


Monoterpenoid Epoxidiol Ameliorates the Pathological Phenotypes of the Rotenone-Induced Parkinson’s Disease Model by Alleviating Mitochondrial Dysfunction

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Rotenone-Induced Neurotoxicity on the SH-SY5Y Cell Line
2.2. Rotenone-Mediated Depolarization of Isolated Rat Liver Mitochondria
2.3. Bioenergetic Profile of the SH-SY5Y Cell Line under Conditions of Reduced Mitochondrial Function Caused by Rotenone
2.4. In Vivo Study of Motor Activity and Endurance of Mice Simulating Parkinson’s Disease
2.5. In Vivo Study of Hippocampus-Dependent Spatial Memory of Mice Simulating Parkinson’s Disease
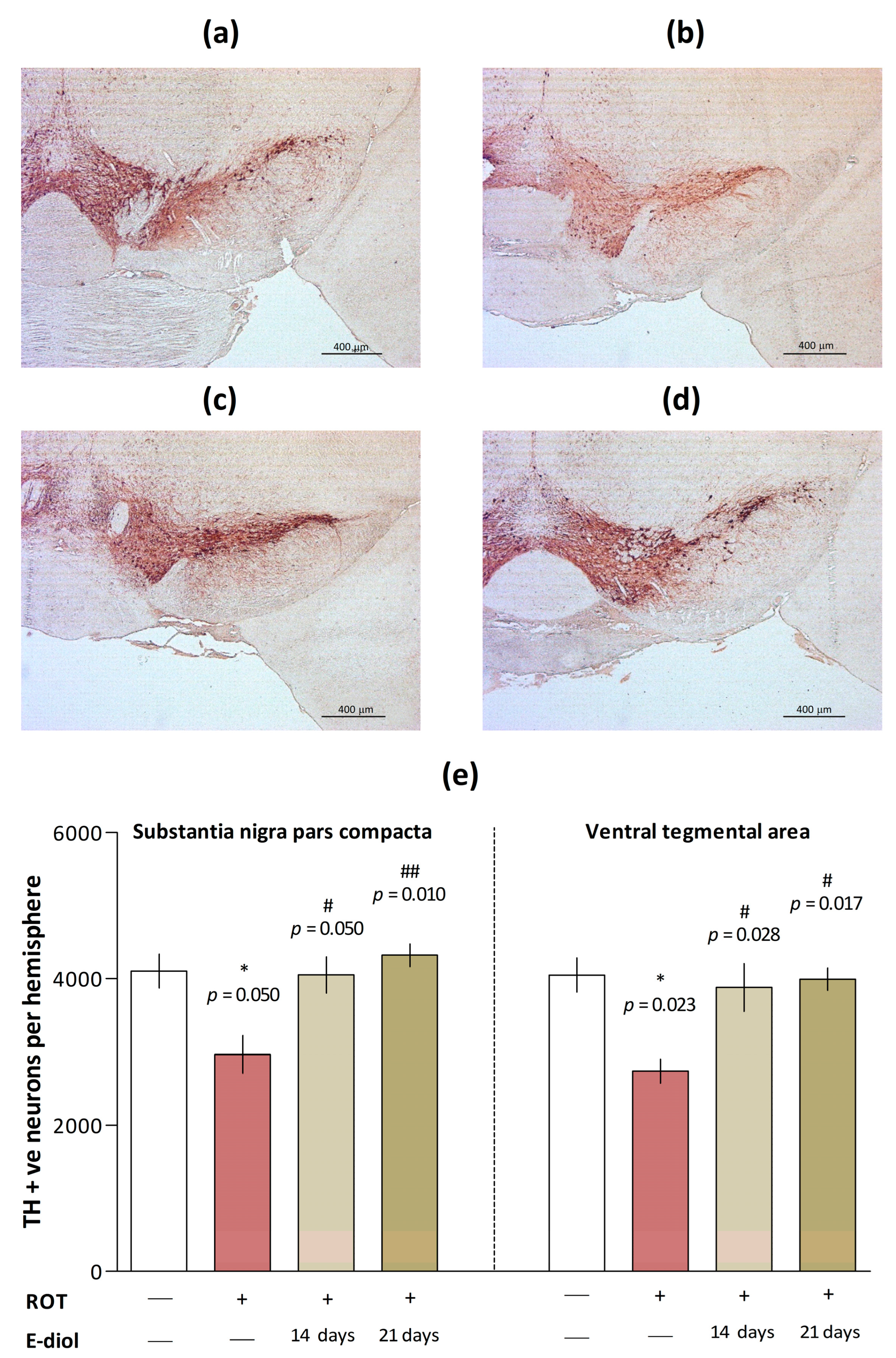
2.6. The Level of Dopaminergic Neurons in Brain Samples of Mice Modeling Parkinson’s Disease
2.7. Dynamics of Mitochondrial Respiration and Oxidative Stress in Brain Samples of Mice Modeling Parkinson’s Disease
3. Discussion
4. Materials and Methods
4.1. Agents
4.2. Preparation of Working Solutions
4.3. Cell Lines and Cultivation
4.4. Cell Viability Assay
4.5. Mitochondrial Membrane Potential Measurements
4.6. Automatic Measurement of Energy Metabolism in Real Time by Registering Oxygen Consumption Rate
4.7. Experimental Animals
- (1)
- Control—a group of mice that were intraperitoneally injected with bidistilled water and a solution of NaCl + 10% DMSO—1 µL/g/day (for 21 days);
- (2)
- ROT—a group of mice receiving intraperitoneal injections of bidistilled water and rotenone (1 mg/kg)—1 µL/g/day (for 21 days);
- (3)
- ROT + E-diol (I)—a group of mice treated intraperitoneally with epoxidiol (15 mg/kg) and NaCl + 10% DMSO solution—1 µL/g/day (for 14 days (starting from the 8th day of the experiment);
- (4)
- ROT + E-diol (II)—a group of mice treated intraperitoneally with epoxidiol (15 mg/kg) and NaCl + 10% DMSO solution—1 µL/g/day (for 21 days (starting from day 1 of the experiment).
4.8. Analysis of the Motor Activity of Mice in the Open Field Test
4.9. Assessment of the Motor Function and Endurance of Animals in the Accelerating Speed Rotarod Test
4.10. Analysis of Hippocampus-Dependent Spatial Memory of Mice in the Y-Maze Test
4.11. Preparation of Histological Sections, Immunohistochemistry and Counting of Neuronal Cells
4.12. Evaluation of Bioenergetic Parameters of the Mitochondrial p2 Fraction
4.13. Study of the Intensity of Lipid Peroxidation in Mouse Brain Homogenates
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cabreira, V.; Massano, J. Parkinson’s Disease: Clinical Review and Update. Acta Med. Port. 2019, 32, 661–670. [Google Scholar] [CrossRef]
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021, 20, 385–397. [Google Scholar] [CrossRef]
- Pleen, J.; Townley, R. Alzheimer’s disease clinical trial update 2019–2021. J. Neurol. 2022, 269, 1038–1051. [Google Scholar] [CrossRef]
- Marino, B.L.B.; de Souza, L.R.; Sousa, K.P.A.; Ferreira, J.V.; Padilha, E.C.; da Silva, C.; Taft, C.A.; Hage-Melim, L.I.S. Parkinson’s Disease: A Review from Pathophysiology to Treatment. Mini Rev. Med. Chem. 2020, 20, 754–767. [Google Scholar] [CrossRef]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol. Neurodegener. 2019, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Latif, S.; Jahangeer, M.; Maknoon Razia, D.; Ashiq, M.; Ghaffar, A.; Akram, M.; El Allam, A.; Bouyahya, A.; Garipova, L.; Ali Shariati, M.; et al. Dopamine in Parkinson’s disease. Clin. Chim. Acta 2021, 522, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Borsche, M.; Pereira, S.L.; Klein, C.; Grunewald, A. Mitochondria and Parkinson’s Disease: Clinical, Molecular, and Translational Aspects. J. Parkinsons Dis. 2021, 11, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial Respiratory Chain Complexes. Subcell Biochem. 2018, 87, 167–227. [Google Scholar]
- Gonzalez-Rodriguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Ohnishi, S.T.; Salerno, J.C. Five decades of research on mitochondrial NADH-quinone oxidoreductase (complex I). Biol. Chem. 2018, 399, 1249–1264. [Google Scholar] [CrossRef]
- Ahmed, S.; Panda, S.R.; Kwatra, M.; Sahu, B.D.; Naidu, V. Perillyl Alcohol Attenuates NLRP3 Inflammasome Activation and Rescues Dopaminergic Neurons in Experimental In Vitro and In Vivo Models of Parkinson’s Disease. ACS Chem. Neurosci. 2022, 13, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Volcho, K.P.; Laev, S.S.; Ashraf, G.M.; Aliev, G.; Salakhutdinov, N.F. Application of Monoterpenoids and their Derivatives for Treatment of Neurodegenerative Disorders. Curr. Med. Chem. 2018, 25, 5327–5346. [Google Scholar] [CrossRef] [PubMed]
- Kotliarova, A.; Podturkina, A.V.; Pavlova, A.V.; Gorina, D.S.; Lastovka, A.V.; Ardashov, O.V.; Rogachev, A.D.; Izyurov, A.E.; Arefieva, A.B.; Kulikov, A.V.; et al. A Newly Identified Monoterpenoid-Based Small Molecule Able to Support the Survival of Primary Cultured Dopamine Neurons and Alleviate MPTP-Induced Toxicity In Vivo. Molecules 2022, 27, 8286. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Azimullah, S.; Parekh, K.A.; Ojha, S.K.; Beiram, R. Effect of citronellol on oxidative stress, neuroinflammation and autophagy pathways in an in vivo model of Parkinson’s disease. Heliyon 2022, 8, e11434. [Google Scholar] [CrossRef]
- Ardashov, O.V.; Pavlova, A.V.; Il’ina, I.V.; Morozova, E.A.; Korchagina, D.V.; Karpova, E.V.; Volcho, K.P.; Tolstikova, T.G.; Salakhutdinov, N.F. Highly potent activity of (1R,2R,6S)-3-methyl-6-(prop-1-en-2-yl)cyclohex-3-ene-1,2-diol in animal models of Parkinson’s disease. J. Med. Chem. 2011, 54, 3866–3874. [Google Scholar] [CrossRef]
- Valdman, E.; Kapitsa, I.; Ivanova, E.; Voronina, T.; Ardashov, O.; Volcho, K.; Khazanov, V.; Salakhutdinov, N. Evolution of anti-parkinsonian activity of monoterpenoid (1R,2R,6S)-3-methyl-6-(prop-1-en-2-yl)cyclohex-3-ene-1,2-diol in various in vivo models. Eur. J. Pharmacol. 2017, 815, 351–363. [Google Scholar] [CrossRef]
- Ardashov, O.V.; Pavlova, A.V.; Mahato, A.K.; Sidorova, Y.; Morozova, E.A.; Korchagina, D.V.; Salnikov, G.E.; Genaev, A.M.; Patrusheva, O.S.; Li-Zhulanov, N.S.; et al. A Novel Small Molecule Supports the Survival of Cultured Dopamine Neurons and May Restore the Dopaminergic Innervation of the Brain in the MPTP Mouse Model of Parkinson’s Disease. ACS Chem. Neurosci. 2019, 10, 4337–4349. [Google Scholar] [CrossRef]
- Dodiya, H.B.; Forsyth, C.B.; Voigt, R.M.; Engen, P.A.; Patel, J.; Shaikh, M.; Green, S.J.; Naqib, A.; Roy, A.; Kordower, J.H.; et al. Chronic stress-induced gut dysfunction exacerbates Parkinson’s disease phenotype and pathology in a rotenone-induced mouse model of Parkinson’s disease. Neurobiol. Dis. 2020, 135, 104352. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Li, C.; Han, B.; Wang, Z.; Meng, X.; Zhang, L.; He, J.; Fu, F. Neuroprotective effects of Danshensu on rotenone-induced Parkinson’s disease models in vitro and in vivo. BMC Complement. Med. Ther. 2020, 20, 20. [Google Scholar] [CrossRef]
- Innos, J.; Hickey, M.A. Using Rotenone to Model Parkinson’s Disease in Mice: A Review of the Role of Pharmacokinetics. Chem. Res. Toxicol. 2021, 34, 1223–1239. [Google Scholar] [CrossRef]
- Thirugnanam, T.; Santhakumar, K. Chemically induced models of Parkinson’s disease. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2022, 252, 109213. [Google Scholar] [CrossRef] [PubMed]
- Nistico, R.; Mehdawy, B.; Piccirilli, S.; Mercuri, N. Paraquat- and rotenone-induced models of Parkinson’s disease. Int. J. Immunopathol. Pharmacol. 2011, 24, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Ruan, Z.; Zhang, D.; Liu, X.; Hou, L.; Wang, Q. Rotenone impairs learning and memory in mice through microglia-mediated blood brain barrier disruption and neuronal apoptosis. Chemosphere 2022, 291, 132982. [Google Scholar] [CrossRef]
- Jing, L.; Hou, L.; Zhang, D.; Li, S.; Ruan, Z.; Zhang, X.; Hong, J.S.; Wang, Q. Microglial Activation Mediates Noradrenergic Locus Coeruleus Neurodegeneration via Complement Receptor 3 in a Rotenone-Induced Parkinson’s Disease Mouse Model. J. Inflamm. Res. 2021, 14, 1341–1356. [Google Scholar] [CrossRef]
- Thong-Asa, W.; Jedsadavitayakol, S.; Jutarattananon, S. Benefits of betanin in rotenone-induced Parkinson mice. Metab. Brain Dis. 2021, 36, 2567–2577. [Google Scholar] [CrossRef] [PubMed]
- Hettiarachchi, P.; Niyangoda, S.S.; Jarosova, R.; Johnson, M.A. Dopamine Release Impairments Accompany Locomotor and Cognitive Deficiencies in Rotenone-Treated Parkinson’s Disease Model Zebrafish. Chem. Res. Toxicol. 2022, 35, 1974–1982. [Google Scholar] [CrossRef]
- Zhao, X.; Kong, D.; Zhou, Q.; Wei, G.; Song, J.; Liang, Y.; Du, G. Baicalein alleviates depression-like behavior in rotenone- induced Parkinson’s disease model in mice through activating the BDNF/TrkB/CREB pathway. Biomed. Pharmacother. 2021, 140, 111556. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.L.; Tsai, C.H.; Lu, M.K.; Liu, H.J.; Chen, Y.C.; Chang, F.C. Interleukin-1beta mediates sleep alteration in rats with rotenone-induced parkinsonism. Sleep 2007, 30, 413–425. [Google Scholar] [CrossRef]
- Babatunde, B.R.; Adeyeye, T.A.; Johnson, V.F.; Shallie, P.D. Rotenone induced olfactory deficit in Parkinson’s disease rat model: The protective role of adenosine A(2A) receptors antagonist. J. Chem. Neuroanat. 2023, 127, 102188. [Google Scholar] [CrossRef]
- Kadigamuwa, C.C.; Mapa, M.S.; Wimalasena, K. Lipophilic Cationic Cyanines Are Potent Complex I Inhibitors and Specific in Vitro Dopaminergic Toxins with Mechanistic Similarities to Both Rotenone and MPP+. Chem. Res. Toxicol. 2016, 29, 1468–1479. [Google Scholar] [CrossRef] [PubMed]
- Maegawa, H.; Niwa, H. Generation of Mitochondrial Toxin Rodent Models of Parkinson’s Disease Using 6-OHDA, MPTP, and Rotenone. Methods Mol. Biol. 2021, 2322, 95–110. [Google Scholar] [PubMed]
- Troshev, D.; Berezhnoy, D.; Kulikova, O.; Abaimov, D.; Muzychuk, O.; Nalobin, D.; Stvolinsky, S.; Fedorova, T. The dynamics of nigrostriatal system damage and neurobehavioral changes in the rotenone rat model of Parkinson’s disease. Brain Res. Bull. 2021, 173, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cimdins, K.; Waugh, H.S.; Chrysostomou, V.; Lopez Sanchez, M.I.G.; Johannsen, V.A.; Cook, M.J.; Crowston, J.G.; Hill, A.F.; Duce, J.A.; Bush, A.I.; et al. Amyloid Precursor Protein Mediates Neuronal Protection from Rotenone Toxicity. Mol. Neurobiol. 2019, 56, 5471–5482. [Google Scholar] [CrossRef]
- Beneventano, M.; Spampinato, S.F.; Merlo, S.; Chisari, M.; Platania, P.; Ragusa, M.; Purrello, M.; Nicoletti, F.; Sortino, M.A. Shedding of Microvesicles from Microglia Contributes to the Effects Induced by Metabotropic Glutamate Receptor 5 Activation on Neuronal Death. Front. Pharmacol. 2017, 8, 812. [Google Scholar] [CrossRef]
- Perevoshchikova, I.V.; Sorochkina, A.I.; Zorov, D.B.; Antonenko, Y.N. Safranine O as a fluorescent probe for mitochondrial membrane potential studied on the single particle level and in suspension. Biochemistry 2009, 74, 663–671. [Google Scholar] [CrossRef]
- Jaber, S.M.; Yadava, N.; Polster, B.M. Mapping mitochondrial respiratory chain deficiencies by respirometry: Beyond the Mito Stress Test. Exp. Neurol. 2020, 328, 113282. [Google Scholar] [CrossRef]
- Nagatsu, T.; Nakashima, A.; Watanabe, H.; Ito, S.; Wakamatsu, K. Neuromelanin in Parkinson’s Disease: Tyrosine Hydroxylase and Tyrosinase. Int. J. Mol. Sci. 2022, 23, 4176. [Google Scholar] [CrossRef]
- Jadiya, P.; Garbincius, J.F.; Elrod, J.W. Reappraisal of metabolic dysfunction in neurodegeneration: Focus on mitochondrial function and calcium signaling. Acta Neuropathol. Commun. 2021, 9, 124. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Wright, R. Mitochondrial dysfunction and Parkinson’s disease. Nat. Neurosci. 2022, 25, 2. [Google Scholar] [CrossRef]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 216–231. [Google Scholar] [CrossRef]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Delgado, A.; Ortiz, G.G.; Delgado-Lara, D.L.; Gonzalez-Usigli, H.A.; Gonzalez-Ortiz, L.J.; Cid-Hernandez, M.; Cruz-Serrano, J.A.; Pacheco-Moises, F.P. Effect of Melatonin Administration on Mitochondrial Activity and Oxidative Stress Markers in Patients with Parkinson’s Disease. Oxid. Med. Cell. Longev. 2021, 2021, 5577541. [Google Scholar] [CrossRef] [PubMed]
- Pienaar, I.S.; Elson, J.L.; Racca, C.; Nelson, G.; Turnbull, D.M.; Morris, C.M. Mitochondrial abnormality associates with type-specific neuronal loss and cell morphology changes in the pedunculopontine nucleus in Parkinson disease. Am. J. Pathol. 2013, 183, 1826–1840. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef]
- Li, H.; Ham, A.; Ma, T.C.; Kuo, S.H.; Kanter, E.; Kim, D.; Ko, H.S.; Quan, Y.; Sardi, S.P.; Li, A.; et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 2019, 15, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Lou, D.; Charli, A.; Kong, D.; Jin, H.; Zenitsky, G.; Anantharam, V.; Kanthasamy, A.; Wang, Z.; Kanthasamy, A.G. Mitochondrial dysfunction-induced H3K27 hyperacetylation perturbs enhancers in Parkinson’s disease. JCI Insight 2021, 6, e138088. [Google Scholar] [CrossRef]
- Toomey, C.E.; Heywood, W.E.; Evans, J.R.; Lachica, J.; Pressey, S.N.; Foti, S.C.; Al Shahrani, M.; D’Sa, K.; Hargreaves, I.P.; Heales, S.; et al. Mitochondrial dysfunction is a key pathological driver of early stage Parkinson’s. Acta Neuropathol. Commun. 2022, 10, 134. [Google Scholar] [CrossRef] [PubMed]
- Nataraj, J.; Manivasagam, T.; Justin Thenmozhi, A.; Essa, M.M. Neuroprotective effect of asiatic acid on rotenone-induced mitochondrial dysfunction and oxidative stress-mediated apoptosis in differentiated SH-SYS5Y cells. Nutr. Neurosci. 2017, 20, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.H.; Barron, A.M.; Abdullah, J.M. Mitoprotective Effects of Centella asiatica (L.) Urb.: Anti-Inflammatory and Neuroprotective Opportunities in Neurodegenerative Disease. Front. Pharmacol. 2021, 12, 687935. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, B.; Yang, J.; Chen, Z.; Li, Z.; Zhang, N.; Li, H.; Shen, L. Comparison of the effect of rotenone and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine on inducing chronic Parkinson’s disease in mouse models. Mol. Med. Rep. 2022, 25, 91. [Google Scholar] [CrossRef]
- Scheid, S.; Lejarre, A.; Wollborn, J.; Buerkle, H.; Goebel, U.; Ulbrich, F. Argon preconditioning protects neuronal cells with a Toll-like receptor-mediated effect. Neural Regen. Res. 2023, 18, 1371–1377. [Google Scholar]
- Heo, G.; Sun, M.H.; Jiang, W.J.; Li, X.H.; Lee, S.H.; Guo, J.; Zhou, D.; Cui, X.S. Rotenone causes mitochondrial dysfunction and prevents maturation in porcine oocytes. PLoS ONE 2022, 17, e0277477. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Gangras, P.; Gelfanova, V.; Williams, G.D.; Handelman, S.K.; Smith, R.M.; Debets, M.F. Investigating SH-SY5Y Neuroblastoma Cell Surfaceome as a Model for Neuronal-Targeted Novel Therapeutic Modalities. Int. J. Mol. Sci. 2022, 23, 15062. [Google Scholar] [CrossRef]
- Bernheimer, H.; Birkmayer, W.; Hornykiewicz, O.; Jellinger, K.; Seitelberger, F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 1973, 20, 415–455. [Google Scholar] [CrossRef]
- Ma, K.Y.; Fokkens, M.R.; Reggiori, F.; Mari, M.; Verbeek, D.S. Parkinson’s disease-associated VPS35 mutant reduces mitochondrial membrane potential and impairs PINK1/Parkin-mediated mitophagy. Transl. Neurodegener. 2021, 10, 19. [Google Scholar] [CrossRef]
- Li, J.; Lai, M.; Zhang, X.; Li, Z.; Yang, D.; Zhao, M.; Wang, D.; Sun, Z.; Ehsan, S.; Li, W.; et al. PINK1-parkin-mediated neuronal mitophagy deficiency in prion disease. Cell Death Dis. 2022, 13, 162. [Google Scholar] [CrossRef]
- Ramkumar, M.; Rajasankar, S.; Gobi, V.V.; Dhanalakshmi, C.; Manivasagam, T.; Justin Thenmozhi, A.; Essa, M.M.; Kalandar, A.; Chidambaram, R. Neuroprotective effect of Demethoxycurcumin, a natural derivative of Curcumin on rotenone induced neurotoxicity in SH-SY 5Y Neuroblastoma cells. BMC Complement. Altern. Med. 2017, 17, 217. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.; Zickermann, V. Binding of Natural Inhibitors to Respiratory Complex I. Pharmaceuticals 2022, 15, 1088. [Google Scholar] [CrossRef] [PubMed]
- Valdez, L.B.; Zaobornyj, T.; Bandez, M.J.; Lopez-Cepero, J.M.; Boveris, A.; Navarro, A. Complex I syndrome in striatum and frontal cortex in a rat model of Parkinson disease. Free Radic. Biol. Med. 2019, 135, 274–282. [Google Scholar] [CrossRef]
- Nguyen, M.; Wong, Y.C.; Ysselstein, D.; Severino, A.; Krainc, D. Synaptic, Mitochondrial, and Lysosomal Dysfunction in Parkinson’s Disease. Trends Neurosci. 2019, 42, 140–149. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Romano, R.; Coelho-Junior, H.J.; Bucci, C.; Marzetti, E. Mitochondrial Dysfunction, Protein Misfolding and Neuroinflammation in Parkinson’s Disease: Roads to Biomarker Discovery. Biomolecules 2021, 11, 1508. [Google Scholar] [CrossRef]
- Juarez-Flores, D.L.; Ezquerra, M.; Gonzalez-Casacuberta, I.; Ormazabal, A.; Moren, C.; Tolosa, E.; Fucho, R.; Guitart-Mampel, M.; Casado, M.; Valldeoriola, F.; et al. Disrupted Mitochondrial and Metabolic Plasticity Underlie Comorbidity between Age-Related and Degenerative Disorders as Parkinson Disease and Type 2 Diabetes Mellitus. Antioxidants 2020, 9, 1063. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Casacuberta, I.; Juarez-Flores, D.L.; Ezquerra, M.; Fucho, R.; Catalan-Garcia, M.; Guitart-Mampel, M.; Tobias, E.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Tolosa, E.; et al. Mitochondrial and autophagic alterations in skin fibroblasts from Parkinson disease patients with Parkin mutations. Aging 2019, 11, 3750–3767. [Google Scholar] [CrossRef] [PubMed]
- Formosa, L.E.; Dibley, M.G.; Stroud, D.A.; Ryan, M.T. Building a complex complex: Assembly of mitochondrial respiratory chain complex I. Semin. Cell Dev. Biol. 2018, 76, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Curtabbi, A.; Enriquez, J.A. The ins and outs of the flavin mononucleotide cofactor of respiratory complex I. IUBMB Life 2022, 74, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Ten, V.; Galkin, A. Mechanism of mitochondrial complex I damage in brain ischemia/reperfusion injury. A hypothesis. Mol. Cell. Neurosci. 2019, 100, 103408. [Google Scholar] [CrossRef] [PubMed]
- Murai, M. Exploring the binding pocket of quinone/inhibitors in mitochondrial respiratory complex I by chemical biology approaches. Biosci. Biotechnol. Biochem. 2020, 84, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Feng, X.; Huang, H.; Liu, L.; Qiao, L.; Zhang, B.; Yu, W. The effects of rotenone-induced toxicity via the NF-kappaB-iNOS pathway in rat liver. Toxicol. Mech. Methods 2017, 27, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Kaur, J.; Rakshe, S.; Sharma, N.; Khunt, D.; Khairnar, A. Intranasal Exposure to Low-Dose Rotenone Induced Alpha-Synuclein Accumulation and Parkinson’s Like Symptoms Without Loss of Dopaminergic Neurons. Neurotox. Res. 2022, 40, 215–229. [Google Scholar] [CrossRef]
- Miyazaki, I.; Isooka, N.; Imafuku, F.; Sun, J.; Kikuoka, R.; Furukawa, C.; Asanuma, M. Chronic Systemic Exposure to Low-Dose Rotenone Induced Central and Peripheral Neuropathology and Motor Deficits in Mice: Reproducible Animal Model of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 3254. [Google Scholar] [CrossRef] [PubMed]
- Borland, M.K.; Trimmer, P.A.; Rubinstein, J.D.; Keeney, P.M.; Mohanakumar, K.; Liu, L.; Bennett, J.P., Jr. Chronic, low-dose rotenone reproduces Lewy neurites found in early stages of Parkinson’s disease, reduces mitochondrial movement and slowly kills differentiated SH-SY5Y neural cells. Mol. Neurodegener. 2008, 3, 21. [Google Scholar] [CrossRef]
- Opara, J.; Malecki, A.; Malecka, E.; Socha, T. Motor assessment in Parkinson;s disease. Ann. Agric. Environ. Med. 2017, 24, 411–415. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Aarsland, D.; Batzu, L.; Halliday, G.M.; Geurtsen, G.J.; Ballard, C.; Ray Chaudhuri, K.; Weintraub, D. Parkinson disease-associated cognitive impairment. Nat. Rev. Dis. Primers 2021, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.G.; Sieg, E. Cognitive Impairment and Dementia in Parkinson Disease. Clin. Geriatr. Med. 2020, 36, 365–377. [Google Scholar] [CrossRef]
- Brandao, P.R.P.; Munhoz, R.P.; Grippe, T.C.; Cardoso, F.E.C.; de Almeida, E.C.B.M.; Titze-de-Almeida, R.; Tomaz, C.; Tavares, M.C.H. Cognitive impairment in Parkinson’s disease: A clinical and pathophysiological overview. J. Neurol. Sci. 2020, 419, 117177. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Hwang, J.J.; Poston, K.L. Episodic recognition memory and the hippocampus in Parkinson’s disease: A review. Cortex 2019, 113, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Villar-Conde, S.; Astillero-Lopez, V.; Gonzalez-Rodriguez, M.; Villanueva-Anguita, P.; Saiz-Sanchez, D.; Martinez-Marcos, A.; Flores-Cuadrado, A.; Ubeda-Banon, I. The Human Hippocampus in Parkinson’s Disease: An Integrative Stereological and Proteomic Study. J. Parkinsons Dis. 2021, 11, 1345–1365. [Google Scholar] [CrossRef]
- Smith, D.T.; Casteau, S.; Archibald, N. Spatial attention and spatial short term memory in PSP and Parkinson’s disease. Cortex 2021, 137, 49–60. [Google Scholar] [CrossRef]
- Salazar, R.D.; Moon, K.L.M.; Neargarder, S.; Cronin-Golomb, A. Spatial judgment in Parkinson’s disease: Contributions of attentional and executive dysfunction. Behav. Neurosci. 2019, 133, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J. Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, K.; Evans, R.C.; Lopez, E.; Sun, L.; Kumar, M.; Ding, J.; Khaliq, Z.M.; Cai, H. Function and Regulation of ALDH1A1-Positive Nigrostriatal Dopaminergic Neurons in Motor Control and Parkinson’s Disease. Front. Neural Circuits 2021, 15, 644776. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Chen, J. Salidroside Protects Dopaminergic Neurons by Enhancing PINK1/Parkin-Mediated Mitophagy. Oxid. Med. Cell. Longev. 2019, 2019, 9341018. [Google Scholar] [CrossRef]
- Han, K.; Jin, X.; Guo, X.; Cao, G.; Tian, S.; Song, Y.; Zuo, Y.; Yu, P.; Gao, G.; Chang, Y.Z. Nrf2 knockout altered brain iron deposition and mitigated age-related motor dysfunction in aging mice. Free Radic. Biol. Med. 2021, 162, 592–602. [Google Scholar] [CrossRef]
- Ahn, E.H.; Lei, K.; Kang, S.S.; Wang, Z.H.; Liu, X.; Hong, W.; Wang, Y.T.; Edgington-Mitchell, L.E.; Jin, L.; Ye, K. Mitochondrial dysfunction triggers the pathogenesis of Parkinson’s disease in neuronal C/EBPbeta transgenic mice. Mol. Psychiatry 2021, 26, 7838–7850. [Google Scholar] [CrossRef]
- Theofanous, T.; Kourti, M. Abrogating Oxidative Stress as a Therapeutic Strategy Against Parkinson’s Disease: A Mini Review of the Recent Advances on Natural Therapeutic Antioxidant and Neuroprotective Agents. Med. Chem. 2022, 18, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Biosa, A.; Arduini, I.; Soriano, M.E.; Giorgio, V.; Bernardi, P.; Bisaglia, M.; Bubacco, L. Dopamine Oxidation Products as Mitochondrial Endotoxins, a Potential Molecular Mechanism for Preferential Neurodegeneration in Parkinson’s Disease. ACS Chem. Neurosci. 2018, 9, 2849–2858. [Google Scholar] [CrossRef]
- Tretter, L.; Sipos, I.; Adam-Vizi, V. Initiation of neuronal damage by complex I deficiency and oxidative stress in Parkinson’s disease. Neurochem. Res. 2004, 29, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Han, B.; Zhao, Y.; Li, G.; Wang, T.; He, J.; Du, W.; Cao, X.; Gan, J.; Wang, Z.; et al. Rosmarinic Acid Attenuates Rotenone-Induced Neurotoxicity in SH-SY5Y Parkinson’s Disease Cell Model through Abl Inhibition. Nutrients 2022, 14, 3508. [Google Scholar] [CrossRef]
- Pulliam, D.A.; Deepa, S.S.; Liu, Y.; Hill, S.; Lin, A.L.; Bhattacharya, A.; Shi, Y.; Sloane, L.; Viscomi, C.; Zeviani, M.; et al. Complex IV-deficient Surf1(−/−) mice initiate mitochondrial stress responses. Biochem. J. 2014, 462, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Connor-Robson, N.; Peters, O.M.; Millership, S.; Ninkina, N.; Buchman, V.L. Combinational losses of synucleins reveal their differential requirements for compensating age-dependent alterations in motor behavior and dopamine metabolism. Neurobiol. Aging 2016, 46, 107–112. [Google Scholar] [CrossRef]
- Goloborshcheva, V.V.; Chaprov, K.D.; Teterina, E.V.; Ovchinnikov, R.; Buchman, V.L. Reduced complement of dopaminergic neurons in the substantia nigra pars compacta of mice with a constitutive “low footprint” genetic knockout of alpha-synuclein. Mol. Brain 2020, 13, 75. [Google Scholar] [CrossRef] [PubMed]
- Ninkina, N.; Tarasova, T.V.; Chaprov, K.D.; Roman, A.Y.; Kukharsky, M.S.; Kolik, L.G.; Ovchinnikov, R.; Ustyugov, A.A.; Durnev, A.D.; Buchman, V.L. Alterations in the nigrostriatal system following conditional inactivation of alpha-synuclein in neurons of adult and aging mice. Neurobiol. Aging 2020, 91, 76–87. [Google Scholar] [CrossRef]
- Abercrombie, M. Estimation of nuclear population from microtome sections. Anat. Rec. 1946, 94, 239–247. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aleksandrova, Y.; Chaprov, K.; Podturkina, A.; Ardashov, O.; Yandulova, E.; Volcho, K.; Salakhutdinov, N.; Neganova, M. Monoterpenoid Epoxidiol Ameliorates the Pathological Phenotypes of the Rotenone-Induced Parkinson’s Disease Model by Alleviating Mitochondrial Dysfunction. Int. J. Mol. Sci. 2023, 24, 5842. https://doi.org/10.3390/ijms24065842
Aleksandrova Y, Chaprov K, Podturkina A, Ardashov O, Yandulova E, Volcho K, Salakhutdinov N, Neganova M. Monoterpenoid Epoxidiol Ameliorates the Pathological Phenotypes of the Rotenone-Induced Parkinson’s Disease Model by Alleviating Mitochondrial Dysfunction. International Journal of Molecular Sciences. 2023; 24(6):5842. https://doi.org/10.3390/ijms24065842
Chicago/Turabian StyleAleksandrova, Yulia, Kirill Chaprov, Alexandra Podturkina, Oleg Ardashov, Ekaterina Yandulova, Konstantin Volcho, Nariman Salakhutdinov, and Margarita Neganova. 2023. "Monoterpenoid Epoxidiol Ameliorates the Pathological Phenotypes of the Rotenone-Induced Parkinson’s Disease Model by Alleviating Mitochondrial Dysfunction" International Journal of Molecular Sciences 24, no. 6: 5842. https://doi.org/10.3390/ijms24065842
APA StyleAleksandrova, Y., Chaprov, K., Podturkina, A., Ardashov, O., Yandulova, E., Volcho, K., Salakhutdinov, N., & Neganova, M. (2023). Monoterpenoid Epoxidiol Ameliorates the Pathological Phenotypes of the Rotenone-Induced Parkinson’s Disease Model by Alleviating Mitochondrial Dysfunction. International Journal of Molecular Sciences, 24(6), 5842. https://doi.org/10.3390/ijms24065842