
HBV Infection and Host Interactions: The Role in Viral Persistence and Oncogenesis
 , , ,
, , ,  , , , and
, , , and
Abstract
1. Introduction
2. From Infection to Viral Persistence
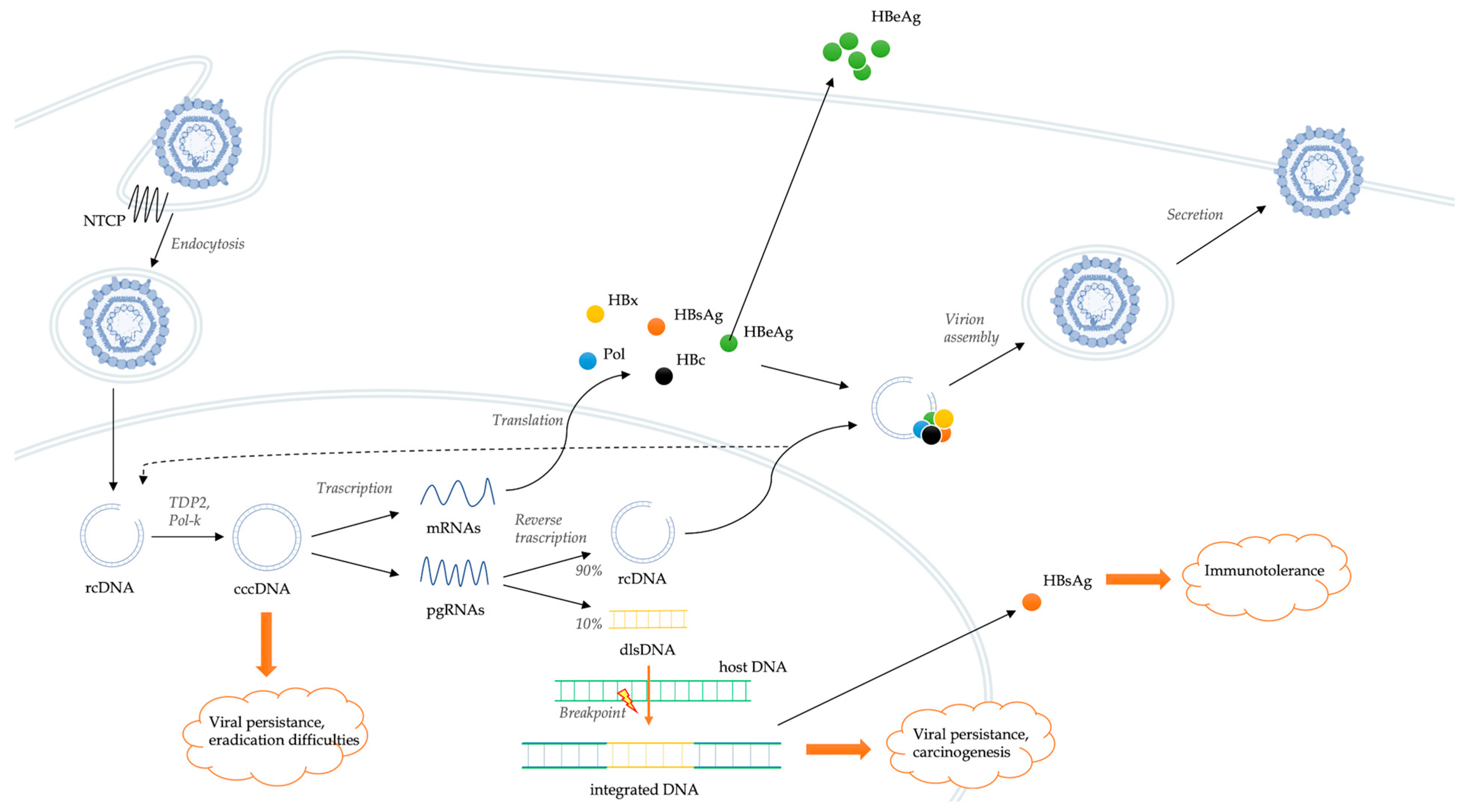
2.1. HBV Replication in the Host Cells
2.2. Integration of Viral Genome
2.3. HBV and Host Immune System
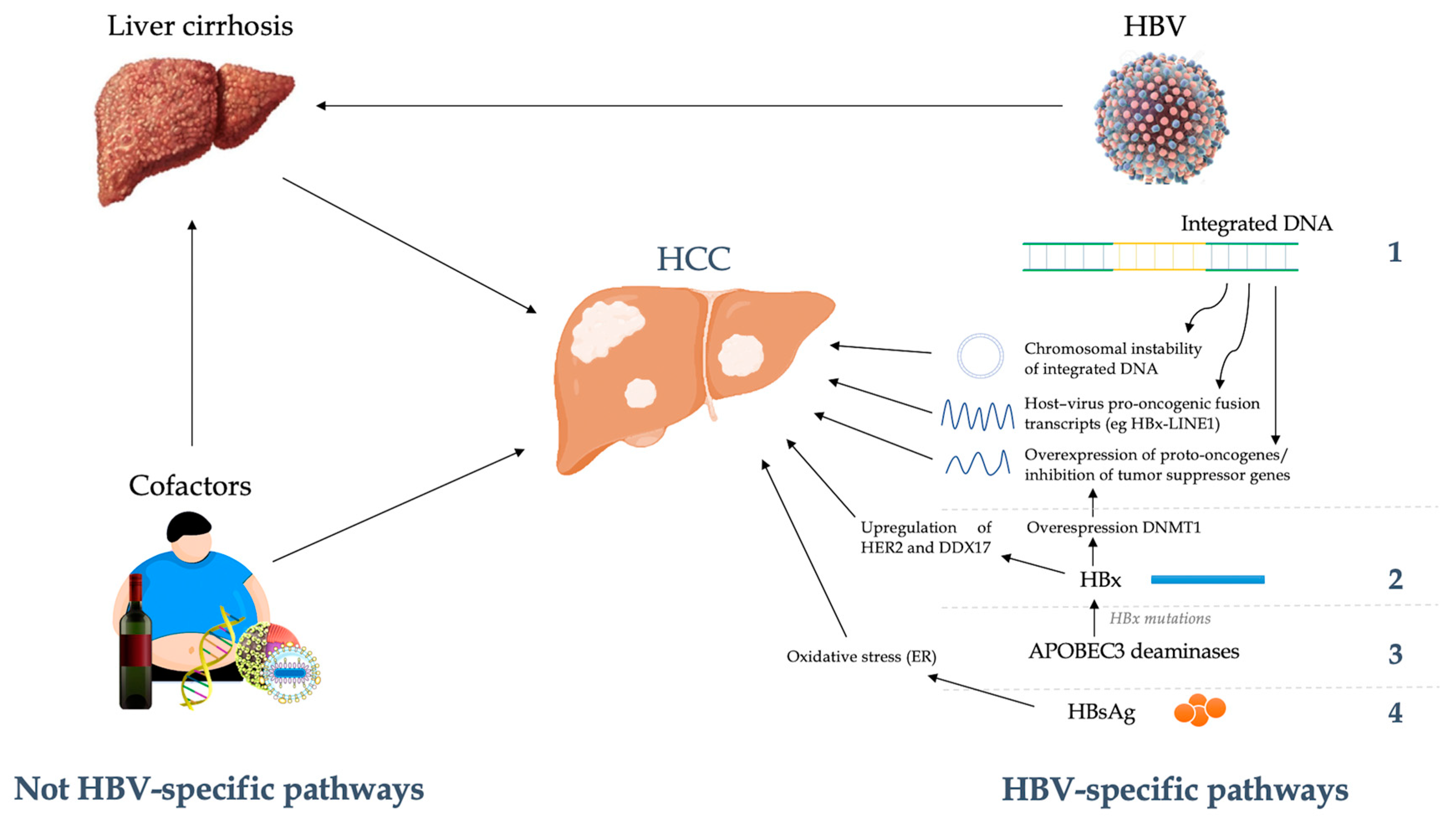
3. HBV-Host Interactions in Oncogenesis
4. Therapeutic Implications

{kind=link}
{kind=link}
| Drug Class | Mechanism of Action | Molecule | Ref. | First Author | Year | Study Typology | Sample Size | Results |
|---|---|---|---|---|---|---|---|---|
| NTCP-inhibitor | Prevent virus entry into the cell | Myrcludex-B | [133] | Bogomolov | 2016 | Phase Ib/IIa trial | An amout of 24 patients (HBV/HDV) | Myrcludex-B plus PegIFNα-2a significantly reduce HBV DNA compared to monotherapy. HBsAg levels remained unchanged. |
| Myrcludex-B | [134] | Wedemeyer | 2019 | Phase II trial | An amount of 60 patients (HBV/HDV) | Myrcludex-B 2 mg plus PegIFNα-2a induced HBsAg loss in a substantial proportion of patients | ||
| Anti-HBsAg monoclonal Abs | Direct inhibition of HBsAg | E6F6 | [135] | Zhang | 2016 | Pre-clinical trial | HBV-transgenic mice | Single-dose of E6F6 suppressed HBsAg and HBV DNA levels for several weeks |
| Immuno-modulators | Stimulation of adaptive immune response | IL-2 | [136] | Tilg | 1993 | Phase I + Phase II trial | An amount of 10 patients | No efficacy on HBeAg clearance |
| IL-12 | [137] | Carreño | 2000 | Phase I/II trial | An amount of 46 patients | IL-12 reduces significantly HBV DNA levels | ||
| Thymosine-alpha-1 | [138] | Iino | 2005 | 72-week multicentre, randomized trial | An amount of 316 patients | Thymosine-alpha-1 therapy is associated with a biochemical and virological response (HBV DNA and HBeAg clearance) in a minority of patients | ||
| [139] | You | 2006 | RCT | An amount of 62 HBeAg+ patients | Thymosine-alpha-1 induce more sustained ALT normalization and HBV DNA and HBeAg loss than IFN-alpha (48% vs. 27%, respectively) | |||
| Levamisole | [140] | Ruiz-Moreno | 1993 | RCT | An amount of 38 children | No significant differences (biochemical and virological) were observed between Levamisole + IFN and IFN alone groups | ||
| Therapeutic vaccines | NASVAC | [149] | Al Mahtab | 2018 | Phase III RCT | An amount of 160 patients | NASVAC induced a greater HBV DNA reduction and more frequent clearance rate of HBeAg compared to Peg-IFN | |
| NASVAC | [151] | Akbar | 2021 | Phase III RCT | An amount of 160 patients | NASVAC was capable of reducing HBV DNA and normalizing ALT 3 years after the EOT. No reported impact on HBsAg | ||
| Anti-PD-1 | Nivolumab | [153] | Gane | 2019 | Phase Ib trial | An amount of 24 patients | Nivolumab (with or without HBV therapeutic vaccine) was well-tolerated and led to HBsAg decline in most patients and sustained HBsAg loss in 1 patient | |
| Enrichment of CD8+ T cells with CARs | N/A | [154] | Krebs | 2013 | Pre-clinical trial | HBV transgenic mice | Engineering of T cells were able to effectively control viral replication | |
| T cells engineered with HBV-specific TCR | N/A | [155] | Kah | 2017 | Pre-clinical trial | HBV transgenic mice | T cells engineered to express a HBV-specific T cell receptor leads to a progressive reduction of viraemia in absence of persistent organ damage | |
| TLR-7 agonism | Vesatolimod | [157] | Janssen | 2018 | Phase II RCT | An amount of 162 patients | Vesatolimod plus antiviral therapy did not demonstrate significantly higher HBsAg declines than placebo | |
| STING agonism | N/A | [159] | Li | 2022 | Pre-clinical trial | HBV mouse model | The activation of STING signaling could inhibit HBV replication and alleviate HBV-induced liver fibrosis | |
| cccDNA silencers | NQO1 inhibitors | Dicoumarol | [160] | Cheng | 2021 | Pre-clinical trial | Humanized liver mouse model | Potent antiviral activity against HBV DNA, HBsAg, HBV RNAs and HBc protein |
| Rnase H inhibitors | 3 compounds | [161] | Chauhan | 2021 | Pre-clinical trial | HBV-infected HepG2-NTCP cells | Rnase H inhibitors effectively suppresses cccDNA formation, as well as HBV RNA, HBV DNA and HbsAg secretion | |
| Genetic editing technologies | TALENs | N/A | [164] | Bloom | 2013 | Pre-clinical trial | HepG2.2.15 cells | TALENs were able to induce disruption of HBV cccDNA |
| CRISPR/Cas9 system | N/A | [171] | Yang | 2020 | Pre-clinical trial | In vitro HBV infection system | CRISPR/Cas9 system were able to modify episomal cccDNA and suppress viral gene expression |
5. Open Issues and Perspectives in HBV Infection Therapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Polaris Observatory Collaborators. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar] [CrossRef] [PubMed]
- Nevola, R.; Messina, V.; Marrone, A.; Coppola, N.; Rescigno, C.; Esposito, V.; Sangiovanni, V.; Claar, E.; Pisaturo, M.; Fusco, F.M.; et al. Epidemiology of HCV and HBV in a High Endemic Area of Southern Italy: Opportunities from the COVID-19 Pandemic-Standardized National Screening or One Tailored to Local Epidemiology? Biology 2022, 11, 609. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.S.; Nguyen, M.H. Epidemiology of hepatitis B and the role of vaccination. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 239–247. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Tsai, K.N.; Ou, J.J. Pathogenicity and virulence of Hepatitis B virus. Virulence 2022, 13, 258–296. [Google Scholar] [CrossRef] [PubMed]
- Mazzaro, C.; Adinolfi, L.E.; Pozzato, G.; Nevola, R.; Zanier, A.; Serraino, D.; Andreone, P.; Fenoglio, R.; Sciascia, S.; Gattei, V.; et al. Extrahepatic Manifestations of Chronic HBV Infection and the Role of Antiviral Therapy. J. Clin. Med. 2022, 11, 6247. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Han, Q.; Zhao, H.; Zhang, J. The Mechanisms of HBV-Induced Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2021, 8, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Ascione, A.; Fontanella, L.; Imparato, M.; Rinaldi, L.; De Luca, M. Mortality from cirrhosis and hepatocellular carcinoma in Western Europe over the last 40 years. Liver Int. 2017, 37, 1193–1201. [Google Scholar] [CrossRef]
- Tong, S.; Revill, P. Overview of hepatitis B viral replication and genetic variability. J. Hepatol. 2016, 64, S4–S16. [Google Scholar] [CrossRef]
- Nguyen, M.H.; Wong, G.; Gane, E.; Kao, J.H.; Dusheiko, G. Hepatitis B Virus: Advances in Prevention, Diagnosis, and Therapy. Clin. Microbiol. Rev. 2020, 33, e00046-19. [Google Scholar] [CrossRef]
- Xu, R.; Hu, P.; Li, Y.; Tian, A.; Li, J.; Zhu, C. Advances in HBV infection and replication systems in vitro. Virol. J. 2021, 18, 105. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, J.; Li, T. Regulation of the HBV Entry Receptor NTCP and its Potential in Hepatitis B Treatment. Front. Mol. Biosci. 2022, 9, 879817. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Blondot, M.L.; Bruss, V.; Kann, M. Intracellular transport and egress of hepatitis B virus. J. Hepatol. 2016, 64, S49–S59. [Google Scholar] [CrossRef] [PubMed]
- Koniger, C.; Wingert, I.; Marsmann, M.; Rosler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA polymerase kappa is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef]
- Teng, Y.; Xu, Z.; Zhao, K.; Zhong, Y.; Wang, J.; Zhao, L.; Zheng, Z.; Hou, W.; Zhu, C.; Chen, X.; et al. Novel function of SART1 in HNF4α transcriptional regulation contributes to its antiviral role during HBV infection. J. Hepatol. 2021, 75, 1072–1082. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef]
- Hu, J.; Liu, K. Complete and incomplete hepatitis B virus particles: Formation, function, and application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef]
- Tu, T.; Zhang, H.; Urban, S. Hepatitis B Virus DNA Integration: In Vitro Models for Investigating Viral Pathogenesis and Persistence. Viruses 2021, 13, 180. [Google Scholar] [CrossRef]
- Svicher, V.; Salpini, R.; Piermatteo, L.; Carioti, L.; Battisti, A.; Colagrossi, L.; Scutari, R.; Surdo, M.; Cacciafesta, V.; Nuccitelli, A.; et al. Whole exome HBV DNA integration is independent of the intrahepatic HBV reservoir in HBeAg-negative chronic hepatitis B. Gut 2021, 70, 2337–2348. [Google Scholar] [CrossRef]
- Pollicino, T.; Caminiti, G. HBV-Integration Studies in the Clinic: Role in the Natural History of Infection. Viruses 2021, 13, 368. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Mason, W.S.; Clouston, A.D.; Shackel, N.A.; McCaughan, G.W.; Yeh, M.M.; Schiff, E.R.; Ruszkiewicz, A.R.; Chen, J.W.; Harley, H.A.; et al. Clonal expansion of hepatocytes with a selective advantage occurs during all stages of chronic hepatitis B virus infection. J. Viral Hepat. 2015, 22, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B Virus DNA Integration Occurs Early in the Viral Life Cycle in an In Vitro Infection Model via Sodium Taurocholate Cotransporting Polypeptide-Dependent Uptake of Enveloped Virus Particles. J. Virol. 2018, 92, e02007-17. [Google Scholar] [CrossRef]
- Zhao, X.L.; Yang, J.R.; Lin, S.Z.; Ma, H.; Guo, F.; Yang, R.F.; Zhang, H.H.; Han, J.C.; Wei, L.; Pan, X.B. Serum viral duplex-linear DNA proportion increases with the progression of liver disease in patients infected with HBV. Gut 2016, 65, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Bill, C.A.; Summers, J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc. Natl. Acad. Sci. USA 2004, 101, 11135–11140, Erratum in: Proc. Natl. Acad. Sci. USA 2004, 101, 15271. [Google Scholar] [CrossRef]
- Tu, T.; Zehnder, B.; Levy, M.; Micali, G.; Tran, L.; Dabere, O.; Main, N.; Shackel, N.; Urban, S. Hepatitis B virus (HBV) DNA integration is not driven by viral proteins. Zeitschrift Für Gastroenterologie 2019, 57, 5–46. [Google Scholar]
- Zhao, L.H.; Liu, X.; Yan, H.X.; Li, W.Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.P.; Zhuang, X.H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992, Erratum in: Nat. Commun. 2016, 7, 13591. [Google Scholar] [CrossRef]
- Decorsiere, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef]
- Bousali, M.; Papatheodoridis, G.; Paraskevis, D.; Karamitros, T. Hepatitis B Virus DNA Integration, Chronic Infections and Hepatocellular Carcinoma. Microorganisms 2021, 9, 1787. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Yuen, M.F.; Chan, H.L.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HbsAg. Sci. Transl. Med. 2017, 9, eaan0241. [Google Scholar] [CrossRef]
- Le Bert, N.; Gill, U.S.; Hong, M.; Kunasegaran, K.; Tan, D.Z.M.; Ahmad, R.; Cheng, Y.; Dutertre, C.A.; Heinecke, A.; Rivino, L.; et al. Effects of Hepatitis B Surface Antigen on Virus-Specific and Global T Cells in Patients with Chronic Hepatitis B Virus infection. Gastroenterology 2020, 159, 652–664. [Google Scholar] [CrossRef]
- Bertoletti, A.; Ferrari, C. Adaptive immunity in HBV infection. J. Hepatol. 2016, 64, S71–S83. [Google Scholar] [CrossRef]
- Tu, T.; Zehnder, B.; Qu, B.; Urban, S. De novo synthesis of hepatitis B virus nucleocapsids is dispensable for the maintenance and transcriptional regulation of cccDNA. JHEP Rep. 2020, 3, 100195. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Chen, D.S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.L. Hepatitis B virus infection. Nat. Rev. Dis. Prim. 2018, 4, 18035. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.; Guidotti, L.G. Immunobiology and pathogenesis of hepatitis B virus infection. Nat. Rev. Immunol. 2022, 22, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Fioravanti, J.; Di Lucia, P.; Magini, D.; Moalli, F.; Boni, C.; Benechet, A.P.; Fumagalli, V.; Inverso, D.; Vecchi, A.; Fiocchi, A.; et al. Effector CD8+ T cell-derived interleukin-10 enhances acute liver immunopathology. J. Hepatol. 2017, 67, 543–548. [Google Scholar] [CrossRef]
- Asabe, S.; Wieland, S.F.; Chattopadhyay, P.K.; Roederer, M.; Engle, R.E.; Purcell, R.H.; Chisari, F.V. The size of the viral inoculum contributes to the outcome of hepatitis B virus infection. J. Virol. 2009, 83, 9652–9662. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Isogawa, M.; Chisari, F.V. Host-virus interactions in hepatitis B virus infection. Curr. Opin. Immunol. 2015, 36, 61–66. [Google Scholar] [CrossRef]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef]
- Thimme, R.; Wieland, S.; Steiger, C.; Ghrayeb, J.; Reimann, K.A.; Purcell, R.H.; Chisari, F.V. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J. Virol. 2003, 77, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Isogawa, M.; Chung, J.; Murata, Y.; Kakimi, K.; Chisari, F.V. CD40 activation rescues antiviral CD8(+) T cells from PD-1-mediated exhaustion. PLoS Pathog. 2013, 9, e1003490, Erratum in: PLoS Pathog. 2016, 12, e1006086; Erratum in: PLoS Pathog. 2017, 13, e1006416. [Google Scholar] [CrossRef] [PubMed]
- Bénéchet, A.P.; De Simone, G.; Di Lucia, P.; Cilenti, F.; Barbiera, G.; Le Bert, N.; Fumagalli, V.; Lusito, E.; Moalli, F.; Bianchessi, V.; et al. Dynamics and genomic landscape of CD8+ T cells undergoing hepatic priming. Nature 2019, 574, 200–205. [Google Scholar] [CrossRef]
- Chen, J.; Chen, H.; Mai, H.; Lou, S.; Luo, M.; Xie, H.; Zhou, B.; Hou, J.; Jiang, D.K. A functional variant of CD40 modulates clearance of hepatitis B virus in hepatocytes via regulation of the ANXA2/CD40/BST2 axis. Hum. Mol. Genet. 2022, 16, ddac284. [Google Scholar] [CrossRef]
- Maier, H.; Isogawa, M.; Freeman, G.J.; Chisari, F.V. PD-1:PD-L1 Interactions Contribute to the Functional Suppression of Virus-Specific CD8 + T Lymphocytes in the Liver. J. Immunol. 2007, 178, 2714–2720. [Google Scholar] [CrossRef] [PubMed]
- Wykes, M.N.; Lewin, S.R. Immune Checkpoint Blockade in Infectious Diseases. Nat. Rev. Immunol. 2018, 18, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring Function in Exhausted CD8 T Cells During Chronic Viral Infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Isogawa, M.; Tanaka, Y. Immunobiology of Hepatitis B Virus Infection. Hepatol. Res. 2015, 45, 179–189. [Google Scholar] [CrossRef]
- Crispe, I.N. Hepatic T Cells and Liver Tolerance. Nat. Rev. Immunol. 2003, 3, 51–62. [Google Scholar] [CrossRef]
- Kawashima, K.; Isogawa, M.; Hamada-Tsutsumi, S.; Baudi, I.; Saito, S.; Nakajima, A.; Tanaka, Y. Type I Interferon Signaling Prevents Hepatitis B Virus-Specific T Cell Responses by Reducing Antigen Expression. J. Virol. 2018, 92, e01099-18. [Google Scholar] [CrossRef]
- Chen, M.; Sallberg, M.; Hughes, J.; Jones, J.; Guidotti, L.G.; Chisari, F.V.; Billaud, J.N.; Milich, D.R. Immune tolerance split between hepatitis B virus precore and core proteins. J. Virol. 2005, 79, 3016–3027. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Kuo, C.F.; Akbari, O.; Ou, J.H. Maternal-Derived Hepatitis B Virus e Antigen Alters Macrophage Function in Offspring to Drive Viral Persistence after Vertical Transmission. Immunity 2016, 44, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Op den Brouw, M.L.; Binda, R.S.; van Roosmalen, M.H.; Protzer, U.; Janssen, H.L.; van der Molen, R.G.; Woltman, A.M. Hepatitis B virus surface antigen impairs myeloid dendritic cell function: A possible immune escape mechanism of hepatitis B virus. Immunology 2009, 126, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, Z.; Hu, C.; Qian, F.; Cheng, Y.; Wu, M.; Shi, B.; Chen, J.; Hu, Y.; Yuan, Z. Hepatitis B virus surface antigen selectively inhibits TLR2 ligand-induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J. Immunol. 2013, 190, 5142–5151. [Google Scholar] [CrossRef]
- Fumagalli, V.; Di Lucia, P.; Venzin, V.; Bono, E.B.; Jordan, R.; Frey, C.R.; Delaney, W.; Chisari, F.V.; Guidotti, L.G.; Iannacone, M. Serum HBsAg clearance has minimal impact on CD8+ T cell responses in mouse models of HBV infection. J. Exp. Med. 2020, 217, e20200298. [Google Scholar] [CrossRef]
- Sadeghpour, S.; Khodaee, S.; Rahnama, M.; Rahimi, H.; Ebrahimi, D. Human APOBEC3 Variations and Viral Infection. Viruses 2021, 13, 1366. [Google Scholar] [CrossRef]
- Chen, Z.; Eggerman, T.L.; Bocharov, A.V.; Baranova, I.N.; Vishnyakova, T.G.; Patterson, A.P. APOBEC3-induced mutation of the hepatitis virus B DNA genome occurs during its viral RNA reverse transcription into (-)-DNA. J. Biol. Chem. 2021, 297, 100889. [Google Scholar] [CrossRef]
- Maini, M.K.; Reignat, S.; Boni, C.; Ogg, G.S.; King, A.S.; Malacarne, F.; Webster, G.J.; Bertoletti, A. T cell receptor usage of virus-specific CD8 cells and recognition of viral mutations during acute and persistent hepatitis B virus infection. Eur. J. Immunol. 2000, 30, 3067–3078. [Google Scholar] [CrossRef]
- Li, Q.; Sun, B.; Zhuo, Y.; Jiang, Z.; Li, R.; Lin, C.; Jin, Y.; Gao, Y.; Wang, D. Interferon and interferon-stimulated genes in HBV treatment. Front. Immunol. 2022, 13, 1034968. [Google Scholar] [CrossRef]
- Sato, S.; Li, K.; Kameyama, T.; Hayashi, T.; Ishida, Y.; Murakami, S.; Watanabe, T.; Iijima, S.; Sakurai, Y.; Watashi, K.; et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 2015, 42, 123–132. [Google Scholar] [CrossRef]
- Robek, M.D.; Boyd, B.S.; Chisari, F.V. Lambda interferon inhibits hepatitis B and C virus replication. J. Virol. 2005, 79, 3851–3854. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Morris, A.; Mendez, H.; Koch, R.; Silverman, R.H.; Williams, B.R.; Chisari, F.V. Interferon-regulated pathways that control hepatitis B virus replication in transgenic mice. J. Virol. 2002, 76, 2617–2621. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.F.; Guidotti, L.G.; Chisari, F.V. Intrahepatic induction of alpha/beta interferon eliminates viral RNA-containing capsids in hepatitis B virus transgenic mice. J. Virol. 2000, 74, 4165–4173. [Google Scholar] [CrossRef] [PubMed]
- Kakimi, K.; Guidotti, L.G.; Koezuka, Y.; Chisari, F.V. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J. Exp. Med. 2000, 192, 921–930. [Google Scholar] [CrossRef]
- Du, Y.; Wu, J.; Liu, J.; Zheng, X.; Yang, D.; Lu, M. Toll-like receptor-mediated innate immunity orchestrates adaptive immune responses in HBV infection. Front. Immunol. 2022, 13, 965018. [Google Scholar] [CrossRef]
- Lanford, R.E.; Guerra, B.; Chavez, D.; Giavedoni, L.; Hodara, V.L.; Brasky, K.M.; Fosdick, A.; Frey, C.R.; Zheng, J.; Wolfgang, G.; et al. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 2013, 144, 1508–1517.e10. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Tan, A.T.; Ussher, J.E.; Sandalova, E.; Tang, X.Z.; Tan-Garcia, A.; To, N.; Hong, M.; Chia, A.; Gill, U.S.; et al. Toll-like receptor 8 agonist and bacteria trigger potent activation of innate immune cells in human liver. PLoS Pathog. 2014, 10, e1004210. [Google Scholar] [CrossRef]
- Huang, L.R.; Wohlleber, D.; Reisinger, F.; Jenne, C.N.; Cheng, R.L.; Abdullah, Z.; Schildberg, F.A.; Odenthal, M.; Dienes, H.P.; van Rooijen, N.; et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8+ T cells and successful immunotherapy against chronic viral liver infection. Nat. Immunol. 2013, 14, 574–583. [Google Scholar] [CrossRef]
- Lin, Y.C.; Hsu, C.Y.; Huang, S.K.; Fan, Y.H.; Huang, C.H.; Yang, C.K.; Su, W.T.; Chang, P.C.; Dutta, A.; Liu, Y.J.; et al. Induction of liver-specific intrahepatic myeloid cells aggregation (iMATEs) expands CD8 T cell and inhibits growth of murine hepatoma. Oncoimmunology 2018, 7, e1502129. [Google Scholar] [CrossRef]
- Li, M.; Sun, R.; Xu, L.; Yin, W.; Chen, Y.; Zheng, X.; Lian, Z.; Wei, H.; Tian, Z. Kupffer Cells Support Hepatitis B Virus-Mediated CD8+ T Cell Exhaustion via Hepatitis B Core Antigen-TLR2 Interactions in Mice. J. Immunol. 2015, 195, 3100–3109. [Google Scholar] [CrossRef]
- Tsai, K.N.; Kuo, C.F.; Ou, J.J. Mechanisms of Hepatitis B Virus Persistence. Trends Microbiol. 2018, 26, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ryu, W.S. Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: Implications for immune evasion. PLoS Pathog. 2010, 6, e1000986. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Jung, S.Y.; Hodgson, A.J.; Madden, C.R.; Qin, J.; Slagle, B.L. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J. Virol. 2011, 85, 987–995. [Google Scholar] [CrossRef]
- Visvanathan, K.; Skinner, N.A.; Thompson, A.J.; Riordan, S.M.; Sozzi, V.; Edwards, R.; Rodgers, S.; Kurtovic, J.; Chang, J.; Lewin, S.; et al. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology 2007, 45, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Tjwa, E.T.; van Oord, G.W.; Hegmans, J.P.; Janssen, H.L.; Woltman, A.M. Viral load reduction improves activation and function of natural killer cells in patients with chronic hepatitis B. J. Hepatol. 2011, 54, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Isogawa, M.; Furuichi, Y.; Chisari, F.V. Oscillating CD8(+) T cell effector functions after antigen recognition in the liver. Immunity 2005, 23, 53–63. [Google Scholar] [CrossRef]
- Nevola, R.; Rinaldi, L.; Giordano, M.; Marrone, A.; Adinolfi, L.E. Mechanisms and clinical behavior of hepatocellular carcinoma in HBV and HCV infection and alcoholic and non-alcoholic fatty liver disease. Hepatoma Res. 2018, 4, 55. [Google Scholar] [CrossRef]
- Sagnelli, E.; Macera, M.; Russo, A.; Coppola, N.; Sagnelli, C. Epidemiological and etiological variations in hepatocellular carcinoma. Infection 2020, 48, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Sagnelli, C.; Sagnelli, E.; Russo, A.; Pisaturo, M.; Occhiello, L.; Coppola, N. HBV/HDV Co-Infection: Epidemiological and Clinical Changes, Recent Knowledge and Future Challenges. Life 2021, 11, 169. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; Mathurin, P.; Cortez-Pinto, H.; Loomba, R. Global epidemiology of alcohol-associated cirrhosis and HCC: Trends, projections and risk factors. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Zampino, R.; Coppola, N.; Cirillo, G.; Boemio, A.; Minichini, C.; Marrone, A.; Stanzione, M.; Starace, M.; Durante-Mangoni, E.; Sagnelli, E.; et al. Insulin resistance and steatosis in HBV-HCV co-infected patients: Role of PNPLA3 polymorphisms and impact on liver fibrosis progression. World J. Hepatol. 2014, 6, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Tang, Z.; Zou, M.; Tan, T.; Tang, Y.; Chen, Y.; Liang, B.; Xie, D.; Yang, Y.; Xie, S.; et al. Correlation of DEPDC5 rs1012068 and rs5998152 Polymorphisms with Risk of Hepatocellular Carcinoma: A Systematic Review and Meta-Analysis. J. Oncol. 2023, 2023, 5957481. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, L.; Nascimbeni, F.; Giordano, M.; Masetti, C.; Guerrera, B.; Amelia, A.; Fascione, M.C.; Ballestri, S.; Romagnoli, D.; Zampino, R.; et al. Clinical features and natural history of cryptogenic cirrhosis compared to hepatitis C virus-related cirrhosis. World J. Gastroenterol. 2017, 23, 1458–1468. [Google Scholar] [CrossRef]
- Vetrano, E.; Rinaldi, L.; Mormone, A.; Giorgione, C.; Galiero, R.; Caturano, A.; Nevola, R.; Marfella, R.; Sasso, F.C. Non-alcoholic Fatty Liver Disease (NAFLD), Type 2 Diabetes, and Non-viral Hepatocarcinoma: Pathophysiological Mechanisms and New Therapeutic Strategies. Biomedicines 2023, 11, 468. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, E.G.; Demeulemeester, J.; Otero, P.; Jolly, C.; García-Souto, D.; Pequeño-Valtierra, A.; Zamora, J.; Tojo, M.; Temes, J.; Baez-Ortega, A.; et al. Aberrant integration of Hepatitis B virus DNA promotes major restructuring of human hepatocellular carcinoma genome architecture. Nat. Commun. 2021, 12, 6910. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.H.; Li, C.L.; Lin, Y.Y.; Ho, M.C.; Wang, Y.C.; Tseng, S.T.; Chen, P.J. Hepatitis B Virus DNA Integration Drives Carcinogenesis and Provides a New Biomarker for HBV-related HCC. Cell Mol. Gastroenterol. Hepatol. 2023, 15, 921–929. [Google Scholar] [CrossRef]
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.L.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA Integration and Clonal Hepatocyte Expansion in Chronic Hepatitis B Patients Considered Immune Tolerant. Gastroenterology 2016, 151, 986–998.e4. [Google Scholar] [CrossRef]
- An, P.; Xu, J.; Yu, Y.; Winkler, C.A. Host and Viral Genetic Variation in HBV-Related Hepatocellular Carcinoma. Front. Genet. 2018, 9, 261. [Google Scholar] [CrossRef]
- Zheng, B.; Liu, X.L.; Fan, R.; Bai, J.; Wen, H.; Du, L.T.; Jiang, G.Q.; Wang, C.-Y.; Fan, X.T.; Ye, Y.N.; et al. The Landscape of Cell-Free HBV Integrations and Mutations in Cirrhosis and Hepatocellular Carcinoma Patients. Clin. Cancer Res. 2021, 27, 3772–3783. [Google Scholar] [CrossRef]
- Jiang, S.; Yang, Z.; Li, W.; Li, X.; Wang, Y.; Zhang, J.; Xu, C.; Chen, P.J.; Hou, J.; McCrae, M.A.; et al. Re-evaluation of the carcinogenic significance of hepatitis B virus integration in hepatocarcinogenesis. PLoS ONE 2012, 7, e40363. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Jiang, X.; Li, M.; Luo, Y. Hepatitis Virus and Hepatocellular Carcinoma: Recent Advances. Cancers 2023, 15, 533. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Budzinska, M.A.; Shackel, N.A.; Urban, S.; Tu, T. Cellular Genomic Sites of Hepatitis B Virus DNA Integration. Genes 2018, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, J.; Yang, Z.; Kang, J.; Jiang, S.; Zhang, T.; Chen, T.; Li, M.; Lv, Q.; Chen, X.; et al. The function of targeted host genes determines the oncogenicity of HBV integration in hepatocellular carcinoma. J. Hepatol. 2014, 60, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Zhang, A.; Lian, J.; Wang, J.; Chang, T.T.; Lin, Y.J.; Song, W.; Su, Y.H. Recurrent HBV Integration Targets as Potential Drivers in Hepatocellular Carcinoma. Cells 2021, 10, 1294. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, Y.; Tian, T.; Rao, X.; Dong, W.; Zhang, J.; Yang, Y.; Tao, Q.; Peng, F.; Shen, C.; et al. Analysis of viral integration reveals new insights of oncogenic mechanism in HBV-infected intrahepatic cholangiocarcinoma and combined hepatocellular-cholangiocarcinoma. Hepatol. Int. 2022, 16, 1339–1352. [Google Scholar] [CrossRef]
- Péneau, C.; Imbeaud, S.; La Bella, T.; Hirsch, T.Z.; Caruso, S.; Calderaro, J.; Paradis, V.; Blanc, J.F.; Letouzé, E.; Nault, J.C.; et al. Hepatitis B virus integrations promote local and distant oncogenic driver alterations in hepatocellular carcinoma. Gut 2022, 71, 616–626. [Google Scholar] [CrossRef]
- Sze, K.M.; Ho, D.W.; Chiu, Y.T.; Tsui, Y.M.; Chan, L.K.; Lee, J.M.; Chok, K.S.; Chan, A.C.; Tang, C.N.; Tang, V.W.; et al. Hepatitis B Virus-Telomerase Reverse Transcriptase Promoter Integration Harnesses Host ELF4, Resulting in Telomerase Reverse Transcriptase Gene Transcription in Hepatocellular Carcinoma. Hepatology 2021, 73, 23–40. [Google Scholar] [CrossRef]
- Jang, J.W.; Kim, H.S.; Kim, J.S.; Lee, S.K.; Han, J.W.; Sung, P.S.; Bae, S.H.; Choi, J.Y.; Yoon, S.K.; Han, D.J.; et al. Distinct Patterns of HBV Integration and TERT Alterations between in Tumor and Non-Tumor Tissue in Patients with Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 7056. [Google Scholar] [CrossRef]
- Nault, J.C.; Ningarhari, M.; Rebouissou, S.; Zucman-Rossi, J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Saigo, K.; Yoshida, K.; Ikeda, R.; Sakamoto, Y.; Murakami, Y.; Urashima, T.; Asano, T.; Kenmochi, T.; Inoue, I. Integration of hepatitis B virus DNA into the myeloid/lymphoid or mixed-lineage leukemia (MLL4) gene and rearrangements of MLL4 in human hepatocellular carcinoma. Hum. Mutat. 2008, 29, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Bousali, M.; Karamitros, T. Hepatitis B Virus Integration into Transcriptionally Active Loci and HBV-Associated Hepatocellular Carcinoma. Microorganisms 2022, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Seo, D.; Andersen, J.B.; Gillen, M.C.; Kim, M.S.; Conner, E.A.; Galle, P.R.; Factor, V.M.; Park, Y.N.; Thorgeirsson, S.S. Sequential transcriptome analysis of human liver cancer indicates late stage acquisition of malignant traits. J. Hepatol. 2014, 60, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R. Review on hepatitis B virus precore/core promoter mutations and their correlation with genotypes and liver disease severity. World J. Hepatol. 2022, 14, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Lee, W.Y.; Toh, S.T.; Tennakoon, C.; Toh, H.C.; Chow, P.K.; Chung, A.Y.; Chong, S.S.; Ooi, L.L.; Sung, W.K.; et al. Comprehensive analysis of transcriptome profiles in hepatocellular carcinoma. J. Transl. Med. 2019, 17, 273. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Nault, J.-C.; Roberts, L.R.; Zucman-Rossi, J. Genomic Medicine and Implications for Hepatocellular Carcinoma Prevention and Therapy. Gastroenterology 2019, 156, 492–509. [Google Scholar] [CrossRef]
- Lau, C.C.; Sun, T.; Ching, A.K.; He, M.; Li, J.W.; Wong, A.M.; Co, N.N.; Chan, A.W.; Li, P.S.; Lung, R.W.; et al. Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell 2014, 25, 335–349. [Google Scholar] [CrossRef]
- Tseng, T.C.; Liu, C.J.; Yang, H.C.; Su, T.H.; Wang, C.C.; Chen, C.L.; Hsu, C.A.; Kuo, S.F.; Liu, C.H.; Chen, P.J.; et al. Serum hepatitis B surface antigen levels help predict disease progression in patients with low hepatitis B virus loads. Hepatology 2013, 57, 441–450. [Google Scholar] [CrossRef]
- Kawanaka, M.; Nishino, K.; Nakamura, J.; Oka, T.; Urata, N.; Goto, D.; Suehiro, M.; Kawamoto, H.; Kudo, M.; Yamada, G. Quantitative levels of hepatitis B virus DNA and surface antigen and the risk of hepatocellular carcinoma in patients with hepatitis B receiving long-term nucleos(t)ide analogue therapy. Liver Cancer 2014, 3, 41–52. [Google Scholar] [CrossRef]
- Liu, J.; Yang, H.I.; Lee, M.H.; Lu, S.N.; Jen, C.L.; Batrla-Utermann, R.; Wang, L.Y.; You, S.L.; Hsiao, C.K.; Chen, P.J.; et al. -HBV Study Group. Spontaneous seroclearance of hepatitis B seromarkers and subsequent risk of hepatocellular carcinoma. Gut 2014, 63, 1648–1657. [Google Scholar] [CrossRef] [PubMed]
- Su, I.J.; Wang, H.C.; Wu, H.C.; Huang, W.Y. Ground glass hepatocytes contain pre-S mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J. Gastroenterol. Hepatol. 2008, 23, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Wu, H.C.; Chen, C.F.; Fausto, N.; Lei, H.Y.; Su, I.J. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am. J. Pathol. 2003, 163, 2441–2449. [Google Scholar] [CrossRef]
- Barone, M.; Spano, D.; D′Apolito, M.; Centra, M.; Lasalandra, C.; Capasso, M.; Di Leo, A.; Volinia, S.; Arcelli, D.; Rosso, N.; et al. Gene expression analysis in HBV transgenic mouse liver: A model to study early events related to hepatocarcinogenesis. Mol. Med. 2006, 12, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Na, B.; Huang, Z.; Wang, Q.; Qi, Z.; Tian, Y.; Lu, C.C.; Yu, J.; Hanes, M.A.; Kakar, S.; Huang, E.J.; et al. Transgenic expression of entire hepatitis B virus in mice induces hepatocarcinogenesis independent of chronic liver injury. PLoS ONE 2011, 6, e26240. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Choi, Y.M.; Lee, S.Y.; Kim, B.J. Naturally Occurring Hepatitis B Virus Mutations Leading to Endoplasmic Reticulum Stress and Their Contribution to the Progression of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 597. [Google Scholar] [CrossRef]
- Dong, M.L.; Wen, X.; He, X.; Ren, J.H.; Yu, H.B.; Qin, Y.P.; Yang, Z.; Yang, M.L.; Zhou, C.Y.; Zhang, H.; et al. HBx Mediated Increase of DDX17 Contributes to HBV-Related Hepatocellular Carcinoma Tumorigenesis. Front. Immunol. 2022, 13, 871558. [Google Scholar] [CrossRef]
- Schollmeier, A.; Glitscher, M.; Hildt, E. Relevance of HBx for Hepatitis B Virus-Associated Pathogenesis. Int. J. Mol. Sci. 2023, 24, 4964. [Google Scholar] [CrossRef]
- Jung, J.K.; Arora, P.; Pagano, J.S.; Jang, K.L. Expression of DNAmethyltransferase 1 is activated by hepatitis B virus X protein via a regulatory circuit involving the p16INK4a–cyclin D1–CDK 4/6–pRb–E2F1 pathway. Cancer Res. 2007, 67, 5771–5778. [Google Scholar] [CrossRef]
- You, H.; Zhang, N.; Yu, T.; Ma, L.; Li, Q.; Wang, X.; Yuan, D.; Kong, D.; Liu, X.; Hu, W.; et al. Hepatitis B virus X protein promotes MAN1B1 expression by enhancing stability of GRP78 via TRIM25 to facilitate hepatocarcinogenesis. Br. J. Cancer 2023, 128, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Tarn, C.; Lee, S.; Hu, Y.; Ashendel, C.; Andrisani, O.M. Hepatitis B virus X protein differentially activates RAS–RAF–MAPK and JNK pathways in X-transforming versus non-transforming AML12 hepatocytes. J. Biol. Chem. 2001, 276, 34671–34680. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Yuan, D.; Li, Q.; Zhang, N.; Kong, D.; Yu, T.; Liu, X.; Liu, X.; Zhou, R.; Kong, F.; et al. Hepatitis B virus X protein increases LASP1 SUMOylation to stabilize HER2 and facilitate hepatocarcinogenesis. Int. J. Biol. Macromol. 2023, 226, 996–1009. [Google Scholar] [CrossRef] [PubMed]
- Sze, K.M.; Chu, G.K.; Lee, J.M.; Ng, I.O. C-terminal truncated hepatitis B virus x protein is associated with metastasis and enhances invasiveness by C-Jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology 2013, 57, 131–139. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Cao, Y.; Yang, Z. Roles of APOBEC3 in hepatitis B virus (HBV) infection and hepatocarcinogenesis. Bioengineered 2021, 12, 2074–2086. [Google Scholar] [CrossRef]
- Kim, G.W.; Imam, H.; Khan, M.; Mir, S.A.; Kim, S.J.; Yoon, S.K.; Hur, W.; Siddiqui, A. HBV-Induced Increased N6 Methyladeno- sine Modification of PTEN RNA Affects Innate Immunity and Contributes to HCC. Hepatology 2021, 73, 533–547. [Google Scholar] [CrossRef]
- Roca Suarez, A.A.; Testoni, B.; Zoulim, F. HBV 2021: New therapeutic strategies against an old foe. Liver Int. 2021, 41, 15–23. [Google Scholar] [CrossRef]
- Revill, P.A.; Chisari, F.V.; Block, J.M.; Dandri, M.; Gehring, A.J.; Guo, H.; Hu, J.; Kramvis, A.; Lampertico, P.; Janssen, H.L.A.; et al. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol. Hepatol. 2019, 4, 545–558, Erratum in: Lancet Gastroenterol. Hepatol. 2019, 4, e7. [Google Scholar] [CrossRef]
- Moini, M.; Fung, S. HBsAg Loss as a Treatment Endpoint for Chronic HBV Infection: HBV Cure. Viruses 2022, 14, 657. [Google Scholar] [CrossRef]
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019, 18, 827–844, Erratum in: Nat. Rev. Drug Discov. 2020, 19, 291. [Google Scholar] [CrossRef]
- Jeng, W.J.; Lok, A.S.F. What will it take to cure hepatitis B? Hepatol. Commun. 2023, 7, e0084. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewicz, D.; Geyer, J. Multitasking Na+/Taurocholate Cotransporting Polypeptide (NTCP) as a Drug Target for HBV Infection: From Protein Engineering to Drug Discovery. Biomedicines 2022, 10, 196. [Google Scholar] [CrossRef] [PubMed]
- Bogomolov, P.; Alexandrov, A.; Voronkova, N.; Macievich, M.; Kokina, K.; Petrachenkova, M.; Lehr, T.; Lempp, F.A.; Wedemeyer, H.; Haag, M.; et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J. Hepatol. 2016, 65, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, H.; Schoneweis, K.; Bogomolov, P.O.; Voronkova, N.; Chulanov, V.; Stepanova, T.; Bremer, B.; Allweiss, L.; Dandri, M.; Burhenne, J.; et al. Final results of a multicenter, open-label phase 2 clinical trial (MYR203) to assess safety and efficacy of myrcludex B in cwith PEG-interferon Alpha 2a in patients with chronic HBV/HDV co-infection. J. Hepatol. 2019, 70, 81. [Google Scholar] [CrossRef]
- Zhang, T.Y.; Yuan, Q.; Zhao, J.H.; Zhang, Y.L.; Yuan, L.Z.; Lan, Y.; Lo, Y.C.; Sun, C.P.; Wu, C.R.; Zhang, J.F.; et al. Prolonged suppression of HBV in mice by a novel antibody that targets a unique epitope on hepatitis B surface antigen. Gut 2016, 65, 658–671. [Google Scholar] [CrossRef]
- Tilg, H.; Vogel, W.; Tratkiewicz, J.; Aulitzky, W.E.; Herold, M.; Gruber, M.; Geissler, D.; Umlauft, F.; Judmaier, G.; Schwulera, U. Pilot study of natural human interleukin-2 in patients with chronic hepatitis B: Immunomodulatory and antiviral effects. J. Hepatol. 1993, 19, 259–267. [Google Scholar] [CrossRef]
- Carreño, V.; Zeuzem, S.; Hopf, U.; Marcellin, P.; Cooksley, W.E.; Fevery, J.; Diago, M.; Reddy, R.; Peters, M.; Rittweger, K.; et al. A phase I/II study of recombinant human interleukin-12 in patients with chronic hepatitis B. J. Hepatol. 2000, 32, 317–324. [Google Scholar] [CrossRef]
- Iino, S.; Toyota, J.; Kumada, H.; Kiyosawa, K.; Kakumu, S.; Sata, M.; Suzuki, H.; Martins, E.B. The efficacy and safety of thymosin alpha-1 in Japanese patients with chronic hepatitis B; results from a randomized clinical trial. J. Viral Hepat. 2005, 12, 300–306. [Google Scholar] [CrossRef]
- You, J.; Zhuang, L.; Cheng, H.Y.; Yan, S.M.; Yu, L.; Huang, J.H.; Tang, B.Z.; Huang, M.L.; Ma, Y.L.; Chongsuvivatwong, V.; et al. Efficacy of thymosin alpha-1 and interferon alpha in treatment of chronic viral hepatitis B: A randomized controlled study. World J. Gastroenterol. 2006, 12, 6715–6721. [Google Scholar] [CrossRef]
- Ruiz-Moreno, M.; García, R.; Rua, M.J.; Serrano, B.; Moraleda, G.; Feijoo, E.; Bartolomé, J.; Ortiz, F.; Castillo, I.; Carreño, V. Le-vamisole and interferon in children with chronic hepatitis B. Hepatology 1993, 18, 264–269. [Google Scholar] [CrossRef]
- Yalcin, K.; Acar, M.; Degertekin, H. Specific Hepatitis B Vaccine Therapy in Inactive HbsAg Carriers: A Randomized Controlled Trial. Infection 2003, 31, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Pol, S.; Nalpas, B.; Driss, F.; Michel, M.L.; Tiollais, P.; Denis, J.; Bréchot, C. Efficacy and limitations of a specific immunotherapy in chronic hepatitis B. J. Hepatol. 2001, 34, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.Z.; Wang, X.Y.; Shen, X.L.; Gong, G.Z.; Ren, H.; Guo, L.M.; Sun, A.M.; Xu, M.; Li, L.J.; Guo, X.H.; et al. Results of a phase III clinical trial with an HbsAg-HBIG immunogenic complex therapeutic vaccine for chronic hepatitis B patients: Experiences and findings. J. Hepatol. 2013, 59, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Dahmen, A.; Herzog-Hauff, S.; Böcher, W.O.; Galle, P.R.; Löhr, H.F. Clinical and immunological efficacy of intradermal vaccine plus lamivudine with or without interleukin-2 in patients with chronic hepatitis B. J. Med. Virol. 2002, 66, 452–460. [Google Scholar] [CrossRef]
- Vandepapelière, P.; Lau, G.K.; Leroux-Roels, G.; Horsmans, Y.; Gane, E.; Tawandee, T.; bin Merican, M.S.; Win, K.M.; Treop, C.; Cooksley, G.; et al. Therapeutic vaccination of chronic hepatitis B patients with virus suppression by antiviral therapy: A randomized, controlled study of co-administration of HbsAg/AS02 candidate vaccine and lamivudine. Vaccine 2007, 25, 8585–8597. [Google Scholar] [CrossRef]
- Fontaine, H.; Kahi, S.; Chazallon, C.; Bourgine, M.; Varaut, A.; Buffet, C.; Godon, O.; Meritet, J.F.; Saidi, Y.; Michel, M.L.; et al. Anti-HBV DNA vaccination does not prevent relapse after discontinuation of analogues in the treatment of chronic hepatitis B: A randomized trial-ANRS HB02 VAC-ADN. Gut 2015, 64, 139–147. [Google Scholar] [CrossRef]
- Lobaina, Y.; Palenzuela, D.; García, D.; Rodrfguez, D.; Pichardo, D.; Muzio, V.; Aguilar, J.C. Comparative study of the immunogenicity and immunoenhancing effects of two hepatitis B core antigen variants in mice by nasal administration. Vaccine 2006, 24, S58–S59. [Google Scholar] [CrossRef]
- Al-Mahtab, M.; Akbar, S.M.F.; Aguilar, J.C.; Uddin, H.; Khan, S.I.; Rahman, S. Therapeutic potential of a combined hepatitis B virus surface and core antigen vaccine in patients with chronic hepatitis B. Hepatol. Int. 2013, 7, 981–989. [Google Scholar] [CrossRef]
- Al Mahtab, M.; Akbar, S.M.F.; Aguilar, J.C.; Guillen, G.; Penton, E.; Tuero, A.; Yoshida, O.; Hiasa, Y.; Onji, M. Treatment of chronic hepatitis B naïve patients with a therapeutic vaccine containing HBs and HBc antigens (a randomized, open and treat-ment-controlled phase III clinical trial). PLoS ONE 2018, 13, e0201236. [Google Scholar] [CrossRef]
- Akbar, S.M.F.; Al Mahtab, M.; Aguilar, J.C.; Yoshida, O.; Penton, E.; Guillen, G.; Hiasa, Y. Sustained antiviral and liver protection by a nasal therapeutic vaccine (NASVAC), containing both HBsAg and HBcAg) in patients with chronic hepatitis B; 2-year follow-up of phase III clinical trial. Pathogens 2021, 10, 1440. [Google Scholar] [CrossRef]
- Akbar, S.M.F.; Al Mahtab, M.; Aguilar, J.C.; Yoshida, O.; Khan, S.; Penton, E.; Gerardo, G.N.; Hiasa, Y. The Safety and Efficacy of a Therapeutic Vaccine for Chronic Hepatitis B: A Follow-Up Study of Phase III Clinical Trial. Vaccines 2021, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Balsitis, S.; Gali, V.; Mason, P.J.; Chaniewski, S.; Levine, S.M.; Wichroski, M.J.; Feulner, M.; Song, Y.; Granaldi, K.; Loy, J.K.; et al. Safety and efficacy of anti-PD-L1 therapy in the woodchuck model of HBV infection. PLoS ONE 2018, 13, e0190058. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.; Verdon, D.J.; Brooks, A.E.; Gaggar, A.; Nguyen, A.H.; Subramanian, G.M.; Schwabe, C.; Dunbar, P.R. Anti-PD-1 blockade with nivolumab with and without therapeutic vaccination for virally suppressed chronic hepatitis B: A pilot study. J. Hepatol. 2019, 71, 900–907. [Google Scholar] [CrossRef]
- Krebs, K.; Bottinger, N.; Huang, L.R.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jager, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology 2013, 145, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Kah, J.; Koh, S.; Volz, T.; Ceccarello, E.; Allweiss, L.; Lutgehetmann, M.; Bertoletti, A.; Dandri, M. Lymphocytes transiently expressing virus-specific T cell receptors reduce hepatitis B virus infection. J. Clin. Investig. 2017, 127, 3177–3188. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Cao, Q.; Xiong, Y.; Zhang, E.; Lu, M. Interaction between Hepatitis B Virus and Toll Like Receptors: Current Status and Potential Therapeutic Use for Chronic Hepatitis, B. Vaccines 2018, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.; Brunetto, M.R.; Kim, Y.J.; Ferrari, C.; Massetto, B.; Nguyen, A.H.; Joshi, A.; Woo, J.; Lau, A.H.; Gaggar, A.; et al. Safety, efficacy and pharmacodynamics of vesatolimod (GS-9620) in virally suppressed patients with chronic hepatitis B. J. Hepatol. 2018, 68, 431–440. [Google Scholar] [CrossRef]
- Sun, Z.; Hornung, V. cGAS-STING signaling. Curr. Biol. 2022, 32, R730–R734. [Google Scholar] [CrossRef]
- Li, Y.; He, M.; Wang, Z.; Duan, Z.; Guo, Z.; Wang, Z.; Gong, R.; Chu, T.; Cai, J.; Gao, B. STING signaling activation inhibits HBV replication and attenuates the severity of liver injury and HBV-induced fibrosis. Cell Mol. Immunol. 2022, 19, 92–107. [Google Scholar] [CrossRef]
- Cheng, S.T.; Hu, J.L.; Ren, J.H.; Yu, H.B.; Zhong, S.; Wai Wong, V.K.; Kwan Law, B.Y.; Chen, W.X.; Xu, H.M.; Zhang, Z.Z.; et al. Dicoumarol, an NQO1 inhibitor, blocks cccDNA transcription by promoting degradation of HBx. J. Hepatol. 2021, 74, 522–534. [Google Scholar] [CrossRef]
- Chauhan, R.; Li, Q.; Woodson, M.E.; Gasonoo, M.; Meyers, M.J.; Tavis, J.E. Efficient Inhibition of Hepatitis B Virus (HBV) Replication and cccDNA Formation by HBV Ribonuclease H Inhibitors during Infection. Antimicrob. Agents Chemother. 2021, 65, e0146021. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Davis, K.M.; Liu, D.R. Chemical biology approaches to genome editing: Understanding, controlling, and delivering programmable nucleases. Cell Chem. Biol. 2016, 23, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Cradick, T.J.; Keck, K.; Bradshaw, S.; Jamieson, A.C.; McCaffrey, A.P. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol. Ther. 2010, 18, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; Ely, A.; Mussolino, C.; Cathomen, T.; Arbuthnot, P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol. Ther. 2013, 21, 1889–1897. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, W.; Lin, J.; Wang, F.; Wu, M.; Chen, C.; Zheng, Y.; Peng, X.; Li, J.; Yuan, Z. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol. Ther. 2014, 22, 303–311. [Google Scholar] [CrossRef]
- Yang, Y.C.; Yang, H.C. Recent Progress and Future Prospective in HBV Cure by CRISPR/Cas. Viruses 2021, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.; Shlomai, A.; Cox, D.B.; Schwartz, R.E.; Michailidis, E.; Bhatta, A.; Scott, D.A.; Zhang, F.; Rice, C.M.; Bhatia, S.N. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci. Rep. 2015, 2, 10833. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Jiang, W.; Marraffini, L.A. CRISPR-Cas: New Tools for Genetic Manipulations from Bacterial Immunity Systems. Annu. Rev. Microbiol. 2015, 69, 209–228. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788, Erratum in: Nat. Rev. Genet. 2018, 19, 801. [Google Scholar] [CrossRef]
- Yang, Y.C.; Chen, Y.H.; Kao, J.H.; Ching, C.; Liu, I.J.; Wang, C.C.; Tsai, C.H.; Wu, F.Y.; Liu, C.J.; Chen, P.J.; et al. Permanent Inactivation of HBV Genomes by CRISPR/Cas9-Mediated Non-cleavage Base Editing. Mol. Ther. Nucleic Acids 2020, 20, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, L.; Li, C.; Long, Q.; Yang, Q.; Huang, A.; Tang, H. CRISPR/Cas9 delivery by NIR-responsive biomimetic nanoparticles for targeted HBV therapy. J. Nanobiotechnol. 2022, 20, 27. [Google Scholar] [CrossRef] [PubMed]
- Fung, S.; Choi, H.S.J.; Gehring, A.; Janssen, H.L.A. Getting to HBV cure: The promising paths forward. Hepatology 2022, 76, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Boni, C.; Vecchi, A.; Rossi, M.; Laccabue, D.; Giuberti, T.; Alfieri, A.; Lampertico, P.; Grossi, G.; Facchetti, F.; Brunetto, M.R.; et al. TLR7 Agonist Increases Responses of Hepatitis B Virus-Specific T Cells and Natural Killer Cells in Patients with Chronic Hepatitis B Treated with Nucleos(T)Ide Analogues. Gastroenterology 2018, 154, 1764–1777.e7. [Google Scholar] [CrossRef] [PubMed]
- Boni, C.; Barili, V.; Acerbi, G.; Rossi, M.; Vecchi, A.; Laccabue, D.; Penna, A.; Missale, G.; Ferrari, C.; Fisicaro, P. HBV Immune-Therapy: From Molecular Mechanisms to Clinical Applications. Int. J. Mol. Sci. 2019, 20, 2754. [Google Scholar] [CrossRef]
- Evans, T.; Barnes, E. Phase 1b/2a study of heterologous ChAdOx1-HBV/MVA-HBV therapeutic vaccination (VTP-300) combined with low-dose nivolumab (LDN) in virally-suppressed patients with CHB on nucleos (t)ide analogues. J. Hepatol. 2022, 77, S868. [Google Scholar] [CrossRef]
- Deng, R.; Liu, S.; Shen, S.; Guo, H.; Sun, J. Circulating HBV RNA: From biology to clinical applications. Hepatology 2022, 76, 1520–1530. [Google Scholar] [CrossRef]
- Watanabe, T.; Inoue, T.; Tanaka, Y. Hepatitis B Core-Related Antigen and New Therapies for Hepatitis, B. Microorganisms 2021, 9, 2083. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nevola, R.; Beccia, D.; Rosato, V.; Ruocco, R.; Mastrocinque, D.; Villani, A.; Perillo, P.; Imbriani, S.; Delle Femine, A.; Criscuolo, L.; et al. HBV Infection and Host Interactions: The Role in Viral Persistence and Oncogenesis. Int. J. Mol. Sci. 2023, 24, 7651. https://doi.org/10.3390/ijms24087651
Nevola R, Beccia D, Rosato V, Ruocco R, Mastrocinque D, Villani A, Perillo P, Imbriani S, Delle Femine A, Criscuolo L, et al. HBV Infection and Host Interactions: The Role in Viral Persistence and Oncogenesis. International Journal of Molecular Sciences. 2023; 24(8):7651. https://doi.org/10.3390/ijms24087651
Chicago/Turabian StyleNevola, Riccardo, Domenico Beccia, Valerio Rosato, Rachele Ruocco, Davide Mastrocinque, Angela Villani, Pasquale Perillo, Simona Imbriani, Augusto Delle Femine, Livio Criscuolo, and et al. 2023. "HBV Infection and Host Interactions: The Role in Viral Persistence and Oncogenesis" International Journal of Molecular Sciences 24, no. 8: 7651. https://doi.org/10.3390/ijms24087651
APA StyleNevola, R., Beccia, D., Rosato, V., Ruocco, R., Mastrocinque, D., Villani, A., Perillo, P., Imbriani, S., Delle Femine, A., Criscuolo, L., Alfano, M., La Montagna, M., Russo, A., Marfella, R., Cozzolino, D., Sasso, F. C., Rinaldi, L., Marrone, A., Adinolfi, L. E., & Claar, E. (2023). HBV Infection and Host Interactions: The Role in Viral Persistence and Oncogenesis. International Journal of Molecular Sciences, 24(8), 7651. https://doi.org/10.3390/ijms24087651