estrogens, osteoarthritis and inflammation. Jt. Bone Spine 2013, 80, 368–373. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, F.; Ma, J.; Ding, W. Intervertebral disc ageing and degeneration: The antiapoptotic effect of oestrogen. Ageing Res. Rev. 2020, 57, 100978. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Xu, Y.; Gan, Y.; Wang, L.; Ouyang, B.; Zhang, C.; Luo, L.; Zhao, C.; Zhou, Q. Estrogen enhances matrix synthesis in nucleus pulposus cell through the estrogen receptor β-p38 MAPK pathway. Cell. Physiol. Biochem. 2016, 39, 2216–2226. [Google Scholar] [CrossRef]
- Wang, T.; Yang, S.D.; Liu, S.; Wang, H.; Liu, H.; Ding, W.Y. 17β-Estradiol inhibites tumor necrosis factor-α induced apoptosis of human nucleus pulposus cells via the PI3K/Akt pathway. Med. Sci. Monit. 2016, 22, 4312–4322. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gan, Y.; Xu, Y.; Wang, L.; Ouyang, B.; Zhang, C.; Luo, L.; Zhao, C.; Zhou, Q. 17beta-estradiol attenuates TNF-α-induced premature senescence of nucleus pulposus cells through regulating the ROS/NF-κB pathway. Int. J. Biol. Sci. 2017, 13, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.D.; Yang, D.L.; Sun, Y.P.; Wang, B.L.; Ma, L.; Feng, S.Q.; Ding, W.Y. 17β-estradiol protects against apoptosis induced by interleukin-1β in rat nucleus pulposus cells by down-regulating MMP-3 and MMP-13. Apoptosis 2015, 20, 348–357. [Google Scholar] [CrossRef]
- Wang, H.; Ding, W.; Yang, D.; Gu, T.; Yang, S.; Bai, Z. Different concentrations of 17β-estradiol modulates apoptosis induced by interleukin-1β in rat annulus fibrosus cells. Mol. Med. Rep. 2014, 10, 2745–2751. [Google Scholar] [CrossRef]
- Zhao, C.M.; Chen, Q.; Zhang, W.J.; Huang, A.B.; Zhang, W.; Yang, H.L.; Zhang, Z.M. 17β-estradiol protects rat annulus fibrosus cells against apoptosis via α1 integrin-mediated adhesion to type I collagen: An in-vitro study. Med. Sci. Monit. 2016, 22, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, Z.; Xie, Z.; Chen, Y.; Zheng, Z.; Wei, X.; Huang, B.; Shan, Z.; Liu, J.; Fan, S.; et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic. Biol. Med. 2020, 160, 552–565. [Google Scholar] [CrossRef]
- Katagiri, T.; Watabe, T. Bone Morphogenetic Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021899. [Google Scholar] [CrossRef]
- Wordinger, R.J.; Sharma, T.; Clark, A.F. The role of TGF-β2 and bone morphogenetic proteins in the trabecular meshwork and glaucoma. J. Ocul. Pharmacol. Ther. 2014, 30, 154–162. [Google Scholar] [CrossRef]
- Xie, S.; Zhao, C.; Chen, W.; Li, G.; Xiong, Z.; Tang, X.; Zhang, F.; Xiao, H. Recombinant human bone morphogenetic protein 2 and 7 inhibit the degeneration of intervertebral discs by blocking the Puma-dependent apoptotic signaling. Int. J. Biol. Sci. 2021, 17, 2367–2379. [Google Scholar] [CrossRef]
- Elkholi, R.; Floros, K.V.; Chipuk, J.E. The role of BH3-only proteins in tumor cell development, signaling, and treatment. Genes Cancer. 2011, 2, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Yao, X.; Dai, Z.; Wang, Y.; Lv, G. Bone morphogenetic protein 2 alleviated intervertebral disc degeneration through mediating the degradation of ECM and apoptosis of nucleus pulposus cells via the PI3K/Akt pathway. Int. J. Mol. Med. 2019, 43, 583–592. [Google Scholar] [CrossRef]
- Matta, A.; Erwin, W.M. Injectable biologics for the treatment of degenerative disc disease. Curr. Rev. Musculoskelet. Med. 2020, 13, 680–687. [Google Scholar] [CrossRef]
- Fernandez-Moure, J.; Moore, C.A.; Kim, K.; Karim, A.; Smith, K.; Barbosa, Z.; Van Eps, J.; Rameshwar, P.; Weiner, B. Novel therapeutic strategies for degenerative disc disease: Review of cell biology and intervertebral disc cell therapy. SAGE Open Med. 2018, 6, 2050312118761674. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad signaling pathways of the TGF-β family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef]
- Roskoski, R. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Research. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.T.; Hsu, P.H.; Hsu, W.C.; Chen, N.J.; Tseng, P.H. Polyubiquitination of transforming growth factor β-activated Kinase 1 (TAK1) at lysine 562 residue regulates TLR4-mediated JNK and p38 MAPK activation. Sci. Rep. 2015, 5, 12300. [Google Scholar] [CrossRef]
- Morishima, N.; Mizoguchi, I.; Takeda, K.; Mizuguchi, J.; Yoshimoto, T. TGF-β is necessary for induction of IL-23R and Th17 differentiation by IL-6 and IL-23. Biochem. Biophys. Res. Commun. 2009, 386, 105–110. [Google Scholar] [CrossRef]
- Arsura, M.; Wu, M.; Sonenshein, G.E. TGFβ1 Inhibits NF-κB/Rel Activity Inducing Apoptosis of B Cells: Transcriptional Activation of IκBα. Immunity 1996, 5, 31–40. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef]
- Smythies, L.E.; Sellers, M.; Clements, R.H.; Mosteller-Barnum, M.; Meng, G.; Benjamin, W.H.; Orenstein, J.M.; Smith, P.D. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Investig. 2005, 115, 66–75. [Google Scholar] [CrossRef]
- Wahl, S.M. Transforming growth factor-β: Innately bipolar. Curr. Opin. Immunol. 2007, 19, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.H. Th9 cells: Differentiation and disease. Immunol. Rev. 2013, 252, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. regulatory T cells and human disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef]
- Guo, J.; Zhou, X. Regulatory T cells turn pathogenic. Cell Mol. Immunol. 2015, 12, 525–532. [Google Scholar] [CrossRef]
- Sawant, D.V.; Vignali, D.A. Once a Treg, Always a Treg? Immunol. Rev. 2014, 259, 173–191. [Google Scholar] [CrossRef]
- Kraj, P. Bone morphogenetic proteins shape treg cells. Front. Immunol. 2022, 13, 865546. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, P.; Liu, J.; Zhu, S.; Kroemer, G.; Klionsky, D.J.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 2019, 5, eaaw2238. [Google Scholar] [CrossRef]
- Xie, J.; Li, B.; Yao, B.; Zhang, P.; Wang, L.; Lu, H. Transforming growth factor-β1-regulated Fas/FasL pathway activation suppresses nucleus pulposus cell apoptosis in an inflammatory environment. Biosci. Rep. 2020, 40, BSR20191726. [Google Scholar] [CrossRef]
- Zhang, X.B.; Hu, Y.C.; Cheng, P.; Zhou, H.Y.; Chen, X.Y.; Wu, D.; Zhang, R.H.; Yu, D.C.; Gao, X.D.; Shi, J.T.; et al. Targeted therapy for intervertebral disc degeneration: Inhibiting apoptosis is a promising treatment strategy. Int. J. Med. Sci. 2021, 18, 2799–2813. [Google Scholar] [CrossRef] [PubMed]
- Gruber, H.E.; Hoelscher, G.L.; Leslie, K.; Ingram, J.A.; Hanley, E.N., Jr. Three-dimensional culture of human disc cells within agarose or a collagen sponge: Assessment of proteoglycan production. Biomaterials 2006, 27, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Younis, S.; Schönke, M.; Massart, J.; Hjortebjerg, R.; Sundström, E.; Gustafson, U.; Björnholm, M.; Krook, A.; Frystyk, J.; Zierath, J.R.; et al. The ZBED6-IGF2 axis has a major effect on growth of skeletal muscle and internal organs in placental mammals. Proc. Natl. Acad. Sci. USA 2018, 115, E2048–E2057. [Google Scholar] [CrossRef]
- Gunnell, D.; Miller, L.L.; Rogers, I.; Holly, J.M. Association of insulin-like growth factor I and insulin-like growth factor-binding protein-3 with intelligence quotient among 8- to 9-year-old children in the avon longitudinal study of parents and children. Pediatrics 2005, 116, e681–e686. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.; Dawes, P.J. Childhood hearing is associated with growth rates in infancy and adolescence. Pediatr. Res. 2007, 62, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Pellegrino, G.; Vollery, M.; Hofland, L.J. Role of IGF-1 system in the modulation of longevity: Controversies and new insights from a centenarians’ perspective. Front. Endocrinol. 2019, 10, 27. [Google Scholar] [CrossRef]
- Junnila, R.K.; List, E.O.; Berryman, D.E.; Murrey, J.W.; Kopchick, J.J. The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol. 2013, 9, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; An, H.S. Prevention of disc degeneration with growth factors. Eur. Spine J. 2006, 15 (Suppl. S3), S422–S432. [Google Scholar] [CrossRef] [PubMed]
- Osada, R.; Ohshima, H.; Ishihara, H.; Yudoh, K.; Sakai, K.; Matsui, H.; Tsuji, H. Autocrine/paracrine mechanism of insulin-like growth factor-1 secretion, and the effect of insulin-like growth factor-1 on proteoglycan synthesis in bovine intervertebral discs. J. Orthop. Res. 1996, 14, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Pratsinis, H.; Kletsas, D. PDGF, bFGF and IGF-I stimulate the proliferation of intervertebral disc cells in vitro via the activation of the ERK and Akt signaling pathways. Eur. Spine J. 2007, 16, 1858–1866. [Google Scholar] [CrossRef]
- Kennon, J.C.; Awad, M.E.; Chutkan, N.; DeVine, J.; Fulzele, S. Current insights on use of growth factors as therapy for intervertebral disc degeneration. Biomol. Concepts 2018, 9, 43–52. [Google Scholar] [CrossRef]
- Gruber, H.E.; Hoelscher, G.L.; Ingram, J.A.; Bethea, S.; Hanley, E.N. IGF-1 rescues human intervertebral annulus cells from in vitro stress-induced premature senescence. Growth Factors 2008, 26, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Engström, B.E.; Karlsson, F.A.; Wide, L. Gender differences in diurnal growth hormone and epinephrine values in young adults during ambulation. Clin. Chem. 1999, 45 Pt 1, 1235–1239. [Google Scholar] [CrossRef]
- Ursavas, A.; Karadag, M.; Ilcol, Y.O.; Ercan, I.; Burgazlioglu, B.; Coskun, F.; Gozu, R.O. Low level of IGF-1 in obesity may be related to obstructive sleep apnea syndrome. Lung 2007, 185, 309–314. [Google Scholar] [CrossRef]
- Patil, P.; Dong, Q.; Wang, D.; Chang, J.; Wiley, C.; Demaria, M.; Lee, J.; Kang, J.; Niedernhofer, L.J.; Robbins, P.D.; et al. Systemic clearance of p16INK4a -positive senescent cells mitigates age-associated intervertebral disc degeneration. Aging Cell 2019, 18, e12927. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Bergholz, J.; Zhang, H.; He, H.; Wang, Y.; Zhang, Y.; Li, Q.; Kirkland, J.L.; Xiao, Z.X. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 2014, 13, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Todd, M.D.; Seely, B.L. Prolonged treatment of human osteoarthritic chondrocytes with insulin-like growth factor-I stimulates proteoglycan synthesis but not proteoglycan matrix accumulation in alginate cultures. J. Rheumatol. 2003, 30, 1565–1570. [Google Scholar] [PubMed]
- Osborn, K.D.; Trippel, S.B.; Mankin, H.J. Growth factor stimulation of adult articular cartilage. J. Orthop. Res. 1989, 7, 35–42. [Google Scholar] [CrossRef]
- Kritschil, R.; Zhang, Z.; Lei, C.; Zhong, J.; Dong, Q.; Lee, J.; Conover, C.A.; Sowa, G.; Vallejo, A.N.; Vo, N. Effects of suppressing bioavailability of insulin-like growth factor on age-associated intervertebral disc degeneration. JOR Spine 2020, 3, e1112. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cisewski, S.; Sachs, B.L.; Yao, H. Effect of cartilage endplate on cell based disc regeneration: A finite element analysis. Mol. Cell. Biomech. 2013, 10, 159–182. [Google Scholar]
- Chen, R.S.; Zhang, X.B.; Zhu, X.T.; Wang, C.S. The crosstalk between IGF-1R and ER-α in the proliferation and anti-Inflammation of nucleus pulposus cells. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 5886–5894. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Shen, P.; Li, H.; Yang, Y.; Guo, J.; Chen, S.; Ma, Y.; Sheng, J.; Shen, S.; Liu, G.; et al. Carbonic anhydrase 12 protects endplate cartilage from degeneration regulated by IGF-1/PI3K/CREB signaling pathway. Front. Cell Dev. Biol. 2020, 8, 595969. [Google Scholar] [CrossRef]
- Zhang, M.; Zhou, Q.; Liang, Q.Q.; Li, C.G.; Holz, J.D.; Tang, D.; Sheu, T.J.; Li, T.F.; Shi, Q.; Wang, Y.J. IGF-1 regulation of type II collagen and MMP-13 expression in rat endplate chondrocytes via distinct signaling pathways. Osteoarthr. Cartil. 2009, 17, 100–106. [Google Scholar] [CrossRef]
- Li, B.; Zheng, X.F.; Ni, B.B.; Yang, Y.H.; Jiang, S.D.; Lu, H.; Jiang, L.S. Reduced expression of insulin-like growth factor 1 receptor leads to accelerated intervertebral disc degeneration in mice. Int. J. Immunopathol. Pharmacol. 2013, 26, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, X.; Wang, Y.; Cao, F.; Chen, Z.; Hu, Z.; Yu, B.; Feng, H.; Ba, Z.; Liu, T.; et al. Intervertebral disc degeneration in mice with type II diabetes induced by leptin receptor deficiency. BMC Musculoskelet. Disord. 2020, 21, 77. [Google Scholar] [CrossRef]
- Pattison, S.; Melrose, J.; Ghosh, P.; Taylor, T.K. Regulation of gelatinase-A (MMP-2) production by ovine intervertebral disc nucleus pulposus cells grown in alginate bead culture by transforming growth factor-Β1and insulin like growth factor-I. Cell Biol. Int. 2001, 25, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhou, X.; Liang, C.; Li, H.; Han, B.; Li, F.; Chen, Q. TGF-β3 and IGF-1 synergy ameliorates nucleus pulposus mesenchymal stem cell differentiation towards the nucleus pulposus cell type through MAPK/ERK signaling. Growth Factors 2015, 33, 326–336. [Google Scholar] [CrossRef]
- Zhang, C.C.; Zhou, J.S.; Hu, J.G.; Wang, X.; Zhou, X.S.; Sun, B.A.; Shao, C.; Lin, Q. Effects of IGF-1 on IL-1β-induced apoptosis in rabbit nucleus pulposus cells in vitro. Mol. Med. Rep. 2013, 7, 441–444. [Google Scholar] [CrossRef]
- Zhang, C.C.; Cui, G.P.; Hu, J.G.; Xiao, Y.Z.; Zhou, X.S.; Shao, C.; Lin, Q.; Zhou, J.S. Effects of adenoviral vector expressing hIGF-1 on apoptosis in nucleus pulposus cells in vitro. Int. J. Mol. Med. 2014, 33, 401–405. [Google Scholar] [CrossRef]
- Le Maitre, C.L.; Richardson, S.M.A.; Baird, P.; Freemont, A.J.; Hoyland, J.A. Expression of receptors for putative anabolic growth factors in human intervertebral disc: Implications for repair and regeneration of the disc. J. Pathol. 2005, 207, 445–452. [Google Scholar] [CrossRef]
- Zhu, Z.; Huang, P.; Chong, Y.; George, S.K.; Wen, B.; Han, N.; Liu, Z.; Kang, L.; Lin, N. Nucleus pulposus cells derived IGF-1 and MCP-1 enhance osteoclastogenesis and vertebrae disruption in lumbar disc herniation. Int. J. Clin. Exp. Pathol. 2014, 7, 8520–8531. [Google Scholar] [PubMed]
- Mavrogonatou, E.; Kletsas, D. Effect of varying osmotic conditions on the response of bovine nucleus pulposus cells to growth factors and the activation of the ERK and Akt pathways. J. Orthop. Res. 2010, 28, 1276–1282. [Google Scholar] [CrossRef]
- Guo, Y.; Tian, L.; Liu, X.; He, Y.; Chang, S.; Shen, Y. ERRFI1 inhibits proliferation and inflammation of nucleus pulposus and is negatively regulated by miR-2355-5p in intervertebral disc degeneration. Spine 2019, 44, E873–E881. [Google Scholar] [CrossRef] [PubMed]
- Travascio, F.; Elmasry, S.; Asfour, S. Modeling the role of IGF-1 on extracellular matrix biosynthesis and cellularity in intervertebral disc. J. Biomech. 2014, 47, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Travascio, F.; Gu, W.Y. Quantitative analysis of exogenous IGF-1 administration of intervertebral disc through intradiscal injection. J. Biomech. 2012, 45, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhou, X.; Chen, G. Expression and mechanism of interleukin 1 (IL-1), interleukin 2 (IL-2), interleukin 8 (IL-8), BMP, fibroblast growth factor 1 (FGF1), and insulin-like growth factor (IGF-1) in lumbar disc herniation. Med. Sci. Monit. 2019, 25, 984–990. [Google Scholar] [CrossRef]
- Lin, H.; Tian, S.; Peng, Y.; Wu, L.; Xiao, Y.; Qing, X.; Shao, Z. IGF signaling in intervertebral disc health and disease. Front. Cell Dev. Biol. 2022, 9, 817099. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, S.; Hu, Y.; Wang, N.; Wu, L.; Ding, M. Role of PI3K/AKT signaling pathway in proliferation; migration and odontogenic differentiation of human dental pulp stem cells. J. Hard Tissue Biol. 2020, 29, 99–104. [Google Scholar] [CrossRef]
- Oussaief, L.; Ramírez, V.; Hippocrate, A.; Arbach, H.; Cochet, C.; Proust, A.; Raphaël, M.; Khelifa, R.; Joab, I. NF-kappaB-mediated modulation of inducible nitric oxide synthase activity controls induction of the Epstein-Barr virus productive cycle by transforming growth factor beta 1. J. Virol. 2011, 85, 6502–6512. [Google Scholar] [CrossRef]
- Kondo, T.; Otsuka, Y.; Aoki, H.; Goto, Y.; Kawaguchi, Y.; Waguri-Nagaya, Y.; Miyazawa, K.; Goto, S.; Aoyama, M. The inducible nitric oxide synthase pathway promotes osteoclastogenesis under hypoxic culture conditions. Am. J. Pathol. 2021, 191, 2072–2079. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, T.; Kyumoto-Nakamura, Y.; Kukita, A.; Uehara, N.; Zhang, J.; Koda, K.; Kamiya, M.; Badawy, T.; Tomoda, E.; Xu, X.; et al. IL-1β induces pathologically activated osteoclasts bearing extremely high levels of resorbing activity: A possible pathological subpopulation of osteoclasts, accompanied by suppressed expression of kindlin-3 and talin-1. J. Immunol. 2018, 200, 218–228. [Google Scholar] [CrossRef]
- Kondo, T.; Aoki, H.; Otsuka, Y.; Kawaguchi, Y.; Waguri-Nagaya, Y.; Aoyama, M. Insulin-like growth factor 2 promotes osteoclastogenesis increasing inflammatory cytokine levels under hypoxia. J. Pharmacol. Sci. 2022, 149, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Sobajima, S.; Shimer, A.L.; Chadderdon, R.C.; Kompel, J.F.; Kim, J.S.; Gilbertson, L.G.; Kang, J.D. Quantitative analysis of gene expression in a rabbit model of intervertebral disc degeneration by real-time polymerase chain reaction. Spine J. 2005, 5, 14–23. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, X.; Liu, Z.; Xiao, X.; Hu, W.; Sun, Z. Osteogenic protein-1 attenuates nucleus pulposus cell apoptosis through activating the PI3K/Akt/mTOR pathway in a hyperosmotic culture. Biosci. Rep. 2018, 38, BSR20181708. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, R.; Gan, Y.; Wang, L.; Zhao, C.; Luo, L.; Zhang, C.; Zhou, Q. Effects of osteogenic protein-1 on intervertebral disc regeneration: A systematic review of animal studies. Biomed. Pharmacother. 2017, 88, 260–266. [Google Scholar] [CrossRef]
- An, H.S.; Takegami, K.; Kamada, H.; Nguyen, C.M.; Thonar, E.J.; Singh, K.; Andersson, G.B.; Masuda, K. Intradiscal administration of osteogenic protein-1 increases intervertebral disc height and proteoglycan content in the nucleus pulposus in normal adolescent rabbits. Spine 2005, 30, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.; Brisby, H.; Chung, S.A.; Diwan, A.D. Bone morphogenetic protein-7 protects human intervertebral disc cells in vitro from apoptosis. Spine J. 2008, 8, 466–474. [Google Scholar] [CrossRef]
- Xie, J.; Li, B.; Zhang, P.; Wang, L.; Lu, H.; Song, X. Osteogenic protein-1 attenuates the inflammatory cytokine-induced NP cell senescence through regulating the ROS/NF-κB pathway. Biomed. Pharmacother. 2018, 99, 431–437. [Google Scholar] [CrossRef]
- Cheng, H.; Jiang, W.; Phillips, F.M.; Haydon, R.C.; Peng, Y.; Zhou, L.; Luu, H.H.; An, N.; Breyer, B.; Vanichakarn, P.; et al. Osteogenic activity of the fourteen types of human bone morphogenetic proteins (BMPs). J. Bone Joint Surg. Am. 2003, 85, 1544–1552. [Google Scholar] [CrossRef]
- Miyazono, K.; Kamiya, Y.; Morikawa, M. Bone morphogenetic protein receptors and signal transduction. J. Biochem. 2010, 147, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Mazerbourg, S.; Sangkuhl, K.; Luo, C.-W.; Sudo, S.; Klein, C.; Hsueh, A.J.W. Identification of receptors and signaling pathways for orphan bone morphogenetic protein/growth differentiation factor ligands based on genomic analyses. J. Biol. Chem. 2005, 280, 32122–32132. [Google Scholar] [CrossRef] [PubMed]
- Le Maitre, C.L.; Freemont, A.J.; Hoyland, J.A. Localization of degradative enzymes and their inhibitors in the degenerate human intervertebral disc. J. Pathol. 2004, 204, 47–54. [Google Scholar] [CrossRef]
- Lo, L.; Dormand, E.L.; Anderson, D.J. Late-emigrating neural crest cells in the roof plate are restricted to a sensory fate by GDF7. Proc. Natl. Acad. Sci. USA 2005, 102, 7192–7197. [Google Scholar] [CrossRef]
- Maher, C.; Underwood, M.; Buchbinder, R. Non-specific low back pain. Lancet 2017, 389, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Song, Y.; Luo, R.; Li, S.; Li, G.; Wang, K.; Liao, Z.; Wang, B.; Ke, W.; Xiang, Q.; et al. Ferroportin-dependent iron homeostasis protects against oxidative stress-induced nucleus pulposus cell ferroptosis and ameliorates intervertebral disc degeneration in vivo. Oxidative Med. Cell. Longev. 2021, 2021, 6670497. [Google Scholar] [CrossRef] [PubMed]
- Krut, Z.; Pelled, G.; Gazit, D.; Gazit, Z. Stem cells and exosomes: New therapies for intervertebral disc degeneration. Cells 2021, 10, 2241. [Google Scholar] [CrossRef]
- Williams, F.M.; Popham, M.; Hart, D.J.; de Schepper, E.; Bierma-Zeinstra, S.; Hofman, A.; Uitterlinden, A.G.; Arden, N.K.; Cooper, C.; Spector, T.D.; et al. GDF5 single-nucleotide polymorphism rs143383 is associated with lumbar disc degeneration in northern european women. Arthritis Rheum. 2011, 63, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Leo, B.M.; Beck, G.; Balian, G.; Anderson, G.D. Collagen and proteoglycan abnormalities in the GDF-5-deficient mice and molecular changes when treating disk cells with recombinant growth factor. Spine 2004, 29, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Wan, Y.; Anderson, D.G.; Shen, F.H.; Leo, B.M.; Laurencin, C.T.; Balian, G.; Li, X. Mouse growth and differentiation factor-5 protein and DNA therapy potentiates intervertebral disc cell aggregation and chondrogenic gene expression. Spine J. 2008, 8, 287–295. [Google Scholar] [CrossRef]
- Luo, X.W.; Liu, K.; Chen, Z.; Zhao, M.; Han, X.W.; Bai, Y.G.; Feng, G. Adenovirus-mediated GDF-5 promotes the extracellular matrix expression in degenerative nucleus pulposus cells. J. Zhejiang Univ. Sci. B 2016, 17, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Yang, S.; Sun, H.; Guo, D.; Wu, B.; Ji, F.; Zhou, D. Effects of releasing recombinant human growth and differentiation factor-5 from poly(lactic-co-glycolic acid) microspheres for repair of the rat degenerated intervertebral disc. J. Biomater. Appl. 2014, 29, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.J.; Bradford, D.S.; Lotz, J.C. In vivo growth factor treatment of degenerated intervertebral discs. Spine 2004, 29, 156–163. [Google Scholar] [CrossRef]
- Liang, H.; Ma, S.Y.; Feng, G.; Shen, F.H.; Joshua Li, X. Therapeutic effects of adenovirus-mediated growth and differentiation factor-5 in a mice disc degeneration model induced by annulus needle puncture. Spine J. 2010, 10, 32–41. [Google Scholar] [CrossRef]
- Chujo, T.; An, H.S.; Akeda, K.; Miyamoto, K.; Muehleman, C.; Attawia, M.; Andersson, G.; Masuda, K. Effects of growth differentiation factor-5 on the intervertebral disc--in vitro bovine study and in vivo rabbit disc degeneration model study. Spine 2006, 31, 2909–2917. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, H.B.; Svala, E.; Skioldebrand, E.; Lindahl, A.; Brisby, H. Support of concept that migrating progenitor cells from stem cell niches contribute to normal regeneration of the adult mammal intervertebral disc: A descriptive study in the New Zealand white rabbit. Spine 2012, 37, 722–732. [Google Scholar] [CrossRef]
- Gulati, T.; Chung, S.A.; Wei, A.Q.; Diwan, A.D. Localization of bone morphogenetic protein 13 in human intervertebral disc and its molecular and functional effects in vitro in 3D culture. J. Orthop. Res. 2015, 33, 1769–1775. [Google Scholar] [CrossRef]
- Wei, A.; Williams, L.A.; Bhargav, D.; Shen, B.; Kishen, T.; Duffy, N.; Diwan, A.D. BMP13 prevents the effects of annular injury in an ovine model. Int. J. Biol. Sci. 2009, 5, 388–396. [Google Scholar] [CrossRef]
- Enochson, L.; Stenberg, J.; Brittberg, M.; Lindahl, A. GDF5 reduces MMP13 expression in human chondrocytes via DKK1 mediated canonical Wnt signaling inhibition. Osteoarthr. Cartil. 2014, 22, 566–577. [Google Scholar] [CrossRef]
- Shen, L.; Wu, Y.; Han, L.; Zhang, H. Overexpression of growth and differentiation factor-5 inhibits inflammatory factors released by intervertebral disc cells. Exp. Ther. Med. 2018, 15, 3603–3608. [Google Scholar] [CrossRef] [PubMed]
- Gruber, H.E.; Hoelscher, G.L.; Ingram, J.A.; Bethea, S.; Hanley, E.N., Jr. Growth and differentiation factor-5 (GDF-5) in the human intervertebral annulus cells and its modulation by IL-1ß and TNF-α in vitro. Exp. Mol. Pathol. 2014, 96, 225–229. [Google Scholar] [CrossRef]
- Hisamatsu, D.; Ohno-Oishi, M.; Nakamura, S.; Mabuchi, Y.; Naka-Kaneda, H. Growth differentiation factor 6 derived from mesenchymal stem/stromal cells reduces age-related functional deterioration in multiple tissues. Aging 2016, 8, 1259–1275. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, T.; Shen, B.; Diwan, A.; Hoyland, J.A.; Richardson, S.M. Therapeutic potential of growth differentiation factors in the treatment of degenerative disc diseases. JOR Spine 2019, 2, e1045. [Google Scholar] [CrossRef]
- Guo, S.; Cui, L.; Xiao, C.; Wang, C.; Zhu, B.; Liu, X.; Li, Y.; Liu, X.; Wang, D.; Li, S. The mechanisms and functions of GDF-5 in intervertebral disc degeneration. Orthop. Surg. 2021, 13, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, J.; Röderer, G.; Günther, K.P.; Brenner, R.E. BMP-2; BMP-4; and PDGF-bb stimulate chemotactic migration of primary human mesenchymal progenitor cells. J. Cell. Biochem. 2002, 87, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Henrotin, Y.; Malaise, M.; Wittoek, R.; de Vlam, K.; Brasseur, J.P.; Luyten, F.P.; Jiangang, Q.; Van den Berghe, M.; Uhoda, R.; Bentin, J.; et al. Bio-optimized curcuma longa extract is efficient on knee osteoarthritis pain: A double-blind multicenter randomized placebo controlled three-arm study. Arthritis Res. Ther. 2019, 21, 179. [Google Scholar] [CrossRef]
- Sukhikh, S.; Babich, O.; Prosekov, A.; Patyukov, N.; Ivanova, S. Future of chondroprotectors in the treatment of degenerative processes of connective tissue. Pharmaceuticals 2020, 13, 220. [Google Scholar] [CrossRef] [PubMed]
- Shavlovskaya, O.A.; Zolotovskaya, I.A.; Prokofyeva, Y.S. Antiresorptive activity of pharmacological chondroitin sulfate in the older age group. Ter. Arkhiv 2020, 92, 75–79. [Google Scholar] [CrossRef]
- Shavlovskaya, O.A.; Zolotovskaya, I.A.; Prokofyeva, Y.S. Anti-inflammatory and anti-aging effects of chondroitin sulfate. Neurol. Neuropsychiatry Psychosom. 2020, 12, 111–116. (In Russian) [Google Scholar] [CrossRef]
- Shavlovskaya, O.A.; Zolotovskaya, I.A.; Prokofyeva, Y.A. A new look at back pain treatment in light of the latest ESCEO guidelines. Neurol. Neuropsychiatry Psychosom. 2020, 12, 90–95. (In Russian) [Google Scholar] [CrossRef]
- Gromova, O.A.; Torshin, I.Y.; Lila, A.M.; Shostak, N.A.; Rudakov, K.V. Molecular mechanisms of myoprotective action of chondroitin sulfate and glucosamine sulfate in sarcopenia. Neurol. Neuropsychiatry Psychosom. 2019, 11, 117–124. (In Russian) [Google Scholar] [CrossRef]
- Shavlovskaya, O.A.; Razumov, A.N.; Bokova, I.A.; Shavlovskiy, N.I.; Yukhnovskaya, Y.D. Rol khondroitina sulfata v kompleksnoi reabilitatsii lits pozhilogo vozrasta s khronicheskim bolevym sindromom [Chondroitin sulfate role in the complex rehabilitation of elderly people with chronic pain syndrome]. Vopr Kurortol Fizioter Lech Fiz Kult. 2021, 98, 71–78. (In Russian) [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, L.I.; Taskina, E.A.; Kashevarova, N.G. Osteoarthritis: Epidemiology, classification, risk factors, and progression, clinical presentation, diagnosis, and treatment. Mod. Rheumatol. J. 2019, 13, 9–21. [Google Scholar] [CrossRef]
- Herrero-Beaumont, G.; Marcos, M.E.; Sánchez-Pernaute, O.; Granados, R.; Ortega, L.; Montell, E.; Vergés, J.; Egido, J.; Largo, R. Effect of chondroitin sulphate in a rabbit model of atherosclerosis aggravated by chronic arthritis. Br. J. Pharmacol. 2008, 154, 843–851. [Google Scholar] [CrossRef]
- Zolotovskaya, I.A.; Davydkin, I.L. A antiresorptive-cytokine effects of chondroprotective therapy in patients with lower back pain. S.S. Korsakov J. Neurol. Psychiatry 2020, 120, 65–71. [Google Scholar] [CrossRef]
- Honvo, G.; Bruyère, O.; Geerinck, A.; Veronese, N.; Reginster, J.Y. Efficacy of chondroitin sulfate in patients with knee osteoarthritis: A comprehensive meta-analysis exploring inconsistencies in randomized, placebo-controlled trials. Adv. Ther. 2019, 36, 1085–1099. [Google Scholar] [CrossRef]
- Melgar-Lesmes, P.; Sánchez-Herrero, A.; Lozano-Juan, F.; de la Torre Hernández, J.; Montell, E.; Jiménez, W.; Edelman, E.; Balcells, M. Chondroitin sulphate attenuates atherosclerosis in ApoE knockout mice involving cellular regulation of the inflammatory response. Thromb. Haemost. 2018, 118, 1329–1339. [Google Scholar] [CrossRef]
- Gromova, O.A.; Torshin, I.I.; Lila, A.M.; Naumov, A.V.; Rudakov, K.V. On the safety of glucosamine sulfate in patients with insulin resistance. Cons. Medicum 2019, 21, 75–83. [Google Scholar] [CrossRef]
- Chen, J.T.; Liang, J.B.; Chou, C.L.; Chien, M.W.; Shyu, R.C.; Chou, P.I.; Lu, D.W. Glucosamine sulfate inhibits TNF-α and IFN-γ-induced production of ICAM-1 in human retinal pigment epithelial cells in vitro. Investig. Ophthalmol. Vis. Sci. 2006, 47, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.T.; Chen, P.L.; Chang, Y.H.; Chien, M.W.; Chen, Y.H.; Lu, D.W. Glucosamine sulfate inhibits leukocyte adhesion in response to cytokine stimulation of retinal pigment epithelial cells in vitro. Exp. Eye Res. 2006, 83, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Jerosch, J. Effects of glucosamine and chondroitin sulfate on cartilage metabolism in OA: Outlook on other nutrient partners especially omega-3 fatty acids. Int. J. Rheumatol. 2011, 2011, 969012. [Google Scholar] [CrossRef]
- Naumov, A.V.; Khovasova, N.O.; Moroz, V.I.; Tkacheva, O.N. Mesto khondroitina sulfata i gliukozamina sulfata v terapii boli pri osteoartrite [The place of chondroitin sulfate and glucosamine sulfate in osteoarthritis pain therapy: A practical view from evidence-based medicine]. Zh Nevrol. Psikhiatr. Im S. S. Korsakova 2019, 119, 112–117. (In Russian) [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Farran, A.; Montell, E.; Vergés, J.; Pelletier, J.P. Discrepancies in composition and biological effects of different formulations of chondroitin sulfate. Molecules 2015, 20, 4277–4289. [Google Scholar] [CrossRef] [PubMed]
- Gromova, O.A.; Torshin, I.Y.; Lila, A.M.; Naumov, A.V.; Reier, I.A.; Karateev, A.E. Differential chemoreactome analysis of glucosamine sulfate and non-steroidal anti-inflammatory drugs: Promising synergistic drug combinations. Mod. Rheumatol. J. 2018, 12, 36–43. (In Russian) [Google Scholar] [CrossRef]
- Colotta, F.; Dower, S.K.; Sims, J.E.; Mantovani, A. The type II ‘decoy’ receptor: A novel regulatory pathway for interleukin 1. Immunol. Today 1994, 15, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Orlando, S.; Fadlon, E.J.; Sozzani, S.; Matteucci, C.; Mantovani, A. Chemoattractants induce rapid release of the interleukin 1 type II decoy receptor in human polymorphonuclear cells. J. Exp. Med. 1995, 181, 2181–2186. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Re, F.; Muzio, M.; Bertini, R.; Polentarutti, N.; Sironi, M.; Giri, J.G.; Dower, S.K.; Sims, J.E.; Mantovani, A. Interleukin-1 type II receptor: A decoy target for IL-1 that is regulated by IL-4. Science 1993, 261, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Bertani, A.; Polentarutti, N.; Sica, A.; Rambaldi, A.; Mantovani, A.; Colotta, F. Expression of c-jun protooncogene in human myelomonocytic cells. Blood 1989, 74, 1811–1816. [Google Scholar] [CrossRef]
- Re, F.; Muzio, M.; De Rossi, M.; Polentarutti, N.; Giri, J.G.; Mantovani, A.; Colotta, F. The type II “receptor” as a decoy target for interleukin 1 in polymorphonuclear leukocytes: Characterization of induction by dexamethasone and ligand binding properties of the released decoy receptor. J. Exp. Med. 1994, 179, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Porteu, F.; Nathan, C. Shedding of tumor necrosis factor receptors by activated human neutrophils. J. Exp. Med. 1990, 172, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Chomarat, P.; Banchereau, J. An update on interleukin-4 and its receptor. Eur. Cytokine Net. 1997, 8, 333–344. [Google Scholar]
- DeKruyff, R.H.; Fang, Y.; Wolf, S.F.; Umetsu, D.T. IL-12 inhibits IL-4 synthesis in keyhole limpet hemocyanin-primed CD4+ T-cells through an effect on antigen-presenting cells. J. Immunol. 1995, 154, 2578–2585. [Google Scholar] [CrossRef]
- Vannier, E.; Miller, L.C.; Dinarello, C.A. Coordinated anti-inflammatory effects of interleukin-4: IL-4 suppresses IL-1 production but upregulates gene expression and synthesis of interleukin-1 receptor antagonist. Proc. Natl. Acad. Sci. USA 1992, 89, 4076–4080. [Google Scholar] [CrossRef]
- Cawston, T.E.; Ellis, A.J.; Bigg, H.; Curry, V.; Lean, E.; Ward, D. Interleukin-4 blocks the release of collagen fragments from bovine nasal cartilage treated with cytokines. Biochim. Biophys. Acta 1996, 1314, 226–232. [Google Scholar] [CrossRef]
- Moore, K.W.; O’Garra, A.; De Waal-Malefyt, R.; Vieira, P.; Mosmann, T.R. Interleukin-10. Annu. Rev. Immunol. 1993, 11, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, D.F.; Zlotnik, A.; Mosmann, T.R.; Howard, M.; O’Garra, A. IL-10 inhibits the cytokine production by activated macrophages. J. Immunol. 1991, 147, 3815–3822. [Google Scholar] [CrossRef]
- Joosten, L.A.; Helsen, M.M.; Saxne, T.; Heinegård, D.; van de Putte, L.B.; van den Berg, W.B. Synergistic protection against cartilage destruction by low dose prednisolone and interleukin-10 in established murine collagen arthritis. Inflamm. Res. 1999, 48, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.; Lubberts, E.; Durez, P.; Helsen, M.M.; Jacobs, M.J.; Goldman, M.; van den Berg, W.B. Role of interleukin-4 and interleukin-10 in murine collagen-induced arthritis. Protective effect of interleukin-4 and interleukin-10 treatment on cartilage destruction. Arthritis Rheum. 1997, 40, 249–260. [Google Scholar] [CrossRef]
- Mauri, C.; Williams, R.O.; Walmsley, M.; Feldmann, M. Relationship between Th1/Th2 cytokine pattern and the arthritogenic response in collagen-induced arthritis. Eur. J. Immunol. 1996, 26, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Rocken, M.; Racke, M.; Shevach, E.M. IL-4-induced immune deviation as antigen-specific therapy for inflammatory autoimmune disease. Immunol. Today 1996, 17, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.; Helsen, M.M.; Saxne, T.; van De Loo, F.A.; Heinegard, D.; van Den Berg, W.B. IL-1 alpha beta blockade prevents cartilage and bone destruction in murine type II collagen-induced arthritis, whereas TNF-alpha blockade only ameliorates joint inflammation. J. Immunol. 1999, 163, 5049–5055. [Google Scholar] [CrossRef] [PubMed]
- Horsfall, A.C.; Butler, D.M.; Marinova, L.; Warden, P.J.; Williams, R.O.; Maini, R.N.; Feldmann, M. Suppression of collagen-induced arthritis by continuous administration of IL-4. J. Immunol. 1997, 159, 5687–5696. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.B.; Lubberts, E.; Helsen, M.M.A.; van den Berg, W.B. Dual role of IL-12 in early and late stages of murine collagen type II arthritis. J. Immunol. 1997, 159, 4094–4102. [Google Scholar] [CrossRef]
- Allen, J.B.; Wong, H.L.; Costa, G.L.; Bienkowski, M.J.; Wahl, S.M. Suppression of monocyte function and differential regulation of IL-1 and IL-1Ra by IL-4 contribute to resolution of experimental arthritis. J. Immunol. 1993, 151, 4344–4351. [Google Scholar] [CrossRef]
- Wong, H.L.; Costa, M.T.; Lotze, M.T.; Wahl, S.M. Interleukin (IL) 4 differently regulates monocyte IL-1 family gene expression and synthesis in vitro and in vivo. J. Exp. Med. 1993, 177, 775–781. [Google Scholar] [CrossRef]
- Miossec, P.; Chomarat, P.; Dechanet, J.; Moreau, J.F.; Roux, J.P.; Delmas, P.; Banchereau, J. Interleukin-4 inhibits bone resorption through an effect on osteoclasts and proinflammatory cytokines in an ex vivo model of bone resorption in rheumatoid arthritis. Arthritis Rheum. 1994, 37, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Kunicka, J.E.; Talle, M.A.; Denhardt, G.H.; Brown, M.; Prince, L.A.; Goldstein, G. Immunosuppression by glucocorticoids: Inhibition of production of multiple lymphokines by in vivo administration of dexamethasone. Cell. Immunol. 1993, 149, 39–49. [Google Scholar] [CrossRef]
- Joyce, D.A.; Steer, J.H.; Kloda, A. Dexamethasone antagonizes IL-4 and IL-10-induced release of IL-1RA by monocytes but augments IL-4, IL-10 and TGF β induced suppression of TNFα release. J. Interferon Cytokine Res. 1996, 16, 511–517. [Google Scholar] [CrossRef]
- Van de Loo, F.A.J.; Arntz, O.J.; Enckevort, F.H.J.; van Lent, P.L.E.M.; van den Berg, W.B. Reduced cartilage proteoglycan loss during zymosan-induced gonarthritis in NOS-2 deficient mice and in anti-interleukin-1 treated wild type mice with unabated joint inflammation. Arthritis Rheum. 1998, 41, 634–646. [Google Scholar] [CrossRef]
- Shnayder, N.A.; Khasanova, A.K.; Strelnik, A.I.; Al-Zamil, M.; Otmakhov, A.P.; Neznanov, N.G.; Shipulin, G.A.; Petrova, M.M.; Garganeeva, N.P.; Nasyrova, R.F. Cytokine Imbalance as a Biomarker of Therapeutic Resistance to Antipsychotics. Int. J. Mol. Sci. 2022, 23, 11324. [Google Scholar] [CrossRef] [PubMed]
- Meyaard, L.; Hovenkamp, E.; Otto, S.A.; Miedema, F. IL-12-induced IL-10 production by human T cells as a negative feedback for IL12-induced immune responses. J. Immunol. 1996, 156, 2777–2782. [Google Scholar] [CrossRef]
- Joosten, L.A.; Helsen, M.M.; van de Loo, F.A.; van den Berg, W.B. Anticytokine treatment of established type II collagen-induced arthritis in DBA/1 mice: A comparative study using anti-TNFα, anti-IL-1α/β and IL-1Ra. Arthritis Rheum. 2008, 58 (Suppl. S2), S110–S122. [Google Scholar] [CrossRef] [PubMed]
- Lügering, A.; Schmidt, M.; Lügering, N.; Pauels, H.G.; Domschke, W.; Kucharzik, T. Infliximab induces apoptosis in monocytes from patients with chronic active Crohn’s disease by using a caspase-dependent pathway. Gastroenterology 2001, 121, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Ten Hove, T.; van Montfrans, C.; Peppelenbosch, M.P.; van Deventer, S.J. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn’s disease. Gut 2002, 50, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Sans, M.; Scaldaferri, F.; Sgambato, A.; Rutella, S.; Cittadini, A.; Piqué, J.M.; Panes, J.; Katz, J.A.; Gasbarrini, A.; et al. TNF-α blockade down-regulates the CD40/CD40L pathway in the mucosal microcirculation: A novel anti-inflammatory mechanism of infliximab in Crohn’s disease. J. Immunol. 2006, 176, 2617–2624. [Google Scholar] [CrossRef]
- Cornillie, F.; Shealy, D.; D’Haens, G.; Geboes, K.; Van Assche, G.; Ceuppens, J.; Wagner, C.; Schaible, T.; Plevy, S.E.; Targan, S.R.; et al. Infliximab induces potent anti-inflammatory and local immunomodulatory activity but no systemic immune suppression in patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2001, 15, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Ruud, T.E.; Gundersen, Y.; Krohn, C.D.; Sveen, O.; Aasen, A.O. Effects of infliximab and hydrocortisone on in vitro cytokine responses after stimulation with lipopolysaccharide. Surg. Infect. (Larchmt.) 2013, 14, 30–34. [Google Scholar] [CrossRef]
- Vis, M.; Voskuyl, A.E.; Wolbink, G.J.; Dijkmans, B.A.; Lems, W.F. Bone mineral density in patients with rheumatoid arthritis treated with infliximab. Ann. Rheum. Dis. 2005, 64, 336–337. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J. Adalimumab in the treatment of arthritis. Ther. Clin. Risk. Manag. 2007, 3, 133–148. [Google Scholar] [CrossRef]
- Wijbrandts, C.A.; Klaasen, R.; Dijkgraaf, M.G.; Gerlag, D.M.; van Eck-Smit, B.L.; Tak, P.P. Bone mineral density in rheumatoid arthritis patients 1 year after adalimumab therapy: Arrest of bone loss. Ann. Rheum. Dis. 2009, 68, 373–376. [Google Scholar] [CrossRef]
- Krieckaert, C.L.; Nurmohamed, M.T.; Wolbink, G.; Lems, W.F. Changes in bone mineral density during long-term treatment with adalimumab in patients with rheumatoid arthritis: A cohort study. Rheumatology 2013, 52, 547–553. [Google Scholar] [CrossRef]
- Wong, M.; Ziring, D.; Korin, Y.; Desai, S.; Kim, S.; Lin, J.; Gjertson, D.; Braun, J.; Reed, E.; Singh, R.R. TNFα blockade in human diseases: Mechanisms and future directions. Clin. Immunol. 2008, 126, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Banner, D.W.; D’Arcy, A.; Janes, W.; Gentz, R.; Schoenfeld, H.J.; Broger, C.; Loetscher, H.; Lesslauer, W. Crystal structure of the soluble human 55 kd TNF receptor-human TNFβ complex: Implications for TNF receptor activation. Cell 1993, 73, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Felquer, M.L.; Rosa, J.; Soriano, E.R. An evidence-based review of certolizumab pegol in the treatment of active psoriatic arthritis: Place in therapy. Open Access Rheumatol. 2016, 8, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Gulyás, K.; Horváth, Á.; Végh, E.; Pusztai, A.; Szentpétery, Á.; Pethö, Z.; Váncsa, A.; Bodnár, N.; Csomor, P.; Hamar, A.; et al. Effects of 1-year anti-TNF-α therapies on bone mineral density and bone biomarkers in rheumatoid arthritis and ankylosing spondylitis. Clin. Rheumatol. 2020, 39, 167–175. [Google Scholar] [CrossRef]
- Orsolini, G.; Adami, G.; Adami, S.; Viapiana, O.; Idolazzi, L.; Gatti, D.; Rossini, M. Short-term effects of TNF inhibitors on bone turnover markers and bone mineral density in rheumatoid arthritis. Calcif. Tissue Int. 2016, 98, 580–585. [Google Scholar] [CrossRef]
- Maeda, K.; Yoshida, K.; Nishizawa, T.; Otani, K.; Yamashita, Y.; Okabe, H.; Hadano, Y.; Kayama, T.; Kurosaka, D.; Saito, M. Inflammation and bone metabolism in rheumatoid arthritis: Molecular mechanisms of joint destruction and pharmacological treatments. Int. J. Mol. Sci. 2022, 23, 2871. [Google Scholar] [CrossRef]
- Fleischmann, R.M.; Schechtman, J.; Bennett, R.; Handel, M.L.; Burmester, G.R.; Tesser, J.; Modafferi, D.; Poulakos, J.; Sun, G. Anakinra; a recombinant human interleukin-1 receptor antagonist (r-metHuIL-1ra); in patients with rheumatoid arthritis: A large, international, multicenter, placebo-controlled trial. Arthritis Rheum. 2003, 48, 927–934. [Google Scholar] [CrossRef]
- Giai, C.; Gonzalez, C.D.; Sabbione, F.; Garofalo, A.; Ojeda, D.; Sordelli, D.O.; Trevani, A.S.; Gómez, M.I. Staphylococcus aureus induces shedding of IL-1RII in monocytes and neutrophils. J. Innate Immun. 2016, 8, 284–298. [Google Scholar] [CrossRef]
- Shimizu, K.; Nakajima, A.; Sudo, K.; Liu, Y.; Mizoroki, A.; Ikarashi, T.; Horai, R.; Kakuta, S.; Watanabe, T.; Iwakura, Y. IL-1 receptor type 2 suppresses collagen-induced arthritis by inhibiting IL-1 signal on macrophages. J. Immunol. 2015, 194, 3156–3168. [Google Scholar] [CrossRef]
- Martin, P.; Palmer, G.; Rodriguez, E.; Seemayer, C.A.; Palomo, J.; Talabot-Ayer, D.; Gabay, C. Deficiency in IL-1 receptor type 2 aggravates K/BxN serum transfer-induced arthritis in mice but has no impact on systemic inflammatory responses. J. Immunol. 2017, 198, 2916–2926. [Google Scholar] [CrossRef] [PubMed]
- Rauschmayr, T.; Groves, R.W.; Kupper, T.S. Keratinocyte expression of the type 2 interleukin 1 receptor mediates local and specific inhibition of interleukin 1-mediated inflammation. Proc. Nat. Acad. Sci. USA 1997, 94, 5814–5819. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Xia, L.; Shen, H.; Lu, J. Elevated frequency of IL-37- and IL-18Rα-positive T cells in the peripheral blood of rheumatoid arthritis patients. Cytokine 2018, 110, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Batliwalla, F.M.; Li, W.; Ritchlin, C.T.; Xiao, X.; Brenner, M.; Laragione, T.; Shao, T.; Durham, R.; Kemshetti, S.; Schwarz, E.; et al. Microarray analyses of peripheral blood cells identifies unique gene expression signature in psoriatic arthritis. Mol. Med. 2005, 11, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.E.; Stefanska, A.M.; Kubica, M.; Horan, R.M.; Mantovani, A.; Garlanda, C.; Fallon, P.G.; Walsh, P.T. Toll IL-1R8/single Ig IL-1-related receptor regulates psoriasiform inflammation through direct inhibition of innate IL-17A expression by γδ T cells. J. Immunol. 2013, 191, 3337–3346. [Google Scholar] [CrossRef]
- Giannoudaki, E.; Stefanska, A.M.; Lawler, H.; Leon, G.; Hernandez Santana, Y.E.; Hassan, N.; Russell, S.E.; Horan, R.; Sweeney, C.; Preston, R.S.; et al. SIGIRR negatively regulates IL-36-driven psoriasiform inflammation and neutrophil infiltration in the skin. J. Immunol. 2021, 207, 651–660. [Google Scholar] [CrossRef]
- Supino, D.; Minute, L.; Mariancini, A.; Riva, F.; Magrini, E.; Garlanda, C. Negative regulation of the IL-1 system by IL-1R2 and IL-1R8: Relevance in pathophysiology and disease. Front. Immunol. 2022, 13, 804641. [Google Scholar] [CrossRef]
- Takeuchi, T.; Yoshida, H.; Tanaka, S. Role of interleukin-6 in bone destruction and bone repair in rheumatoid arthritis. Autoimmun. Rev. 2021, 20, 102884. [Google Scholar] [CrossRef]
- Yoshida, H.; Suzuki, M.; Tanaka, K.; Takeda, S.; Yogo, K.; Matsumoto, Y. Anti-interleukin-6 receptor antibody prevents loss of bone structure and bone strength in collagen-induced arthritis mice. Scand. J. Rheumatol. 2018, 47, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Abu-Shakra, M.; Zisman, D.; Balbir-Gurman, A.; Amital, H.; Levy, Y.; Langevitz, P.; Tishler, M.; Molad, Y.; Aamar, S.; Roser, I.; et al. Effect of tocilizumab on fatigue and bone mineral density in patients with rheumatoid arthritis. Isr. Med. Assoc. J. 2018, 20, 239–244. [Google Scholar]
- Kume, K.; Amano, K.; Yamada, S.; Kanazawa, T.; Ohta, H.; Hatta, K.; Amano, K.; Kuwaba, N. The effect of tocilizumab on bone mineral density in patients with methotrexate-resistant active rheumatoid arthritis. Rheumatology 2014, 53, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Briot, K.; Rouanet, S.; Schaeverbeke, T.; Etchepare, F.; Gaudin, P.; Perdriger, A.; Vray, M.; Steinberg, G.; Roux, C. The effect of tocilizumab on bone mineral density, serum levels of Dickkopf-1 and bone remodeling markers in patients with rheumatoid arthritis. Jt. Int. Bone Spine 2015, 82, 109–115. [Google Scholar] [CrossRef]
- Huizinga, T.W.; Fleischmann, R.M.; Jasson, M.; Radin, A.R.; van Adelsberg, J.; Fiore, S.; Huang, X.; Yancopoulos, G.D.; Stahl, N.; Genovese, M.C. Sarilumab, a fully human monoclonal antibody against IL-6Rα in patients with rheumatoid arthritis and an inadequate response to methotrexate: Efficacy and safety results from the randomised SARIL-RA-MOBILITY Part A trial. Ann. Rheum. Dis. 2014, 73, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Msihid, J.; Zilberstein, M.; Paccard, C.; Lin, Y.; Graham, N.M.H.; Boyapati, A. Identification of sarilumab pharmacodynamic and predictive markers in patients with inadequate response to TNF inhibition: A biomarker substudy of the phase 3 TARGET study. RMD Open 2018, 4, e000607. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Burmester, G.R.; Strand, V.; Msihid, J.; Zilberstein, M.; Kimura, T.; van Hoogstraten, H.; Boklage, S.H.; Sadeh, J.; Graham, N.M.H.; et al. Sarilumab and adalimumab differential effects on bone remodelling and cardiovascular risk biomarkers, and predictions of treatment outcomes. Arthritis Res. Ther. 2020, 22, 70. [Google Scholar] [CrossRef]
- Vidal, B.; Cascão, R.; Finnilä, M.A.J.; Lopes, I.P.; da Glória, V.G.; Saarakkala, S.; Zioupos, P.; Canhão, H.; Fonseca, J.E. Effects of tofacitinib in early arthritis-induced bone loss in an adjuvant-induced arthritis rat model. Rheumatology 2018, 57, 1461–1471. [Google Scholar] [CrossRef]
- LaBranche, T.P.; Jesson, M.I.; Radi, Z.A.; Storer, C.E.; Guzova, J.A.; Bonar, S.L.; Thompson, J.M.; Happa, F.A.; Stewart, Z.S.; Zhan, Y.; et al. JAK inhibition with tofacitinib suppresses arthritic joint structural damage through decreased RANKL production. Arthritis Rheum. 2012, 64, 3531–3542. [Google Scholar] [CrossRef]
- Yokota, K.; Sato, K.; Miyazaki, T.; Aizaki, Y.; Tanaka, S.; Sekikawa, M.; Kozu, N.; Kadono, Y.; Oda, H.; Mimura, T. Characterization and function of tumor necrosis factor and interleukin-6-induced osteoclasts in rheumatoid arthritis. Arthritis Rheumatol. 2021, 73, 1145–1154. [Google Scholar] [CrossRef]
- Gaber, T.; Brinkman, A.C.K.; Pienczikowski, J.; Diesing, K.; Damerau, A.; Pfeiffenberger, M.; Lang, A.; Ohrndorf, S.; Burmester, G.R.; Buttgereit, F.; et al. Impact of janus kinase inhibition with tofacitinib on fundamental processes of bone healing. Int. J. Mol. Sci. 2020, 21, 865. [Google Scholar] [CrossRef]
- Adam, S.; Simon, N.; Steffen, U.; Andes, F.T.; Scholtysek, C.; Müller, D.I.H.; Weidner, D.; Andreev, D.; Kleyer, A.; Culemann, S.; et al. JAK inhibition increases bone mass in steady-state conditions and ameliorates pathological bone loss by stimulating osteoblast function. Sci. Transl. Med. 2020, 12, eaay4447. [Google Scholar] [CrossRef]
- Fridman, J.S.; Scherle, P.A.; Collins, R.; Burn, T.C.; Li, Y.; Li, J.; Covington, M.B.; Thomas, B.; Collier, P.; Favata, M.F.; et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: Preclinical characterization of INCB028050. J. Immunol. 2010, 184, 5298–5307. [Google Scholar] [CrossRef]
- Murakami, K.; Kobayashi, Y.; Uehara, S.; Suzuki, T.; Koide, M.; Yamashita, T.; Nakamura, M.; Takahashi, N.; Kato, H.; Udagawa, N.; et al. A Jak1/2 inhibitor, baricitinib, inhibits osteoclastogenesis by suppressing RANKL expression in osteoblasts in vitro. PLoS ONE 2017, 12, e0181126. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Yamazaki, S.; Yamagami, K.; Kuno, M.; Morita, Y.; Okuma, K.; Nakamura, K.; Chida, N.; Inami, M.; Inoue, T.; et al. A novel JAK inhibitor, peficitinib, demonstrates potent efficacy in a rat adjuvant-induced arthritis model. J. Pharmacol. Sci. 2017, 133, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Biggioggero, M.; Becciolini, A.; Crotti, C.; Agape, E.; Favalli, E.G. Upadacitinib and filgotinib: The role of JAK1 selective inhibition in the treatment of rheumatoid arthritis. Drugs Context 2019, 8, 212595. [Google Scholar] [CrossRef]
- Van Rompaey, L.; Galien, R.; van der Aar, E.M.; Clement-Lacroix, P.; Nelles, L.; Smets, B.; Lepescheux, L.; Christophe, T.; Conrath, K.; Vandeghinste, N.; et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J. Immunol. 2013, 191, 3568–3577. [Google Scholar] [CrossRef]
- Fossiez, F.; Djossou, O.; Chomarat, P.; Flores-Romo, L.; Ait-Yahia, S.; Maat, C.; Pin, J.J.; Garrone, P.; Garcia, E.; Saeland, S.; et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J. Exp. Med. 1996, 183, 2593–2603. [Google Scholar] [CrossRef]
- Yao, Z.; Fanslow, W.C.; Seldin, M.F.; Rousseau, A.M.; Painter, S.L.; Comeau, M.R.; Cohen, J.I.; Spriggs, M.K. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity 1995, 3, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Schwandner, R.; Yamaguchi, K.; Cao, Z. Requirement of tumor necrosis factor receptor-associated factor (TRAF)6 in interleukin 17 signal transduction. J. Exp. Med. 2000, 191, 1233–1240. [Google Scholar] [CrossRef]
- Jovanovic, D.V.; Di Battista, J.A.; Martel-Pelletier, J.; Jolicoeur, F.C.; He, Y.; Zhang, M.; Mineau, F.; Pelletier, J.P. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J. Immunol. 1998, 160, 3513–3521. [Google Scholar] [CrossRef]
- Chabaud, M.; Fossiez, F.; Taupin, J.L.; Miossec, P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J. Immunol. 1998, 161, 409–414. [Google Scholar] [CrossRef]
- Lubberts, E.; Joosten, L.A.B.; van de Loo, F.A.J.; van den Bersselaar, L.A.; van den Berg, W.B. Reduction of interleukin-17–induced inhibition of chondrocyte proteoglycan synthesis in intact murine articular cartilage by interleukin-4. Arthritis Rheum. 2000, 43, 1300–1306. [Google Scholar] [CrossRef]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.T.; et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Investig. 1999, 103, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Chabaud, M.; Lubberts, E.; Joosten, L.; van den Berg, W.; Miossec, P. IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res. 2001, 3, 168–177. [Google Scholar] [CrossRef]
- Koshy, P.J.; Henderson, N.; Logan, C.; Life, P.F.; Cawston, T.E.; Rowan, A.D. Interleukin 17 induces cartilage collagen breakdown: Novel synergistic effects in combination with proinflammatory cytokines. Ann. Rheum. Dis. 2002, 61, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Lubberts, E.; Joosten, L.A.; Oppers, B.; van den Bersselaar, L.; Coenen-de Roo, C.J.; Kolls, J.K.; Schwarzenberger, P.; van de Loo, F.A.; van den Berg, W.B. IL-1-independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J. Immunol. 2001, 167, 1004–1013. [Google Scholar] [CrossRef]
- Van Bezooijen, R.L.; van der Wee-Pals, L.; Papapoulos, S.E.; Lo¨wik, C.W. Interleukin 17 synergises with tumour necrosis factor to induce cartilage destruction in vitro. Ann. Rheum. Dis. 2002, 61, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Lubberts, E.; Koenders, M.I.; Oppers-Walgreen, B.; van den Bersselaar, L.; Coenen-de Roo, C.J.; Joosten, L.A.; van den Berg, W.B. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004, 50, 650–659. [Google Scholar] [CrossRef]
- Bush, K.A.; Farmer, K.M.; Walker, J.S.; Kirkham, B.W. Reduction of joint inflammation and bone erosion in rat adjuvant arthritis by treatment with interleukin-17 receptor IgG1 Fc fusion protein. Arthritis Rheum. 2002, 46, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Lubberts, E.; van den Bersselaar, L.; Oppers-Walgreen, B.; Schwarzenberger, P.; Coenen-de Roo, C.J.; Kolls, J.K.; Joosten, L.A.; van den Berg, W.B. IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-kappa B ligand/osteoprotegerin balance. J. Immunol. 2003, 170, 2655–2662. [Google Scholar] [CrossRef]
- Romas, E.; Sims, N.A.; Hards, D.K.; Lindsay, M.; Quinn, J.W.; Ryan, P.F.; Dunstan, C.R.; Martin, T.J.; Gillespie, M.T. Osteoprotegerin reduces osteoclast numbers and prevents bone erosion in collagen-induced arthritis. Am. J. Pathol. 2002, 161, 1419–1427. [Google Scholar] [CrossRef]
- Frieder, J.; Kivelevitch, D.; Menter, A. Secukinumab: A review of the anti-IL-17A biologic for the treatment of psoriasis. Ther. Adv. Chronic Dis. 2018, 9, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Aboobacker, S.; Kurn, H.; Al Aboud, A.M. Secukinumab; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Smolen, J.S.; Landewé, R.B.M.; Bijlsma, J.W.J.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; van Vollenhoven, R.F.; de Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 2020, 79, 685–699. [Google Scholar] [CrossRef]
- Inoue, K.; Yuasa, H. Molecular basis for pharmacokinetics and pharmacodynamics of methotrexate in rheumatoid arthritis therapy. Drug Metab. Pharmacokinet. 2014, 29, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Segawa, Y.; Yamaura, M.; Aota, S.; Omata, T.; Tuzuike, N.; Itokazu, Y.; Oka, H.; Tamaki, H.; Nakamura, T. Methotrexate maintains bone mass by preventing both a decrease in bone formation and an increase in bone resorption in adjuvant-induced arthritic rats. Bone 1997, 20, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Kanagawa, H.; Masuyama, R.; Morita, M.; Sato, Y.; Niki, Y.; Kobayashi, T.; Katsuyama, E.; Fujie, A.; Hao, W.; Tando, T.; et al. Methotrexate inhibits osteoclastogenesis by decreasing RANKL-induced calcium influx into osteoclast progenitors. J. Bone Miner. Metab. 2016, 34, 526–531. [Google Scholar] [CrossRef]
- Poutoglidou, F.; Pourzitaki, C.; Manthou, M.E.; Samoladas, E.; Saitis, A.; Malliou, F.; Kouvelas, D. Infliximab prevents systemic bone loss and suppresses tendon inflammation in a collagen-induced arthritis rat model. Inflammopharmacology 2021, 29, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Gowayed, M.A.; El Achy, S.; Kamel, M.A.; El-Tahan, R.A. Polymyxin B prevents the development of adjuvant arthritis via modulation of TLR/Cox-2 signaling pathway. Life Sci. 2020, 259, 118250. [Google Scholar] [CrossRef]
- Mello, S.; Barros, D.; Silva, A.; Laurindo, I.; Novaes, G. Methotrexate as a preferential cyclooxygenase 2 inhibitor in whole blood of patients with rheumatoid arthritis. Rheumatology 2000, 39, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Lina, C.; Conghua, W.; Nan, L.; Ping, Z. Combined treatment of etanercept and MTX reverses Th1/Th2; Th17/Treg imbalance in patients with rheumatoid arthritis. J. Clin. Immunol. 2011, 31, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Azouz, A.A.; Saleh, E.; Abo-Saif, A.A. Aliskiren, tadalafil, and cinnamaldehyde alleviate joint destruction biomarkers, MMP-3 and RANKL, in complete Freund’s adjuvant arthritis model: Downregulation of IL-6/JAK2/STAT3 signaling pathway. Saudi Pharm. J. 2020, 28, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef] [PubMed]
- Kraynik, S.M.; Miyaoka, R.S.; Beavo, J.A. PDE3 and PDE4 isozyme-selective inhibitors are both required for synergistic activation of brown adipose tissue. Mol. Pharmacol. 2013, 83, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Kragstrup, T.W.; Adams, M.; Lomholt, S.; Nielsen, M.A.; Heftdal, L.D.; Schafer, P.; Deleuran, B. IL-12/IL-23p40 identified as a downstream target of apremilast in ex vivo models of arthritis. Ther. Adv. Musculoskelet. Dis. 2019, 11, 1759720X19828669. [Google Scholar] [CrossRef]
- Imam, F.; Al-Harbi, N.O.; Al-Harbi, M.M.; Ansari, M.A.; Al-Asmari, A.F.; Ansari, M.N.; Al-Anazi, W.A.; Bahashwan, S.; Almutairi, M.M.; Alshammari, M.; et al. Apremilast prevent doxorubicin-induced apoptosis and inflammation in heart through inhibition of oxidative stress mediated activation of NF-κB signaling pathways. Pharmacol. Rep. 2018, 70, 993–1000. [Google Scholar] [CrossRef]
- Mazzilli, S.; Lanna, C.; Chiaramonte, C.; Cesaroni, G.M.; Zangrilli, A.; Palumbo, V.; Cosio, T.; Dattola, A.; Gaziano, R.; Galluzzo, M.; et al. Real life experience of apremilast in psoriasis and arthritis psoriatic patients: Preliminary results on metabolic biomarkers. J. Dermatol. 2020, 47, 578–582. [Google Scholar] [CrossRef]
- Otto, M.; Dorn, B.; Grasmik, T.; Doll, M.; Meissner, M.; Jakob, T.; Hrgovic, I. Apremilast effectively inhibits TNFα-induced vascular inflammation in human endothelial cells. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 237–246. [Google Scholar] [CrossRef]
- Kataoka, S.; Takaishi, M.; Nakajima, K.; Sano, S. Phosphodiesterase-4 inhibitors reduce the expression of proinflammatory mediators by human epidermal keratinocytes independent of intracellular cAMP elevation. J. Dermatol. Sci. 2020, 100, 230–233. [Google Scholar] [CrossRef]
- Shenoy, P.; Agarwal, V. Phosphodiesterase inhibitors in the management of autoimmune disease. Autoimmun. Rev. 2010, 9, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Rocha, F.A.; Silva, F.S., Jr.; Leite, A.C.; Leite, A.K.; Girão, V.C.; Castro, R.R.; Cunha, F.Q. Tadalafil analgesia in experimental arthritis involves suppression of intra-articular TNF release. Br. J. Pharmacol. 2011, 164, 828–835. [Google Scholar] [CrossRef]
- Bahadir, F.E.; Köroğlu, M.K.; Yüksel, M.; Ercan, F.; Alican, Y.İ. Effect of phosphodiesterase-5 inhibition on joint and muscle damage in rats with adjuvant arthritis. Turk. J. Med. Sci. 2018, 48, 635–643. [Google Scholar] [CrossRef]
- Kuno, Y.; Lyoda, M.; Shibata, T.; Hirai, Y.; Akizawa, T. Sildenafil, a phosphodiesterase type 5 inhibitor, attenuates diabetic nephropathy in non-insulin-dependent Otsuka Long-Evans Tokushima Fatty rats. Br. J. Pharmacol. 2011, 162, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Mosheimer, B.A.; Kaneider, N.C.; Feistritzer, C.; Sturn, D.H.; Wiedermann, C.J. Expression and function of RANK in human monocyte chemotaxis. Arthritis Rheum. 2004, 50, 2309–2316. [Google Scholar] [CrossRef]
- Toğral, G.; Arıkan, Ş.M.; Korkusuz, P.; Hesar, R.H.; Ekşioğlu, M.F. Positive effect of tadalafil, a phosphodiesterase-5 inhibitor, on fracture healing in rat femur. Jt. Dis. Relat. Surg. 2015, 26, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, J.; Buttery, L.; O’Shaughnessy, M.; Afzal, F.; de Marticorena, I.F.; Hukkanen, M.; Huang, P.; MacIntyre, I.; Polak, J. Endothelial nitric oxide synthase gene-deficient mice demonstrate marked retardation in postnatal bone formation, reduced bone volume, and defects in osteoblast maturation and activity. Am. J. Pathol. 2001, 158, 247–257. [Google Scholar] [CrossRef]
- Bringel, P.H.d.S.F.; Marques, G.F.O.; de Queiroz Martins, M.G.; da Silva, M.T.L.; Nobre, C.A.S.; do Nascimento, K.S.; Cavada, B.S.; Castro, R.R.; Assreuy, A.M.S. The lectin isolated from the alga hypnea cervicornis promotes antinociception in rats subjected to zymosan-induced arthritis: Involvement of cGMP signalization and cytokine expression. Inflammation 2020, 43, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Wei, Y.B.; Strawbridge, R.; Bao, Y.; Chang, S.; Shi, L.; Que, J.; Gadad, B.S.; Trivedi, M.H.; Kelsoe, J.R.; et al. Peripheral cytokine levels and response to antidepressant treatment in depression: A systematic review and meta-analysis. Mol. Psychiatry 2020, 25, 339–350. [Google Scholar] [CrossRef]
- Haapakoski, R.; Mathieu, J.; Ebmeier, K.P.; Alenius, H.; Kivimäki, M. Cumulative meta-analysis of interleukins 6 and 1β, tumour necrosis factor α and C-reactive protein in patients with major depressive disorder. Brain Behav. Immun. 2015, 49, 206–215. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef]
- Maes, M.; Carvalho, A.F. The compensatory immune-regulatory reflex system (CIRS) in depression and bipolar disorder. Mol. Neurobiol. 2018, 55, 8885–8903. [Google Scholar] [CrossRef]
- Harsanyi, S.; Kupcova, I.; Danisovic, L.; Klein, M. Selected biomarkers of depression: What are the effects of cytokines and inflammation? Int. J. Mol. Sci. 2022, 24, 578. [Google Scholar] [CrossRef]
- Baune, B.T.; Konrad, C.; Grotegerd, D.; Suslow, T.; Birosova, E.; Ohrmann, P.; Bauer, J.; Arolt, V.; Heindel, W.; Domschke, K.; et al. Interleukin-6 gene (IL-6): A possible role in brain morphology in the healthy adult brain. J. Neuroinflamm. 2012, 9, 125. [Google Scholar] [CrossRef]
- Tolou-Ghamari, Z.; Zare, M.; Habibabadi, J.M.; Najafi, M.R. A quick review of carbamazepine pharmacokinetics in epilepsy from 1953 to 2012. J. Res. Med. Sci. 2013, 18 (Suppl. S1), S81–S85. [Google Scholar]
- Pottoo, F.H.; Alshayban, D.M.; Joseph, R.; Al-Musa, F.; Al-Jabran, O.; Aljaafari, D. Impact of Adherence to Antiepileptic Medications on Quality of Life of Epileptic Patients in the Eastern Province of Saudi Arabia: A Cross-Sectional Study. Imam J. Appl. Sci. 2020, 5, 1–8. Available online: https://www.e-ijas.org/article.asp?issn=2589-0603;year=2020;volume=5;issue=1;spage=1;epage=8;aulast=Pottoo (accessed on 14 February 2023). [CrossRef]
- Colombi, I.; Mahajani, S.; Frega, M.; Gasparini, L.; Chiappalone, M. Effects of antiepileptic drugs on hippocampal neurons coupled to micro-electrode arrays. Front. Neuroeng. 2013, 6, 10. [Google Scholar] [CrossRef]
- Wang, C.-H.; Hsiao, C.-J.; Lin, Y.-N.; Wu, J.-W.; Kuo, Y.-C.; Lee, C.-K.; Hsiao, G. Carbamazepine Attenuates Inducible Nitric Oxide Synthase Expression through Akt Inhibition in Activated Microglial Cells. Pharm. Biol. 2014, 52, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.D.; Buijs, R.M.; Sitges, M. The anti-seizure drugs vinpocetine and carbamazepine, but not valproic acid, reduce inflammatory IL-1β and TNF-α expression in rat hippocampus. J. Neurochem. 2014, 130, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Pottoo, F.H.; Salahuddin, M.; Khan, F.A.; Al Dhamen, M.A.; Alsaeed, W.J.; Gomaa, M.S.; Vatte, C.; Alomary, M.N. Combinatorial regimen of carbamazepine and imipramine exhibits synergism against grandmal epilepsy in rats: Inhibition of pro-inflammatory cytokines and PI3K/Akt/mTOR signaling pathway. Pharmaceuticals 2021, 14, 1204. [Google Scholar] [CrossRef]
- Tricyclic Antidepressants—StatPearls—NCBI Bookshelf. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557791/ (accessed on 15 February 2023).
- D’Aquila, P.S.; Galistu, A. Further characterization of the effect of the prototypical antidepressant imipramine on the microstructure of licking for sucrose. PLoS ONE 2021, 16, e0245559. [Google Scholar] [CrossRef]
- Heninger, G.R.; Delgado, P.L.; Charney, D.S. The revised monoamine theory of depression: A modulatory role for monoamines, based on new findings from monoamine depletion experiments in humans. Pharmacopsychiatry 1996, 29, 2–11. [Google Scholar] [CrossRef]
- Shelton, R.C. Cellular mechanisms in the vulnerability to depression and response to antidepressants. Psychiatr. Clin. N. Am. 2000, 23, 713–729. [Google Scholar] [CrossRef]
- Fitzgerald, P.J. Is elevated norepinephrine an etiological factor in some cases of epilepsy? Seizure 2010, 19, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Gillman, P.K. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br. J. Pharmacol. 2007, 151, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Low, P.A.; Dotson, R.M. Symptomatic treatment of painful neuropathy. JAMA 1998, 280, 1863–1864. [Google Scholar] [CrossRef] [PubMed]
- Obuchowicz, E.; Bielecka, A.M.; Paul-Samojedny, M.; Pudełko, A.; Kowalski, J. Imipramine and fluoxetine inhibit LPS-induced activation and affect morphology of microglial cells in the rat glial culture. Pharmacol. Rep. 2014, 66, 34–43. [Google Scholar] [CrossRef]
- Ramirez, K.; Sheridan, J.F. Antidepressant imipramine diminishes stress-induced inflammation in the periphery and central nervous system and related anxiety- and depressive-like behaviors. Brain Behav. Immun. 2016, 57, 293–303. [Google Scholar] [CrossRef]
- Alboni, S.; Benatti, C.; Montanari, C.; Tascedda, F.; Brunello, N. Chronic antidepressant treatments resulted in altered expression of genes involved in inflammation in the rat hypothalamus. Eur. J. Pharmacol. 2013, 721, 158–167. [Google Scholar] [CrossRef]
- Bockbrader, H.N.; Wesche, D.; Miller, R.; Chapel, S.; Janiczek, N.; Burger, P. A comparison of the pharmacokinetics and pharmacodynamics of pregabalin and gabapentin. Clin. Pharmacokinet. 2010, 49, 661–669. [Google Scholar] [CrossRef]
- Kukkar, A.; Bali, A.; Singh, N.; Jaggi, A.S. Implications and mechanism of action of gabapentin in neuropathic pain. Arch. Pharm. Res. 2013, 36, 237–251. [Google Scholar] [CrossRef]
- Maneuf, Y.P.; Luo, Z.D.; Lee, K. α2δ and the mechanism of action of gabapentin in the treatment of pain. Semin. Cell Dev. Biol. 2006, 17, 565–570. [Google Scholar] [CrossRef]
- Sills, G.J. The mechanisms of action of gabapentin and pregabalin. Curr. Opin. Pharmacol. 2006, 6, 108–113. [Google Scholar] [CrossRef]
- Gajraj, N.M. Pregabalin: Its pharmacology and use in pain management. Anesth. Analg. 2007, 105, 1805–1815. [Google Scholar] [CrossRef]
- Toth, C. Pregabalin: Latest safety evidence and clinical implications for the management of neuropathic pain. Ther. Adv. Drug Saf. 2014, 5, 38–56. [Google Scholar] [CrossRef]
- Ben-Menachem, E. Pregabalin pharmacology and its relevance to clinical practice. Epilepsia 2004, 45 (Suppl. S6), 13–18. [Google Scholar] [CrossRef] [PubMed]
- Hagen, E.M.; Rekand, T. Management of neuropathic pain associated with spinal cord injury. Pain Ther. 2015, 4, 51–65. [Google Scholar] [CrossRef]
- Kumar, K.P.; Kulkarni, D.K.; Gurajala, I.; Gopinath, R. Pregabalin versus tramadol for postoperative pain management in patients undergoing lumbar laminectomy: A randomized, doubleblinded, placebo-controlled study. J. Pain Res. 2013, 6, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Gianesello, L.; Pavoni, V.; Barboni, E.; Galeotti, I.; Nella, A. Perioperative pregabalin for postoperative pain control and quality of life after major spinal surgery. J. Neurosurg. Anesthesiol. 2012, 24, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Salazar, V.; Dewey, C.W.; Schwark, W.; Badgley, B.L.; Gleed, R.D.; Horne, W.; Ludders, J.W. Pharmacokinetics of single-dose oral pregabalin administration in normal dogs. Vet. Anaesth. Analg. 2009, 36, 574–580. [Google Scholar] [CrossRef]
- Schmierer, P.A.; Tünsmeyer, J.; Tipold, A.; Hartnack-Wilhelm, S.; Lesczuk, P.; Kästner, S.B.R. Randomized controlled trial of pregabalin for analgesia after surgical treatment of intervertebral disc disease in dogs. Vet. Surg. 2020, 49, 905–913. [Google Scholar] [CrossRef]
- Anfuso, C.D.; Olivieri, M.; Fidilio, A.; Lupo, G.; Rusciano, D.; Pezzino, S.; Gagliano, C.; Drago, F.; Bucolo, C. Gabapentin attenuates ocular inflammation: In vitro and in vivo studies. Front. Pharmacol. 2017, 8, 173. [Google Scholar] [CrossRef]
- Bao, Y.H.; Zhou, Q.H.; Chen, R.; Xu, H.; Zeng, L.L.; Zhang, X.; Jiang, W.; Du, D.P. Gabapentin enhances the morphine anti-nociceptive effect in neuropathic pain via the interleukin-10-heme oxygenase-1 signalling pathway in rats. J. Mol. Neurosci. 2014, 54, 137–146. [Google Scholar] [CrossRef]
- Leisengang, S.; Ott, D.; Murgott, J.; Gerstberger, R.; Rummel, C.; Roth, J. Primary cultures from rat dorsal root ganglia: Responses of neurons and glial cells to somatosensory or inflammatory stimulation. Neuroscience 2018, 394, 1–13. [Google Scholar] [CrossRef]
- Nagy, I.; Rang, H. Noxious heat activates all capsaicin-sensitive and also a sub-population of capsaicin-insensitive dorsal root ganglion neurons. Neuroscience 1999, 88, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Julius, D. The vanilloid receptor: A molecular gateway to the pain pathway. Ann. Rev. Neurosci. 2001, 24, 487–517. [Google Scholar] [CrossRef]
- Tachiya, D.; Sato, T.; Ichikawa, H. Nerve injury increases the expression of alpha-2/delta-1 subunit of L-type calcium channel in sensory neurons of rat spinal and trigeminal nerves. Ann. Neurosci. 2017, 24, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Leisengang, S.; Ott, D.; Murgott, J.; Nürnberger, F.; Gerstberger, R.; Rummel, C.; Schmidt, M.; Roth, J. Effects of gabapentinoids on responses of primary cultures from rat dorsal root ganglia to inflammatory or somatosensory stimuli. J. Basic Clin. Physiol. Pharmacol. 2020, 31, 20190261. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Kong, L.Y.; Cai, J.; Li, S.; Liu, X.D.; Han, J.S.; Xing, G.G. Interleukin-6-mediated functional upregulation of TRPV1 receptors in dorsal root ganglion neurons through the activation of JAK/PI3K signaling pathway: Roles in the development of bone cancer pain in a rat model. Pain 2015, 156, 1124–1144. [Google Scholar] [CrossRef]
- Li, X.; Wang, B.; Yu, N.; Yang, L.; Nan, C.; Sun, Z.; Guo, L.; Zhao, Z. Gabapentin alleviates brain injury in intracerebral hemorrhage through suppressing neuroinflammation and apoptosis. Neurochem. Res. 2022, 47, 3063–3075. [Google Scholar] [CrossRef] [PubMed]
- Lynch, B.A.; Lambeng, N.; Nocka, K.; Kensel-Hammes, P.; Bajjalieh, S.M.; Matagne, A.; Fuks, B. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. USA 2004, 101, 9861–9866. [Google Scholar] [CrossRef]
- De Smedt, T.; Raedt, R.; Vonck, K.; Boon, P. Levetiracetam: The profile of a novel anticonvulsant drug-part I: Preclinical data. CNS Drug Rev. 2007, 13, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Johannessen Landmark, C. Antiepileptic drugs in non-epilepsy disorders: Relations between mechanisms of action and clinical efficacy. CNS Drugs 2008, 22, 27–47. [Google Scholar] [CrossRef]
- Srivastava, A.; Dixit, A.B.; Banerjee, J.; Tripathi, M.; Sarat Chandra, P. Role of inflammation and its miRNA based regulation in epilepsy: Implications for therapy. Clin. Chim. Acta 2016, 452, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Stienen, M.N.; Haghikia, A.; Dambach, H.; Thöne, J.; Wiemann, M.; Gold, R.; Chan, A.; Dermietzel, R.; Faustmann, P.M.; Hinkerohe, D.; et al. Antiinflammatory effects of the anticonvulsant drug levetiracetam on electrophysiological properties of astroglia are mediated via TGF b 1 regulation. Br. J. Pharmacol. 2011, 162, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Abed El-Gaphar, O.A.M.; Abo-Youssef, A.M.; Halal, G.K. Levetiracetam mitigates lipopolysaccharide-induced JAK2/STAT3 and TLR4/MAPK signaling pathways activation in a rat model of adjuvant- induced arthritis. Eur. J. Pharmacol. 2018, 826, 85–95. [Google Scholar] [CrossRef]
- Vollmar, P.; Haghikia, A.; Dermietzel, R.; Faustmann, P.M. Venlafaxine exhibits an anti-inflammatory effect in an inflammatory co-culture model. Int. J. Neuropsychopharmacol. 2008, 11, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Vollmar, P.; Nessler, S.; Kalluri, S.R.; Hartung, H.P.; Hemmer, B. The antidepressant venlafaxine ameliorates murine experimental autoimmune encephalomyelitis by suppression of pro-inflammatory cytokines. Int. J. Neuropsychopharmacol. 2009, 12, 525–536. [Google Scholar] [CrossRef]
- Kamel, K.M.; Gad, A.M.; Mansour, S.M.; Safar, M.M.; Fawzy, H.M. Venlafaxine alleviates complete Freund’s adjuvant-induced arthritis in rats: Modulation of STAT-3/IL-17/RANKL axis. Life Sci. 2019, 226, 68–76. [Google Scholar] [CrossRef]
- Juha, A.; Fehe, Z. Impact of venlafaxine on gene expression profile in lymphocytes of the elderly with major depression—Evolution of antidepressants and the role of the ‘“Neuro-Immune”’ system. Neurochem. Res. 2005, 30, 1429–1438. [Google Scholar] [CrossRef]
- Chen, C.X.; Chen, J.Y.; Kou, J.Q.; Xu, Y.L.; Wang, S.Z.; Zhu, Q.; Yang, L.; Qin, Z.H. Suppression of inflammation and arthritis by orally administrated cardiotoxin from Naja naja atra. Evid.-Based Complement. Altern. Med. 2015, 2015, 387094. [Google Scholar] [CrossRef]
- Cho, M.L.; Kang, J.W.; Moon, Y.M.; Nam, H.J.; Jhun, J.Y.; Heo, S.B.; Jin, H.T.; Min, S.Y.; Ju, J.H.; Park, K.S.; et al. STAT3 and NF-kappaB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J. Immunol. 2006, 176, 5652–5661. [Google Scholar] [CrossRef]
- Chen, X.W.; Zhou, S.F. Inflammation, cytokines, the IL-17/IL6/STAT3/NF-κB axis, and tumorigenesis. Drug Des. Devel. Ther. 2015, 9, 2941–2946. [Google Scholar] [CrossRef]
- Gerlag, D.M.; Ransone, L.; Tak, P.P.; Han, Z.; Palanki, M.; Barbosa, M.S.; Boyle, D.; Manning, A.M.; Firestein, G.S. The effect of a T cell-specific NF-kappa B inhibitor on in vitro cytokine production and collagen-induced arthritis. J. Immunol. 2000, 165, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- Badger, A.M.; Bradbeer, J.N.; Votta, B.; Lee, J.C.; Adams, J.L.; Griswold, D.E. Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. J. Pharmacol. Exp. Ther. 1996, 279, 1453–1461. [Google Scholar]
- Lee, J.C.; Young, P.R. Role of CSB/p38/RK stress response kinase in LPS and cytokine signaling mechanisms. J. Leukoc. Biol. 1996, 59, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, J.D.; Bibbs, L.; Ulevitch, R.J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811. [Google Scholar] [CrossRef]
- Rouse, J.; Cohen, P.; Trigon, S.; Morange, M.; Alonso-Llamazares, A.; Zamanillo, D.; Hunt, T.; Nebreda, A.R. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 1994, 78, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Heys, J.R.; Landvatter, S.W.; et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Foey, A.D.; Parry, S.L.; Williams, L.M.; Feldmann, M.; Foxwell, B.M.; Brennan, F.M. Regulation of monocyte IL-10 synthesis by endogenous IL-1 and TNF-alpha: Role of the p38 and p42/44 mitogen-activated protein kinases. J. Immunol. 1998, 160, 920–928. [Google Scholar] [CrossRef]
- Choi, D.E.; Jeong, J.Y.; Lim, B.J.; Chang, Y.K.; Na, K.R.; Shin, Y.T.; Lee, K.W. Aliskiren ameliorates renal inflammation and fibrosis induced by unilateral ureteral obstruction in mice. J. Urol. 2011, 186, 694–701. [Google Scholar] [CrossRef]
- Yan, K.; Shen, Y. Aliskiren has chondroprotective efficacy in a rat model of osteoarthritis through suppression of the local renin-angiotensin system. Mol. Med. Rep. 2017, 16, 3965–3973. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kou, J.; Zhang, H.; Wang, C.; Li, H.; Ren, Y.; Zhang, Y. The renin-angiotensin system in the synovium promotes periarticular osteopenia in a rat model of collagen-induced arthritis. Int. Immunopharmacol. 2018, 65, 550–558. [Google Scholar] [CrossRef]
- Izai, M.; Miyazaki, S.; Murai, R.; Morioka, Y.; Hayashi, H.; Nishiura, M.; Miura, K. Prorenin-renin axis in synovial fluid in patients with rheumatoid arthritis and osteoarthritis. Endocrinol. Jpn. 1992, 39, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Fujii, H.; Awata, R.; Kono, K.; Nakai, K.; Shinohara, M.; Nishi, S. Direct renin inhibitor aliskiren ameliorates low bone turnover in diabetic rat. Nephrol. Dial. Transplant. 2017, 32, iii232–iii233. [Google Scholar] [CrossRef]
- Akpinar, E.; Halici, Z.; Cadirci, E.; Bayir, Y.; Karakus, E.; Calik, M.; Topcu, A.; Polat, B. What is the role of renin inhibition during rat septic conditions: Preventive effect of aliskiren on sepsis-induced lung injury. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2014, 387, 969–978. [Google Scholar] [CrossRef]
- Araújo, A.A.; Souza, T.O.; Moura, L.M.; Brito, G.A.; Aragão, K.S.; Araújo, L.S.; Medeiros, C.A.; Alves, M.S.; Araújo, R.F., Jr. Effect of telmisartan on levels of IL-1, TNF-α, down-regulated COX-2, MMP-2, MMP-9 and RANKL/RANK in an experimental periodontitis model. J. Clin. Periodontol. 2013, 40, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Shuai, B.; Yang, Y.; Shen, L.; Zhu, R.; Xu, X.; Ma, C.; Lv, L.; Zhao, J.; Rong, J. Local renin-angiotensin system is associated with bone mineral density of glucocorticoid-induced osteoporosis patients. Osteoporos. Int. 2015, 26, 1063–1071. [Google Scholar] [CrossRef]
- Chung, S.; Kim, S.; Kim, M.; Koh, E.S.; Shin, S.J.; Park, C.W.; Chang, Y.S.; Kim, H.S. Treatment combining aliskiren with paricalcitol is effective against progressive renal tubulointerstitial fibrosis via dual blockade of intrarenal renin. PLoS ONE 2017, 12, e0181757. [Google Scholar] [CrossRef]
- Ye, S.; Luo, W.; Khan, Z.A.; Wu, G.; Xuan, L.; Shan, P.; Lin, K.; Chen, T.; Wang, J.; Hu, X.; et al. Celastrol attenuates angiotensin II-induced cardiac remodeling by targeting STAT3. Circ. Res. 2020, 126, 1007–1023. [Google Scholar] [CrossRef]
- Ziypak, T.; Halici, Z.; Alkan, E.; Akpinar, E.; Polat, B.; Adanur, S.; Cadirci, E.; Ferah, I.; Bayir, Y.; Karakus, E.; et al. Renoprotective effect of aliskiren on renal ischemia/reperfusion injury in rats: Electron microscopy and molecular study. Ren. Fail. 2015, 37, 343–354. [Google Scholar] [CrossRef]
- Liu, Z.H.; Ma, Y.F.; Wu, J.S.; Gan, J.X.; Xu, S.W.; Jiang, G.Y. Effect of cholinesterase inhibitor galanthamine on circulating tumor necrosis factor alpha in rats with lipopolysaccharide-induced peritonitis. Chin. Med. J. (Engl.) 2010, 123, 1727–1730. [Google Scholar] [PubMed]
- van Maanen, M.A.; Vervoordeldonk, M.J.; Tak, P.P. The cholinergic anti-inflammatory pathway: Towards innovative treatment of rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 229–232. [Google Scholar] [CrossRef]
- Ahsan, H.; Haider, I.; Mushtaq, M.N.; Qaisar, M.N.; Naqvi, F.; Asif, A. Pharmacological support to anti-arthritic prospective of physostigmine: A new approach. Inflammopharmacology 2021, 29, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Jiang, S.; Zhang, Z.; Inserra, P.; Lee, J.; Solkoff, D.; Watson, R.R. Anti-inflammatory effects of theophylline: Modulation of immune functions during murine leukemia virus infection. Immunopharmacol. Immunotoxicol. 2001, 23, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Chaudhary, M.J.; Tiwari, P.C.; Babu, S.; Pant, K.K. Protective role of theophylline and their interaction with nitric oxide (NO) in adjuvant-induced rheumatoid arthritis in rats. Int. Immunopharmacol. 2015, 29, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi-Pirbaluti, M.; Motaghi, E.; Najafi, A.; Hosseini, M.J. The effect of theophylline on acetic acid induced ulcerative colitis in rats. Biomed. Pharmacother. 2017, 90, 153–159. [Google Scholar] [CrossRef]
- Gallelli, L.; Falcone, D.; Cannataro, R.; Perri, M.; Serra, R.; Pelaia, G.; Maselli, R.; Savino, R.; Spaziano, G.; D’Agostino, B. Theophylline action on primary human bronchial epithelial cells under proinflammatory stimuli and steroidal drugs: A therapeutic rationale approach. Drug Des. Dev. Ther. 2017, 11, 265–272. [Google Scholar] [CrossRef]
- Gaafar, A.G.A.; Messiha, B.A.S.; Abdelkafy, A.M.L. Nicorandil and theophylline can protect experimental rats against complete Freund’s adjuvant-induced rheumatoid arthritis through modulation of JAK/STAT/RANKL signaling pathway. Eur. J. Pharmacol. 2018, 822, 177–185. [Google Scholar] [CrossRef]
- Hosseini-Tabatabaei, A.; Abdollahi, M. Potassium channel openers and improvement of toxic stress: Do they have role in the management of inflammatory bowel disease? Inflamm. Allergy Drug Targets 2008, 7, 129–135. [Google Scholar] [CrossRef]
- El-Moselhy, M.A.; Abdel-Hamid, N.M.; Abdel-Raheim, S.R. Gastroprotective effect of nicorandil in indomethacin and alcohol-induced acute ulcers. Appl. Biochem. Biotechnol. 2009, 152, 449–459. [Google Scholar] [CrossRef]
- Zhang, F.; Xuan, Y.; Cui, J.; Liu, X.; Shao, Z.; Yu, B. Nicorandil modulated macrophages activation and polarization via NF-κb signaling pathway. Mol. Immunol. 2017, 88, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.P.; Dong, Y.F.; Liu, W.; Gu, J.; Sun, X.L. Nicorandil inhibits inflammasome activation and Toll-like receptor-4 signal transduction to protect against oxygen-glucose deprivation-induced inflammation in BV-2 cells. CNS Neurosci. Ther. 2014, 20, 147–153. [Google Scholar] [CrossRef]
- Barrett, Y.C.; Wang, J.; Knabb, R.; Mohan, P. Apixaban decreases coagulation activity in patients with acute deep-vein thrombosis. Thromb. Haemost. 2011, 105, 181–189. [Google Scholar] [CrossRef]
- Schuliga, M. The inflammatory actions of coagulant and fibrinolytic proteases in disease. Mediat. Inflamm. 2015, 2015, 437695. [Google Scholar] [CrossRef]
- Morser, J. Thrombomodulin links coagulation to inflammation and immunity. Curr. Drug Targets 2012, 13, 421–431. [Google Scholar] [CrossRef]
- Ito, T.; Maruyama, I. Thrombomodulin: Protectorate God of the vasculature in thrombosis and inflammation. J. Thromb. Haemost. 2011, 9 (Suppl. S1), 168–173. [Google Scholar] [CrossRef]
- El-Ghafar, O.A.M.A.; Helal, G.K.; Abo-Youssef, A.M. Apixaban exhibits anti-arthritic effects by inhibiting activated factor X-mediated JAK2/STAT3 and MAPK phosphorylation pathways. Inflammopharmacology 2020, 28, 1253–1267. [Google Scholar] [CrossRef]
- Bae, J.S.; Yang, L.; Rezaie, A.R. Factor X/Xa elicits protective signaling responses in endothelial cells directly via PAR-2 and indirectly via endothelial protein C receptor-dependent recruitment of PAR-1. J. Biol. Chem. 2010, 285, 34803–34812. [Google Scholar] [CrossRef] [PubMed]
- Hollborn, M.; Kohen, L.; Werschnik, C.; Tietz, L.; Wiedemann, P.; Bringmann, A. Activated blood coagulation factor X (FXa) induces angiogenic growth factor expression in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5930–5939. [Google Scholar] [CrossRef]
- Senden, N.H.; Jeunhomme, T.M.; Heemskerk, J.W.; Wagenvoord, R.; van’t Veer, C.; Hemker, H.C.; Buurman, W.A. Factor Xa induces cytokine production and expression of adhesion molecules by human umbilical vein endothelial cells. J. Immunol. 1998, 161, 4318–4324. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Finotto, S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 2011, 22, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Bromberg, J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J. Clin. Oncol. 2012, 30, 1005–1014. [Google Scholar] [CrossRef]
- Kelso, E.B.; Ferrell, W.R.; Lockhart, J.C.; Elias-Jones, I.; Hembrough, T.; Dunning, L.; Gracie, J.A.; McInnes, I.B. Expression and proinflammatory role of proteinase-activated receptor 2 in rheumatoid synovium: Ex vivo studies using a novel proteinase-activated receptor 2 antagonist. Arthritis Rheum. 2007, 56, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Shnayder, N.A.; Petrova, M.M.; Shesternya, P.A.; Savinova, A.V.; Bochanova, E.N.; Zimnitskaya, O.V.; Pozhilenkova, E.A.; Nasyrova, R.F. Using pharmacogenetics of direct oral anticoagulants to predict changes in their pharmacokinetics and the risk of adverse drug eactions. Biomedicines 2021, 9, 451. [Google Scholar] [CrossRef] [PubMed]
- Al-Zamil, M.K. Efficacy of movalis in marked neurological manifestations of lumbar osteochondrosis. Zh Nevrol. Psikhiatr. Im S.S. Korsakova 2005, 105, 53–54. [Google Scholar] [PubMed]





{kind=link}
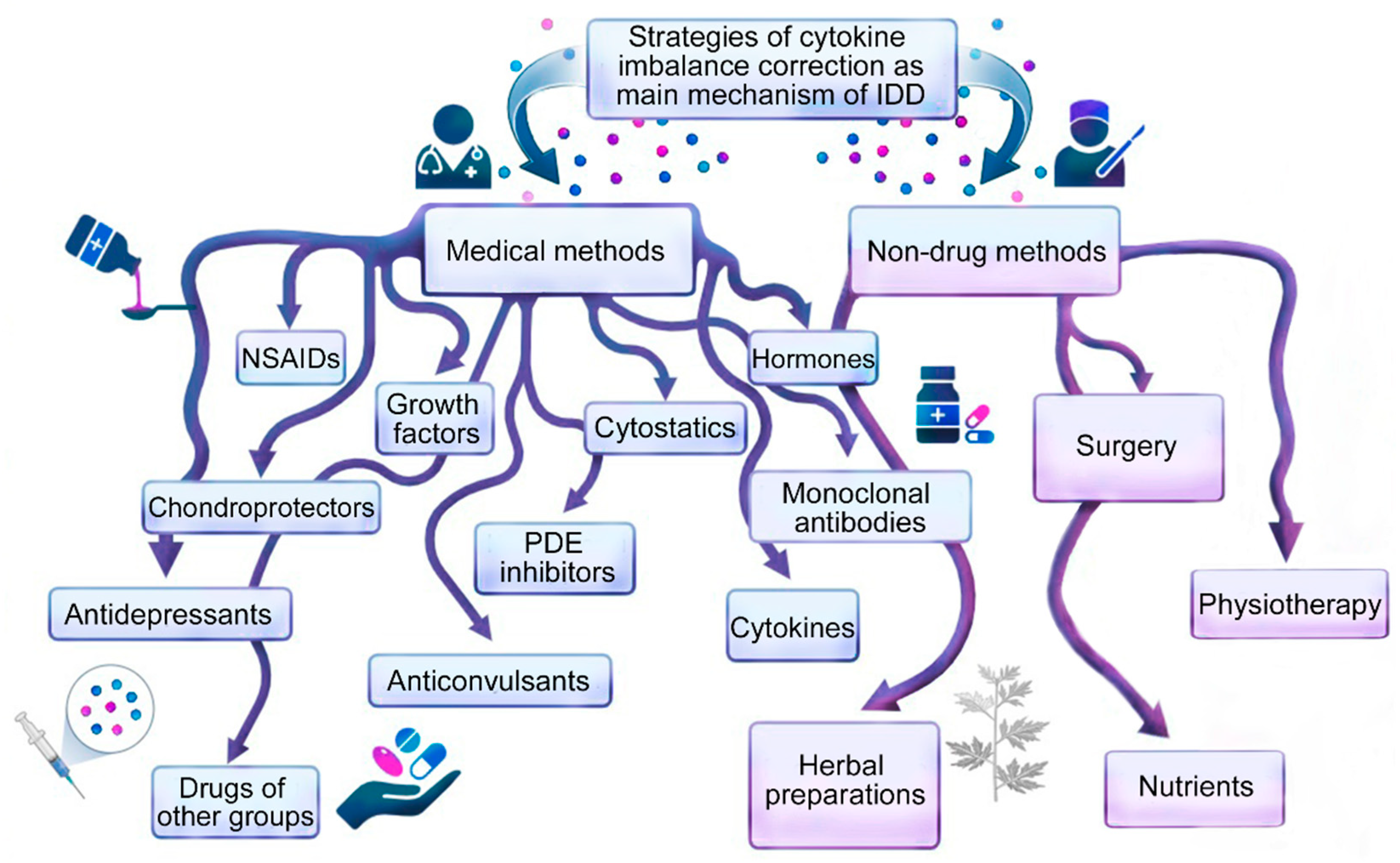{kind=link}
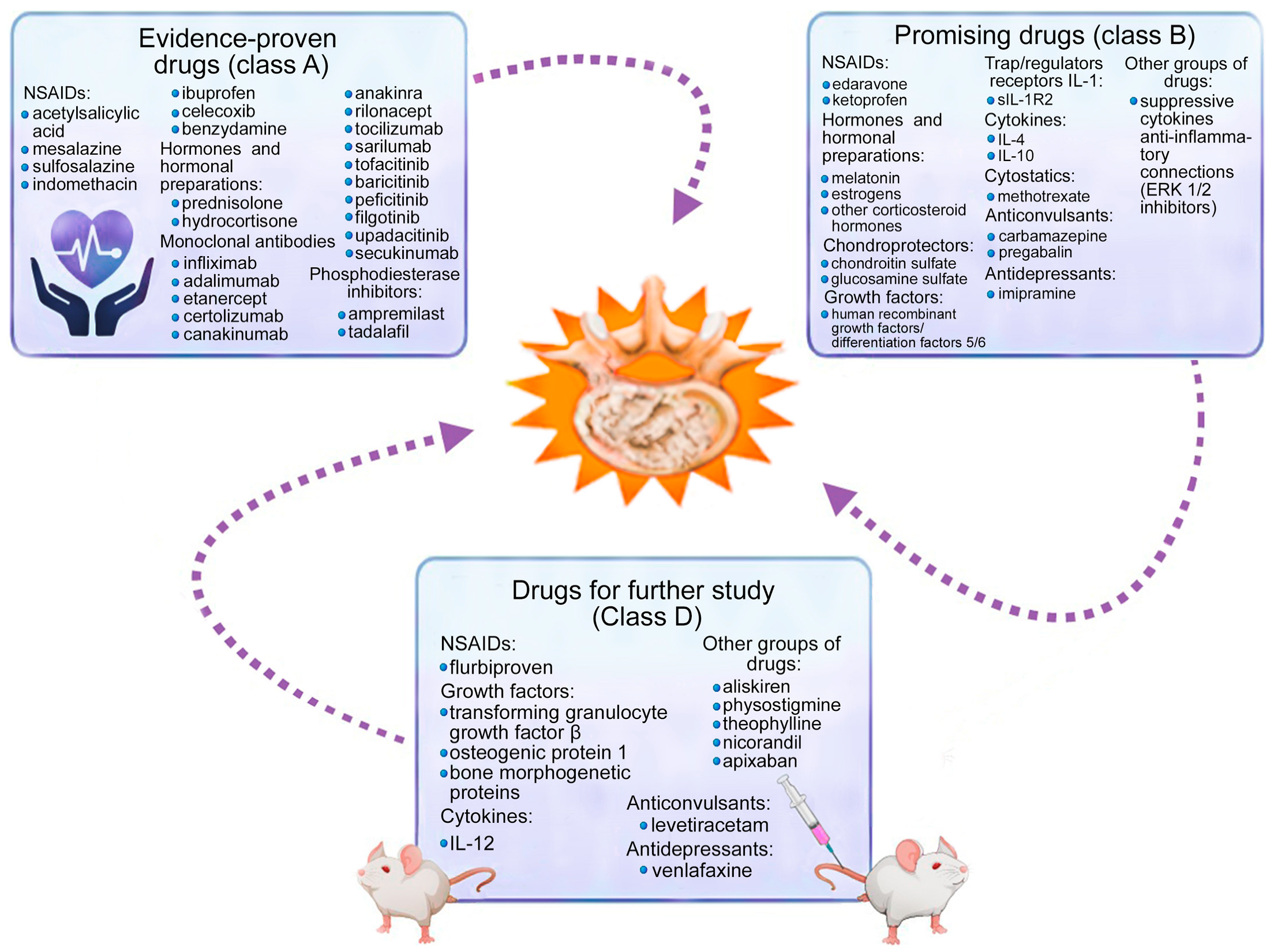{kind=link}
{kind=link}
| Drug | Evidence Class | References |
|---|---|---|
| A. Nonsteroidal Anti-Inflammatory Drugs | ||
| Acetylsalicylic acid | A | [15,16,17,18,19,20,21,22] |
| Mesalazine | A | [23,24] |
| A | [25] | |
| Sulfasalazine | A | [23,28] |
| Edaravone | B | [29] |
| Indomethacin | A | [31,32] |
| B | [31,32] | |
| Ibuprofen | A | [36] |
| Ketoprofen | B | [36,37] |
| Flurbiprofen | D | [36] |
| Celecoxib | A | [41,42] |
| B | [41,42] | |
| Benzydamine | A | [44,45] |
| B | [46] | |
| B. Hormones | ||
| Prednisolone | A | [53,62] |
| Hydrocortisone | A | [63,65,66] |
| B | [64] | |
| Corticosteroids | A | [53,54] |
| B | [55,242] | |
| Melatonin | B | [68,69,70] |
| D | [71,72,73,74] | |
| Estrogens | B | [83,86,94,98] |
| D | [95] | |
| C. Growth Factors | ||
| Bone morphogenetic proteins | D | [105,106,107] |
| Transforming growth factor beta | D | [115,116,118] |
| B | [127] | |
| Insulin-like growth factors | D | [162,163,164,165,166,167,168,283] |
| Osteogenic protein 1 | D | [173,174] |
| Human recombinant growth factors/differentiation 5/6 | B | [194,195,196,197,198,199] |
| D. Chondroprotectors | ||
| Chondroitin sulfate | B | [203,206,207,208,209,211] |
| Glucosamine sulfate | B | [214,215,216,217,219] |
| E. Trap Receptors for Pro-inflammatory Cytokines | ||
| sIL-1R2 | B | [221,265] |
| F. Interleukins Regulating Pro-Inflammatory Cytokines | ||
| Interleukin 4 | B | [53,227,228,229,236] |
| Interleukin 10 | B | [231,232] |
| Interleukin 12 | D | [245] |
| B | [234,246] | |
| G. Monoclonal Antibodies | ||
| Infliximab (antibodies against tumor necrosis factor α) | A | [251] |
| B | [66,252] | |
| Adalimumab (antibodies against tumor necrosis factor α) | A | [38,254] |
| Etanercept (antibodies against tumor necrosis factor α) | A | [258] |
| Certolizumab (antibodies against tumor necrosis factor α) | A | [259] |
| Canakinumab, anakinra, rilonacept (antibodies against interleukin 1 α/β) | A | [247] |
| Tocilizumab (antibodies against interleukin 6) | A | [14] |
| Sarilumab (antibodies against interleukin 6) | A | [278] |
| B | [279,280] | |
| Tofacitinib (antibodies against Janus kinase) | A | [14,283] |
| B | [282] | |
| Baricitinib (antibodies against Janus kinase) | A | [38] |
| B | [287] | |
| Peficitinib (antibodies against Janus kinase) | A | [288] |
| Filgotinib (antibodies against Janus kinase) | A | [289] |
| B | [290] | |
| Upadacitinib (antibodies against Janus kinase) | A | [38] |
| Secukinumab, anakinra, rilonacept (antibodies against interleukin 17) | A | [14,302,306] |
| B | [305] | |
| H. Cytostatics | ||
| Methotrexate | B | [311,312,313,315,316] |
| I. Phosphodiesterase Inhibitors | ||
| Apremilast | A | [319] |
| B | [320,322,323] | |
| Tadalafil | A | [325,328,330] |
| B | [316,327,331] | |
| J. Anticonvulsants | ||
| Carbamazepine | B | [351,355] |
| Gabapentin | B | [368,369,375] |
| A | [357,358,363] | |
| Pregabalin | B | [375] |
| A | [361,363,364] | |
| Levetiracetam | B | [381,382] |
| K. Antidepressants | ||
| Venfalaxine | B | [383,384,385,387,388,389] |
| Imipramine | B | [350,351,353,354,355] |
| L. Other | ||
| Pyridine imidazole | B | [392,393,395,396] |
| Aliskiren | B | [403,404,405,406,407] |
| Physostigmine | B | [408,409,410] |
| Theophylline | B | [412,413,414,415] |
| Nicorandil | B | [415,417,418] |
| Apixaban | B | [424,426,427,428,429] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shnayder, N.A.; Ashkhotov, A.V.; Trefilova, V.V.; Nurgaliev, Z.A.; Novitsky, M.A.; Petrova, M.M.; Narodova, E.A.; Al-Zamil, M.; Chumakova, G.A.; Garganeeva, N.P.; et al. Molecular Basic of Pharmacotherapy of Cytokine Imbalance as a Component of Intervertebral Disc Degeneration Treatment. Int. J. Mol. Sci. 2023, 24, 7692. https://doi.org/10.3390/ijms24097692
Shnayder NA, Ashkhotov AV, Trefilova VV, Nurgaliev ZA, Novitsky MA, Petrova MM, Narodova EA, Al-Zamil M, Chumakova GA, Garganeeva NP, et al. Molecular Basic of Pharmacotherapy of Cytokine Imbalance as a Component of Intervertebral Disc Degeneration Treatment. International Journal of Molecular Sciences. 2023; 24(9):7692. https://doi.org/10.3390/ijms24097692
Chicago/Turabian StyleShnayder, Natalia A., Azamat V. Ashkhotov, Vera V. Trefilova, Zaitun A. Nurgaliev, Maxim A. Novitsky, Marina M. Petrova, Ekaterina A. Narodova, Mustafa Al-Zamil, Galina A. Chumakova, Natalia P. Garganeeva, and et al. 2023. "Molecular Basic of Pharmacotherapy of Cytokine Imbalance as a Component of Intervertebral Disc Degeneration Treatment" International Journal of Molecular Sciences 24, no. 9: 7692. https://doi.org/10.3390/ijms24097692
APA StyleShnayder, N. A., Ashkhotov, A. V., Trefilova, V. V., Nurgaliev, Z. A., Novitsky, M. A., Petrova, M. M., Narodova, E. A., Al-Zamil, M., Chumakova, G. A., Garganeeva, N. P., & Nasyrova, R. F. (2023). Molecular Basic of Pharmacotherapy of Cytokine Imbalance as a Component of Intervertebral Disc Degeneration Treatment. International Journal of Molecular Sciences, 24(9), 7692. https://doi.org/10.3390/ijms24097692