

Novel Functionalized Spiro [Indoline-3,5′-pyrroline]-2,2′dione Derivatives: Synthesis, Characterization, Drug-Likeness, ADME, and Anticancer Potential

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
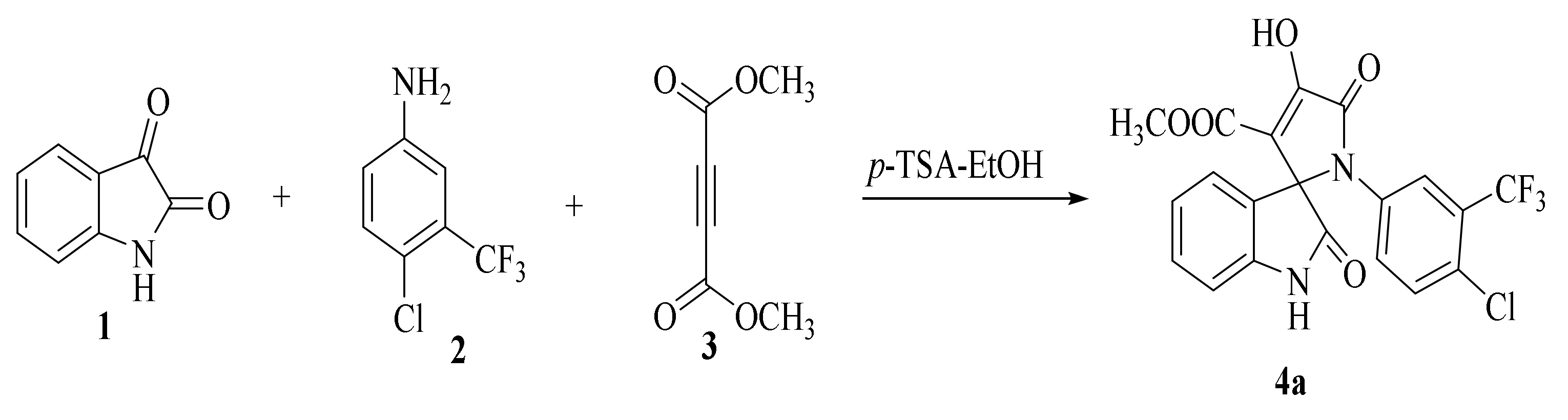
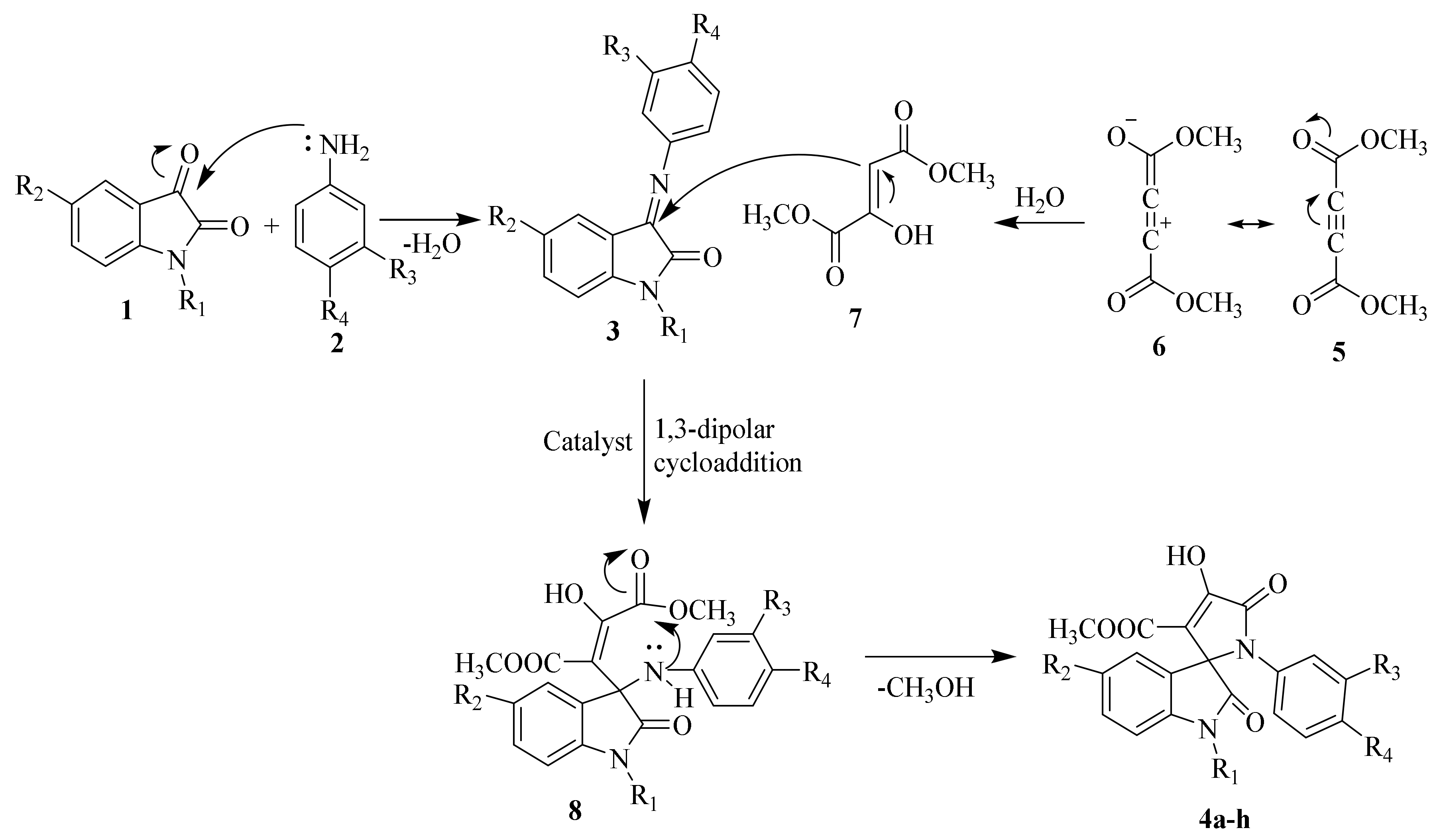
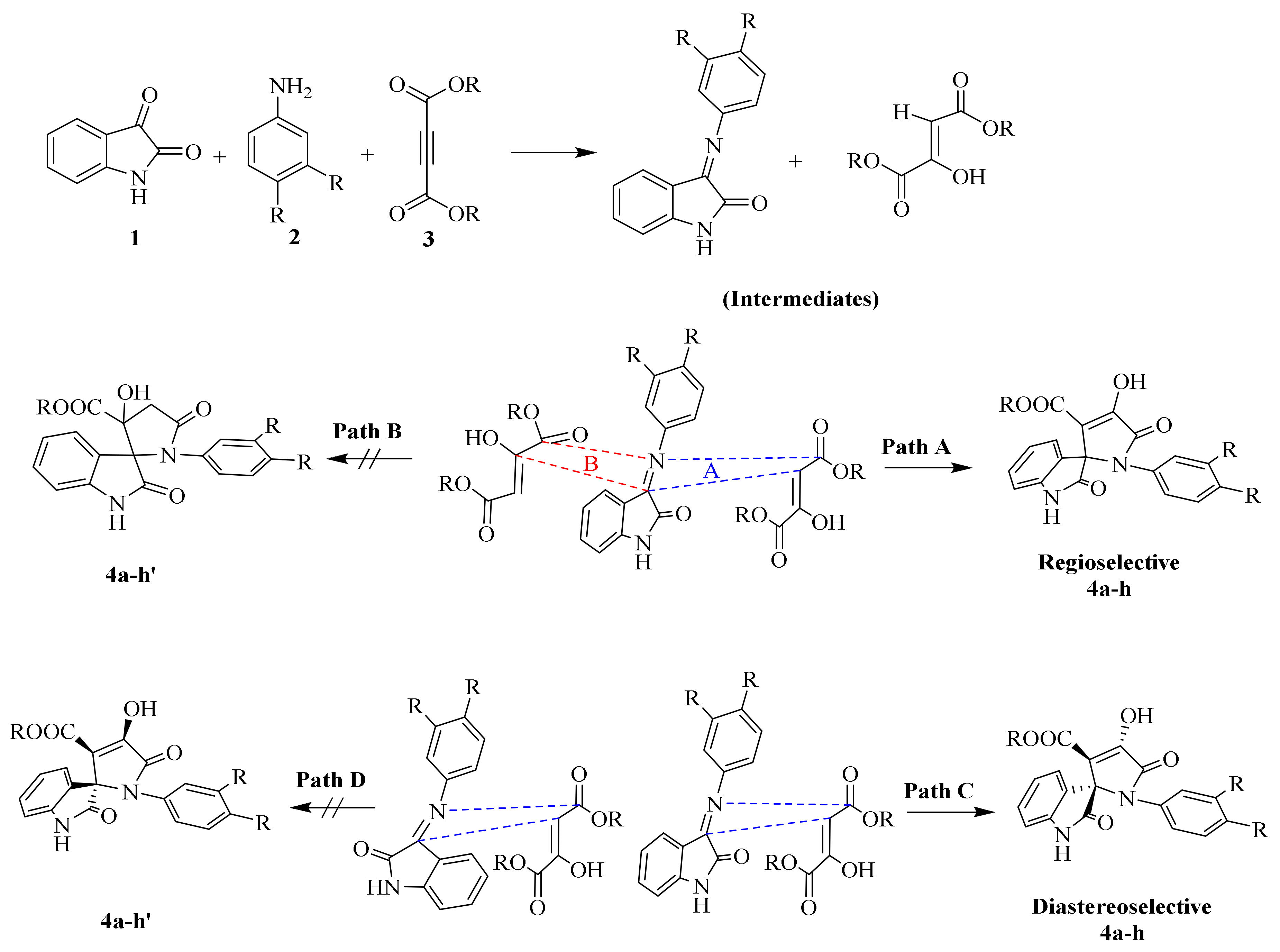
2.1.1. SOX Synthesis
2.1.2. Characterization Details of SOXs (4a–h)
Methyl-1’-(4-chloro-3-(trifluoromethyl)phenyl)-4’-hydroxy-2,5’-dioxo-1’,5’-dihydrospiro[indoline-3,2’-pyrrole]-3’-carboxylate (4a)
Methyl-1’-(3-chloro-4-fluorophenyl)-4’-hydroxy-2,5’-dioxo-1’,5’-dihydrospiro[indoline-3,2’-pyrrole]-3’-carboxylate (4b)
Methyl-4’-hydroxy-2,5’-dioxo-1’-phenyl-1’,5’-dihydrospiro[indoline-3,2’-pyrrole]-3’-carboxylate (4c)
Methyl-5-bromo-4’-hydroxy-2,5’-dioxo-1’-phenyl-1’,5’-dihydrospiro[indoline-3,2’-pyrrole]-3’-carboxylate (4d)
Methyl-5-bromo-1’-(3-chloro-4-fluorophenyl)-4’-hydroxy-2,5’-dioxo-1’,5’-dihydrospiro[indoline-3,2’-pyrrole]-3’-carboxylate (4e)
Methyl-5-bromo-1’-(4-chloro-3-(trifluoromethyl)phenyl)-4’-hydroxy-2,5’-dioxo-1’,5’-dihydrospiro[indoline-3,2’-pyrrole]-3’-carboxylate (4f)
Methyl-4’-hydroxy-5-nitro-2,5’-dioxo-1’-phenyl-1’,5’-dihydrospiro[indoline-3,2’-pyrrole]-3’-carboxylate (4g)
Methyl-1’-(3-chloro-4-fluorophenyl)-4’-hydroxy-5-nitro-2,5’-dioxo-1’,5’-dihydrospiro[indoline-3,2’-pyrrole]-3’-carboxylate (4h)
HPLC Analysis of SOX 4a
2.2. Computational Chemistry
2.2.1. Substituted Spirooxindole-Pyrrolines (4a–h) Exert Drug-like Properties
2.2.2. Substituted Spirooxindole-Pyrrolines (4a–h) Have Acceptable ADME Properties
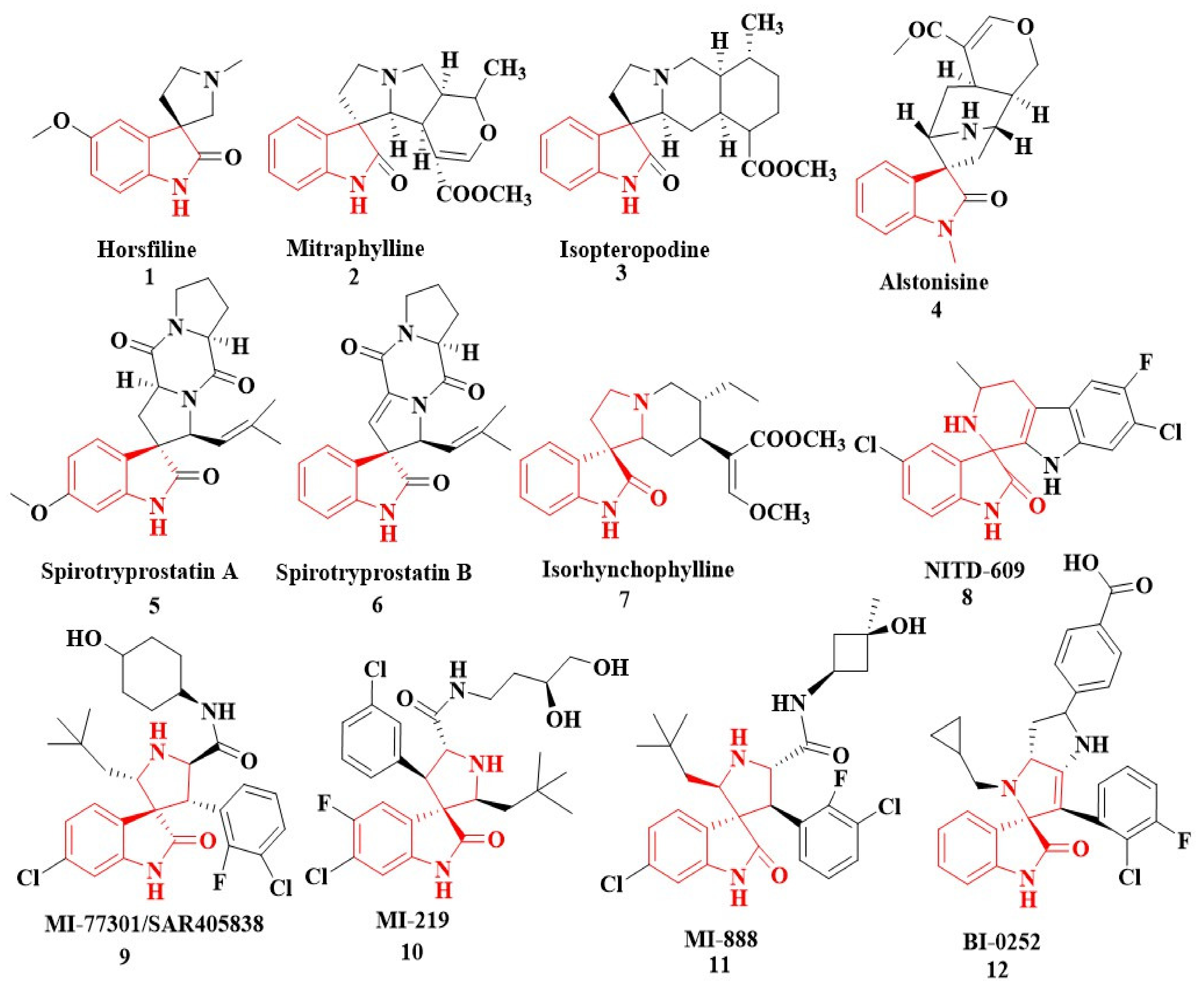
2.3. Newly Synthesized SOXs Exhibit Strong Anticancer Activity via Targeting Multiple Enzymes: Outcomes from the In Silico Molecular Docking Studies
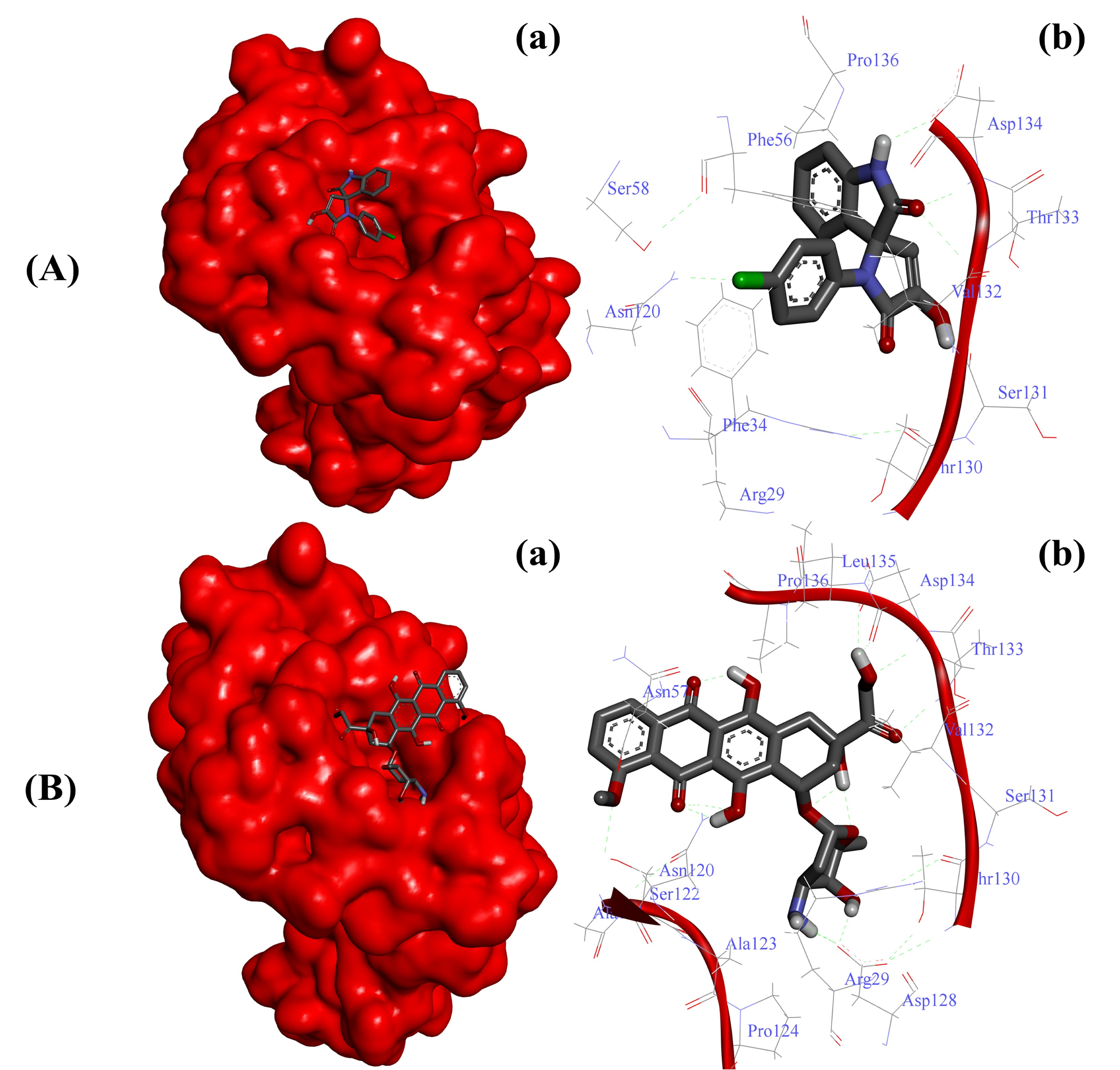
2.3.1. Substituted SOXs (4a–h) Are Potent Inhibitors of CD44
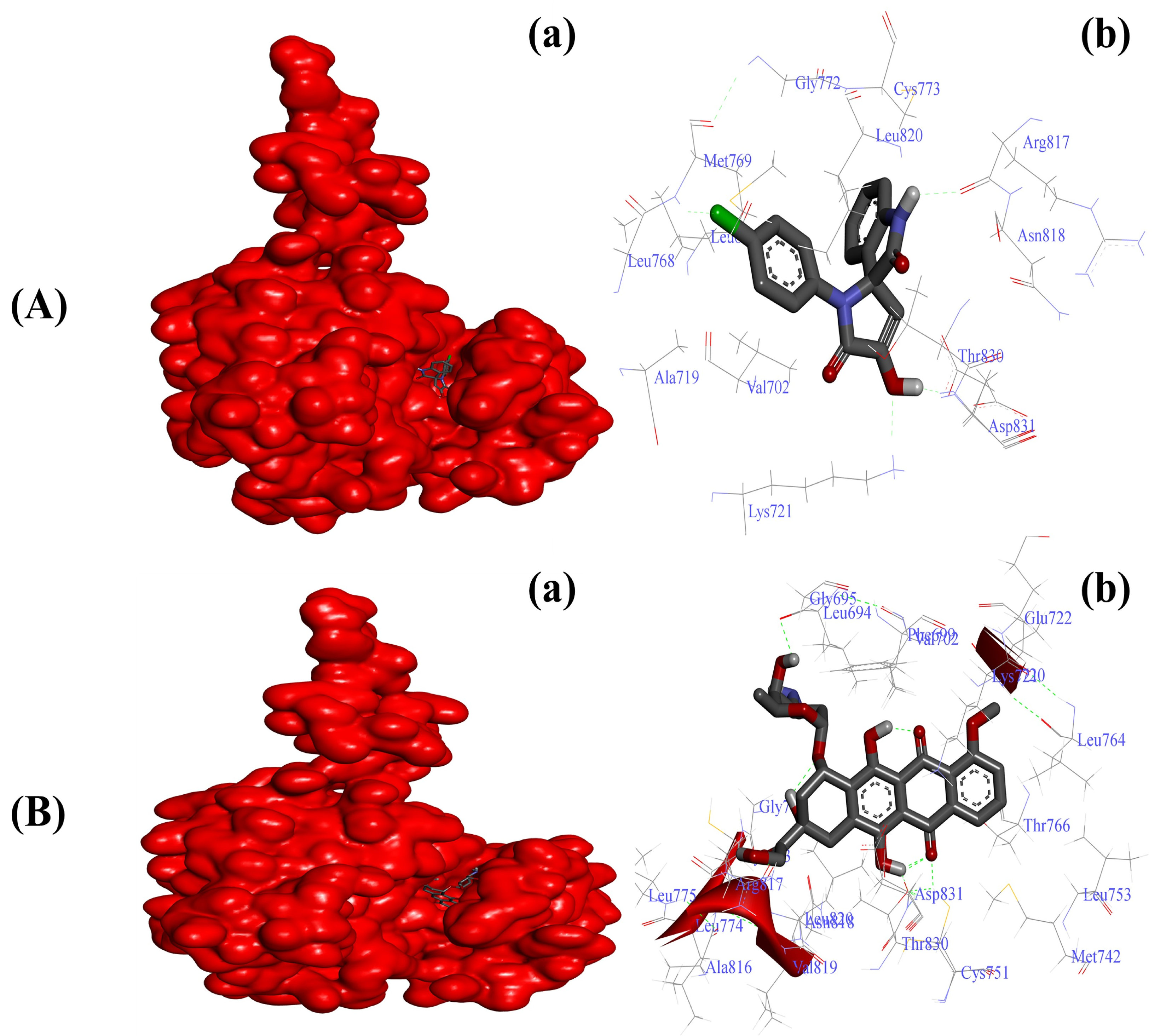
2.3.2. Substituted SOXs (4a–h) Are Potent Inhibitors of the Tyrosine Kinase Domain of the EGFR
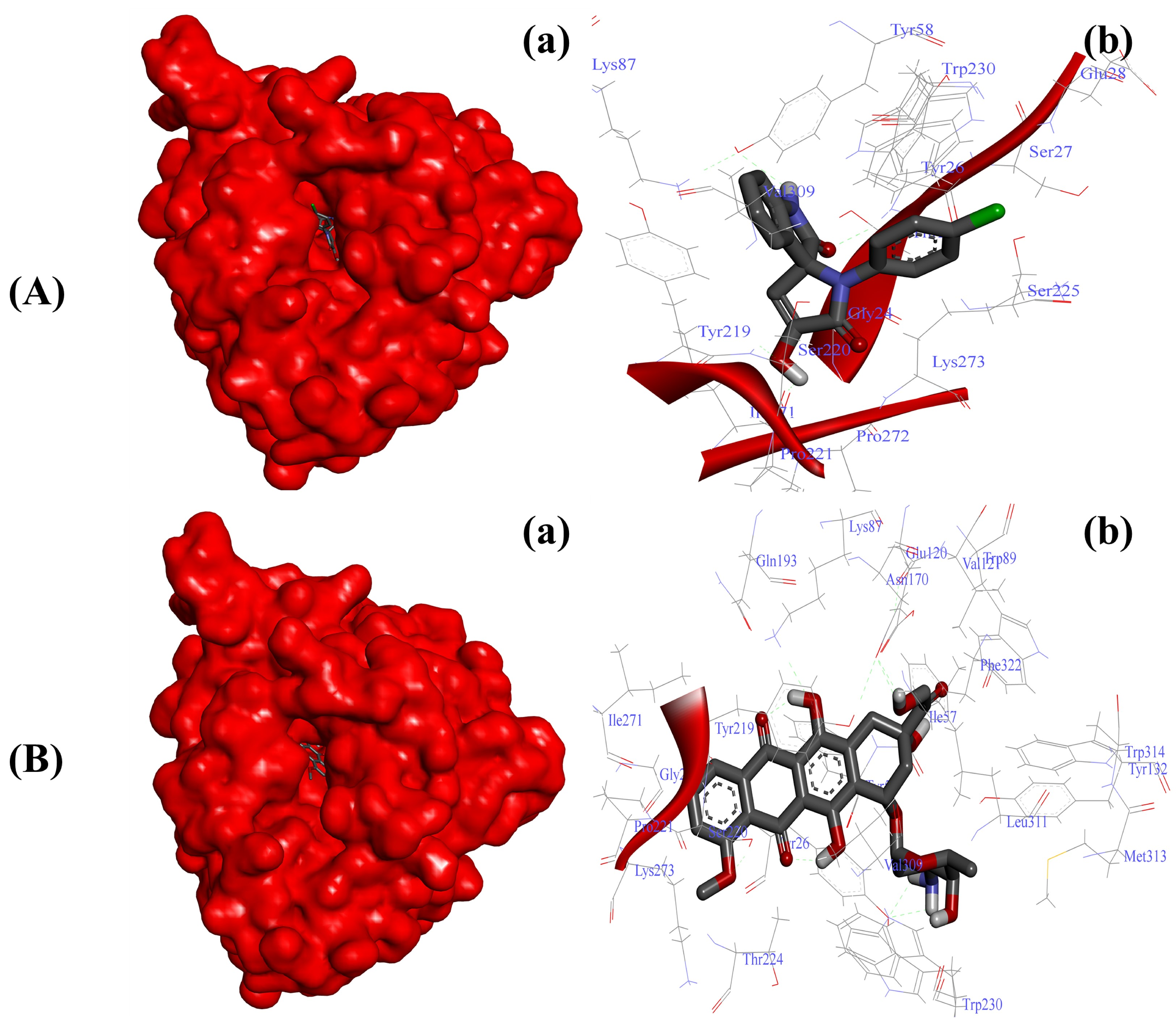
2.3.3. Substituted SOXs (4a–h) Inhibit the Activity of 5-β-Reductase (AKR1D1)
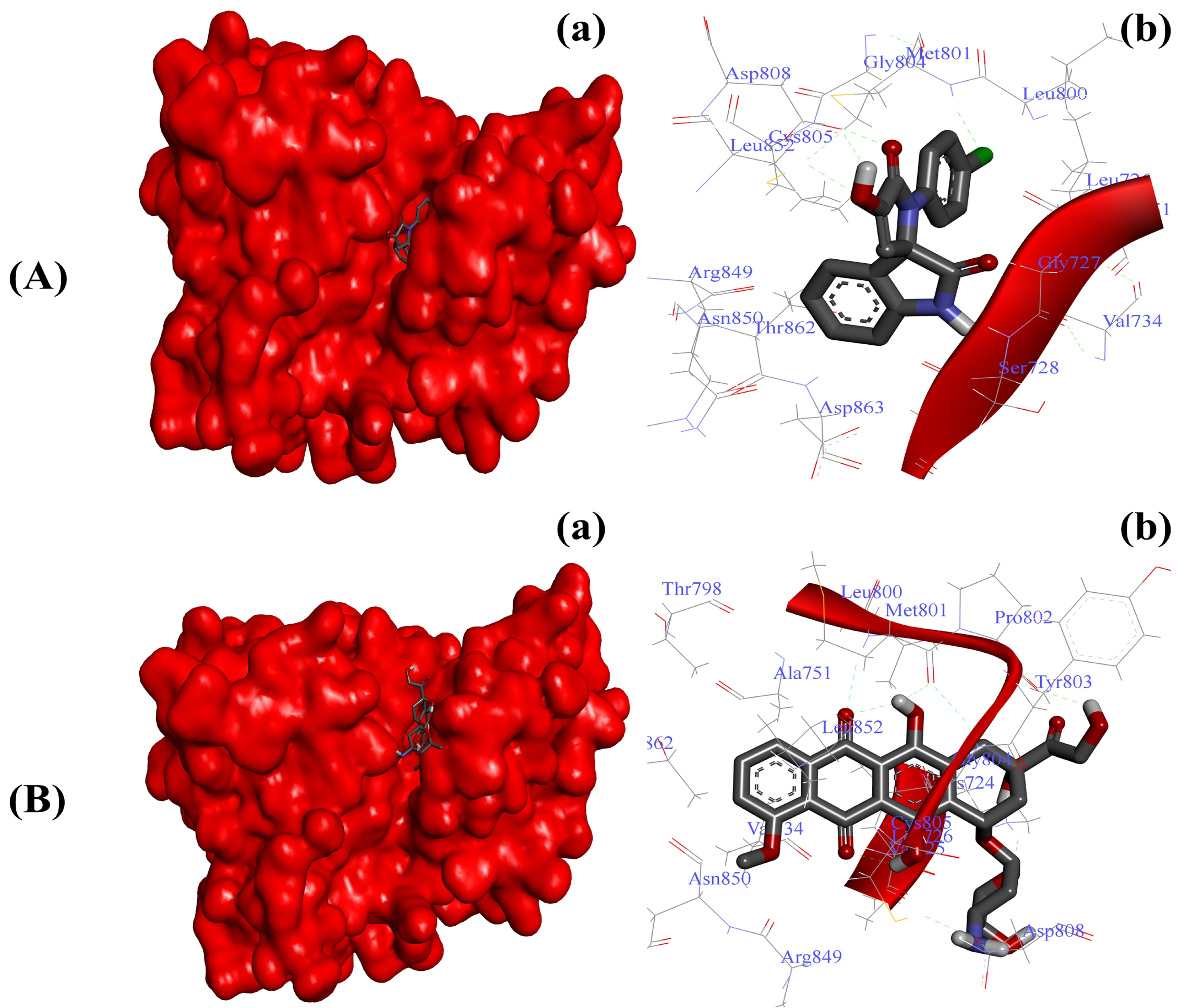
2.3.4. Substituted SOXs (4a–h) Occupy the Binding Pocket of HER-2
2.4. Substituted SOX (4a) Exerts Antiproliferative Effects against Prostate Adenocarcinoma Cells (PC-3)
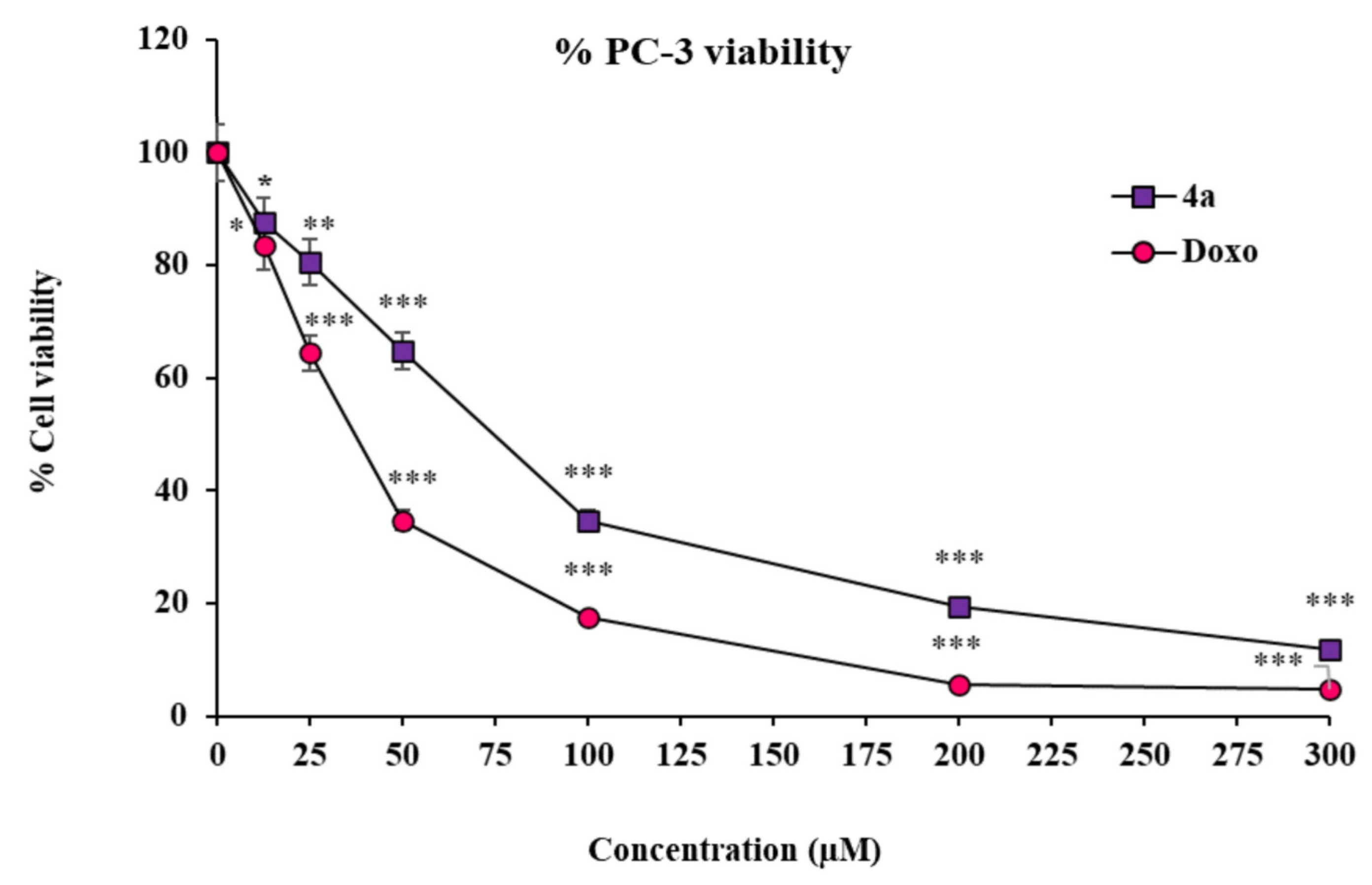
2.4.1. The Substituted SOX (4a) Exhibits Cytotoxic Effects on PC-3 Cells
2.4.2. The Substituted SOX (4a) Triggers ROS Generation in PC-3 Cells
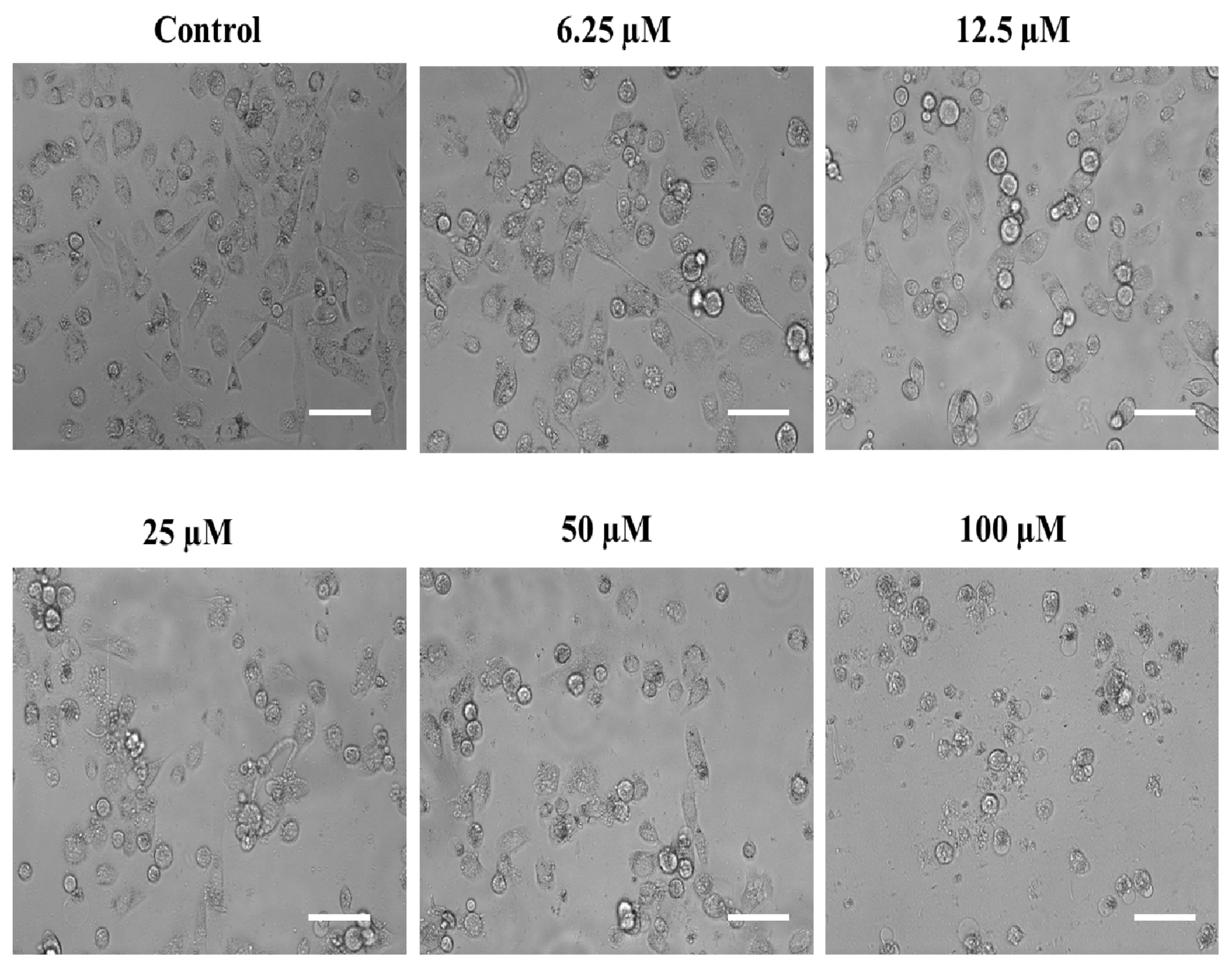
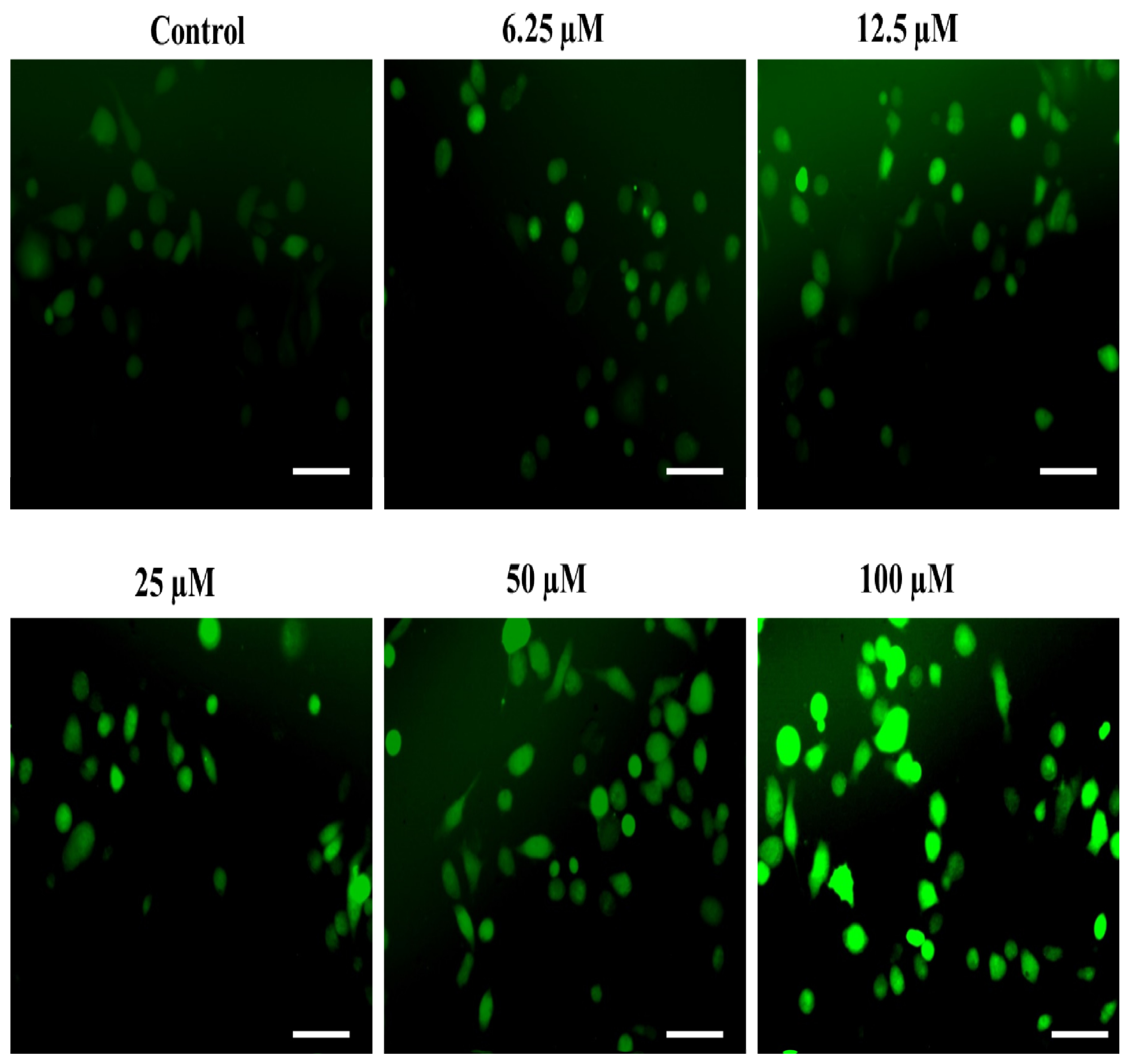
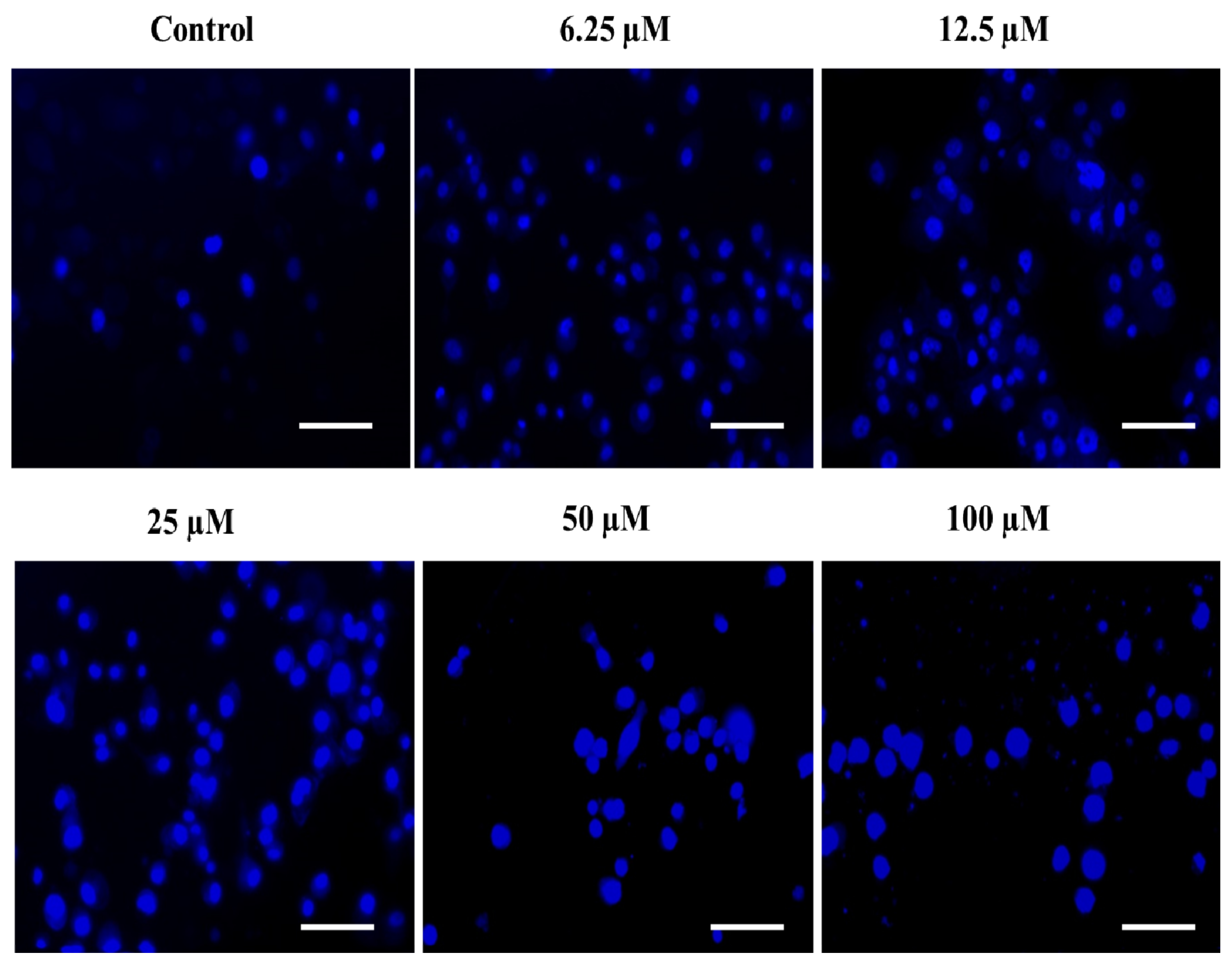
2.4.3. The Substituted SOX (4a) Stimulate the Nuclear Condensation in PC-3 Cells
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of the SOX Derivatives (4a–h)
3.1.2. Apparatus Used for the Characterization
3.2. Computational Chemistry
3.2.1. Ligand Preparation
3.2.2. ADME and Drug-Likeness of Substituted SOXs
3.2.3. Retrieval of the Human CD-44, EGFR, AKR1D1, and HER-2
3.2.4. Molecular Docking of Substituted SOXs against CD-44, EGFR, AKR1D1, and HER-2
3.3. In Vitro Antiproliferative Studies of Substituted SOX (4a) in PC3 Cells
3.3.1. Materials
3.3.2. Cell Culture
3.3.3. Investigations on the Cytotoxic Effects of Substituted SOX (4a) on PC-3 Cells through MTT Assay
3.3.4. Assessment of the Impact of the Substituted SOX (4a) on the Morphological Features of PC-3 Cells
3.3.5. Detection of ROS Generation
3.3.6. DAPI Staining
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer Prostate. Source: Globocan 2020 Number of New Cases in 2020, Both Sexes, All Ages; IARC: Lyon, France, 2020. [Google Scholar]
- Li, W.; Qian, L.; Lin, J.; Huang, G.; Hao, N.; Wei, X.; Wang, W.; Liang, J. CD44 Regulates Prostate Cancer Proliferation, Invasion and Migration via PDK1 and PFKFB4. Oncotarget 2017, 8, 65143. [Google Scholar] [CrossRef] [PubMed]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Li, H.; Bhatia, B.; Tang, S.; Reilly, J.G.; Chandra, D.; Zhou, J.; Claypool, K.; et al. Highly Purified CD44+ Prostate Cancer Cells from Xenograft Human Tumors Are Enriched in Tumorigenic and Metastatic Progenitor Cells. Oncogene 2006, 25, 1696–1708. [Google Scholar] [CrossRef] [PubMed]
- Bucay, N.; Sekhon, K.; Yang, T.; Majid, S.; Shahryari, V.; Hsieh, C.; Mitsui, Y.; Deng, G.; Tabatabai, Z.L.; Yamamura, S.; et al. MicroRNA-383 Located in Frequently Deleted Chromosomal Locus 8p22 Regulates CD44 in Prostate Cancer. Oncogene 2017, 36, 2667–2679. [Google Scholar] [CrossRef] [PubMed]
- Moasser, M.M. The Oncogene HER2: Its Signaling and Transforming Functions and Its Role in Human Cancer Pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef]
- Hyman, D.M.; Piha-Paul, S.A.; Won, H.; Rodon, J.; Saura, C.; Shapiro, G.I.; Juric, D.; Quinn, D.I.; Moreno, V.; Doger, B.; et al. HER Kinase Inhibition in Patients with HER2-and HER3-Mutant Cancers. Nature 2018, 554, 189–194. [Google Scholar] [CrossRef]
- Majumder, A.; Sandhu, M.; Banerji, D.; Steri, V.; Olshen, A.; Moasser, M.M. The Role of HER2 and HER3 in HER2-Amplified Cancers beyond Breast Cancers. Sci. Rep. 2021, 11, 9091. [Google Scholar] [CrossRef]
- Joshi, S.K.; Keck, J.M.; Eide, C.A.; Bottomly, D.; Traer, E.; Tyner, J.W.; McWeeney, S.K.; Tognon, C.E.; Druker, B.J. ERBB2/HER2 Mutations Are Transforming and Therapeutically Targetable in Leukemia. Leukemia 2020, 34, 2798–2804. [Google Scholar] [CrossRef]
- Wen, W.; Chen, W.S.; Xiao, N.; Bender, R.; Ghazalpour, A.; Tan, Z.; Swensen, J.; Millis, S.Z.; Basu, G.; Gatalica, Z.; et al. Mutations in the Kinase Domain of the HER2/ERBB2 Gene Identified in a Wide Variety of Human Cancers. J. Mol. Diagn. 2015, 17, 487–495. [Google Scholar] [CrossRef]
- Schlam, I.; Swain, S.M. HER2-Positive Breast Cancer and Tyrosine Kinase Inhibitors: The Time Is Now. npj Breast Cancer 2021, 7, 56. [Google Scholar] [CrossRef]
- Moy, B.; Goss, P.E. Lapatinib-Associated Toxicity and Practical Management Recommendations. Oncologist 2007, 12, 756–765. [Google Scholar] [CrossRef]
- Nikolaou, N.; Gathercole, L.L.; Marchand, L.; Althari, S.; Dempster, N.J.; Green, C.J.; van de Bunt, M.; McNeil, C.; Arvaniti, A.; Hughes, B.A.; et al. AKR1D1 Is a Novel Regulator of Metabolic Phenotype in Human Hepatocytes and Is Dysregulated in Non-Alcoholic Fatty Liver Disease. Metabolism 2019, 99, 67–80. [Google Scholar] [CrossRef]
- Nikolaou, N.; Gathercole, L.L.; Kirkwood, L.; Dunford, J.E.; Hughes, B.A.; Gilligan, L.C.; Oppermann, U.; Penning, T.M.; Arlt, W.; Hodson, L.; et al. AKR1D1 Regulates Glucocorticoid Availability and Glucocorticoid Receptor Activation in Human Hepatoma Cells. J. Steroid Biochem. Mol. Biol. 2019, 189, 218–227. [Google Scholar] [CrossRef]
- Tan, M.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen Receptor: Structure, Role in Prostate Cancer and Drug Discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef]
- Fenner, A. AR Regulates CD44. Nat. Rev. Urol. 2021, 18, 252. [Google Scholar] [CrossRef]
- Basu, S.; Tindall, D.J. Androgen Action in Prostate Cancer. Horm. Cancer 2010, 1, 223–228. [Google Scholar] [CrossRef]
- Feng, Q.; He, B. Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 858. [Google Scholar] [CrossRef]
- Drury, J.E.; Di Costanzo, L.; Penning, T.M.; Christianson, D.W. Inhibition of Human Steroid 5β-Reductase (AKR1D1) by Finasteride and Structure of the Enzyme-Inhibitor Complex. J. Biol. Chem. 2009, 284, 19786–19790. [Google Scholar] [CrossRef]
- Diviccaro, S.; Melcangi, R.C.; Giatti, S. Post-Finasteride Syndrome: An Emerging Clinical Problem. Neurobiol. Stress 2020, 12, 100209. [Google Scholar] [CrossRef]
- Thomas, R.; Weihua, Z. Rethink of EGFR in Cancer with Its Kinase Independent Function on Board. Front. Oncol. 2019, 9, 800. [Google Scholar] [CrossRef]
- Du, Z.; Brown, B.P.; Kim, S.; Ferguson, D.; Pavlick, D.C.; Jayakumaran, G.; Benayed, R.; Gallant, J.N.; Zhang, Y.K.; Yan, Y.; et al. Structure–Function Analysis of Oncogenic EGFR Kinase Domain Duplication Reveals Insights into Activation and a Potential Approach for Therapeutic Targeting. Nat. Commun. 2021, 12, 1382. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.Q.; Peng, Y.; Buckley, M.T.; Wei, J.; Chen, F.; Liebes, L.; Gerald, W.L.; Pincus, M.R.; Osman, I.; Lee, P. Epidermal Growth Factor Receptor Activation in Prostate Cancer by Three Novel Missense Mutations. Oncogene 2008, 27, 3201–3210. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Chmielecki, J. Rational, Biologically Based Treatment of EGFR-Mutant Non-Small-Cell Lung Cancer. Nat. Rev. Cancer 2010, 10, 760–774. [Google Scholar] [CrossRef] [PubMed]
- Dancer, J.; Takei, H.; Ro, J.Y.; Lowery-Nordberg, M. Coexpression of EGFR and HER-2 in Pancreatic Ductal Adenocarcinoma: A Comparative Study Using Immunohistochemistry Correlated with Gene Amplification by Fluorescencent in Situ Hybridization. Oncol. Rep. 2007, 18, 151–155. [Google Scholar] [CrossRef]
- Ahmad, P.; Alvi, S.S.; Iqbal, D.; Khan, M.S. Insights into Pharmacological Mechanisms of Polydatin in Targeting Risk Factors-Mediated Atherosclerosis. Life Sci. 2020, 254, 117756. [Google Scholar] [CrossRef]
- Alvi, S.S.; Ansari, I.A.; Khan, I.; Iqbal, J.; Khan, M.S. Potential Role of Lycopene in Targeting Proprotein Convertase Subtilisin/Kexin Type-9 to Combat Hypercholesterolemia. Free Radic. Biol. Med. 2017, 108, 394–403. [Google Scholar] [CrossRef]
- Ahmad, P.; Alvi, S.S.; Khan, M.S. Functioning of Organosulfur Compounds from Garlic (Allium sativum Linn) in Targeting Risk Factor-Mediated Atherosclerosis: A Cross Talk Between Alternative and Modern Medicine. In Natural Bio-Active Compounds; Springer: Singapore, 2019; pp. 561–585. ISBN 9789811371547. [Google Scholar]
- Alvi, S.S.; Ahmad, P.; Ishrat, M.; Iqbal, D.; Khan, M.S. Secondary Metabolites from Rosemary (Rosmarinus officinalis L.): Structure, Biochemistry and Therapeutic Implications against Neurodegenerative Diseases. In Natural Bio-Active Compounds: Chemistry, Pharmacology and Health Care Practices; Springer: Singapore, 2019; Volume 2, pp. 1–24. ISBN 9789811372056. [Google Scholar]
- Akhter, F.; Alvi, S.S.; Ahmad, P.; Iqbal, D.; Alshehri, B.M.; Khan, M.S. Therapeutic Efficacy of Boerhaavia diffusa (Linn.) Root Methanolic Extract in Attenuating Streptozotocin-Induced Diabetes, Diabetes-Linked Hyperlipidemia and Oxidative-Stress in Rats. Biomed. Res. Ther. 2019, 6, 3293–3306. [Google Scholar] [CrossRef]
- Nabi, R.; Alvi, S.S.; Shah, A.; Chaturvedi, C.P.; Faisal, M.; Alatar, A.A.; Ahmad, S.; Khan, M.S. Ezetimibe Attenuates Experimental Diabetes and Renal Pathologies via Targeting the Advanced Glycation, Oxidative Stress and AGE-RAGE Signalling in Rats. Arch. Physiol. Biochem. 2021, 1–16. [Google Scholar] [CrossRef]
- Nabi, R.; Alvi, S.S.; Alouffi, S.; Khan, S.; Ahmad, A.; Khan, M.; Ahmad, S.; Khan, S. Amelioration of Neuropilin-1 and RAGE/Matrix Metalloproteinase-2 Pathway-Induced Renal Injury in Diabetic Rats by Rosuvastatin. Arch. Biol. Sci. 2021, 73, 265–278. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, F.; Zhao, B.; Yang, J.; Ferrara, J.; Sankaran, B.; Venkataram Prasad, B.V.; Kundu, B.B.; Phillips, G.N.; Gao, Y.; et al. Structural Basis of the Stereoselective Formation of the Spirooxindole Ring in the Biosynthesis of Citrinadins. Nat. Commun. 2021, 12, 4158. [Google Scholar] [CrossRef]
- Hassan, F.; Azad, I.; Asif, M.; Shukla, D.; Husain, A.; Khan, A.R.; Saquib, M.; Nasibullah, M. Isatin Conjugates as Antibacterial Agents: A Brief Review. Med. Chem. 2022, 18, 413–430. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, J.; Sun, T.; Zhang, X.; Yao, J.; Kai, M.; Jiang, X.; Wang, R. Anti-Cancer Small Molecule JP-8g Exhibits Potent in Vivo Anti-Inflammatory Activity. Sci. Rep. 2014, 4, 4372. [Google Scholar] [CrossRef]
- Davis, H.J.; Kavanagh, M.E.; Balan, T.; Abell, C.; Coyne, A.G. Spirooxindoles as Novel 3D-Fragment Scaffolds: Synthesis and Screening against CYP121 from M. Tuberculosis. Bioorganic Med. Chem. Lett. 2016, 26, 3735–3740. [Google Scholar] [CrossRef]
- Dhokne, P.; Sakla, A.P.; Shankaraiah, N. Structural Insights of Oxindole Based Kinase Inhibitors as Anticancer Agents: Recent Advances. Eur. J. Med. Chem. 2021, 216, 113334. [Google Scholar] [CrossRef]
- Zhang, W.H.; Chen, S.; Liu, X.L.; Feng, T.T.; De Yang, W.; Zhou, Y. Design, Synthesis and Evaluation of Structurally Diverse Chrysin-Chromene-Spirooxindole Hybrids as Anticancer Agents. Bioorg. Med. Chem. 2019, 27, 115109. [Google Scholar] [CrossRef]
- Jeba Reeda, V.S.; Bena Jothy, V.; Asif, M.; Nasibullah, M.; Alharbi, N.S.; Abbas, G.; Muthu, S. Synthesis, Solvent Polarity(Polar and Nonpolar), Structural and Electronic Properties with Diverse Solvents and Biological Studies of (E)-3-((3-Chloro-4-Fluorophenyl) Imino) Indolin-2-One. J. Mol. Liq. 2023, 380, 121709. [Google Scholar] [CrossRef]
- Singh, M.; Hazra, A.; Bharitkar, Y.P.; Kalia, R.; Sahoo, A.; Saha, S.; Ravichandiran, V.; Ghosh, S.; Mondal, N.B. Synthesis of Diversely Substituted Bis-Pyrrolizidino/ Thiopyrrolizidino Oxindolo/Acenaphthyleno Curcuminoids: Via Sequential Azomethine Ylide Cycloaddition. RSC Adv. 2018, 8, 18938–18951. [Google Scholar] [CrossRef]
- Ghosh, R.; Vitor, J.B.; Mendes, E.; Paulo, A.; Acharya, P.C. Stereoselective Synthesis of Spirooxindole Derivatives Using One-Pot Multicomponent Cycloaddition Reaction and Evaluation of Their Antiproliferative Efficacy. ACS Omega 2020, 5, 27332–27343. [Google Scholar] [CrossRef]
- Asif, M.; Azaz, T.; Tiwari, B.; Nasibullah, M. Propagative Isatin in Organic Synthesis of Spirooxindoles through Catalysis. Tetrahedron 2023, 134, 133308. [Google Scholar] [CrossRef]
- Islam, M.S.; Ghawas, H.M.; El-Senduny, F.F.; Al-Majid, A.M.; Elshaier, Y.A.M.M.; Badria, F.A.; Barakat, A. Synthesis of New Thiazolo-Pyrrolidine–(Spirooxindole) Tethered to 3-Acylindole as Anticancer Agents. Bioorg. Chem. 2019, 82, 423–430. [Google Scholar] [CrossRef]
- Al-Rashood, S.T.; Hamed, A.R.; Hassan, G.S.; Alkahtani, H.M.; Almehizia, A.A.; Alharbi, A.; Al-Sanea, M.M.; Eldehna, W.M. Antitumor Properties of Certain Spirooxindoles towards Hepatocellular Carcinoma Endowed with Antioxidant Activity. J. Enzym. Inhib. Med. Chem. 2020, 35, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Asif, M.; Saquib, M.; Rahman Khan, A.; Aqil, F.; Salem Almalki, A.; Ali Alasmary, F.; Singh, J.; Nasibullah, M. Synthesis of Functionalized 2′,5-Oxo-spiro[Furan-2,3′-indoline]-3-carboxylate Derivatives as Antiproliferative Agents: ADMET Studies, and Molecular Docking against P2Y12 Inhibitors. ChemistrySelect 2023, 8, e202204536. [Google Scholar] [CrossRef]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S.; et al. Rethinking Drug Design in the Artificial Intelligence Era. Nat. Rev. Drug Discov. 2020, 19, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Hessler, G.; Baringhaus, K.-H. Artificial Intelligence in Drug Design. Molecules 2018, 23, 2520. [Google Scholar] [CrossRef]
- Ahmad, P.; Alvi, S.S.; Iqbal, J.; Khan, M.S. Identification and Evaluation of Natural Organosulfur Compounds as Potential Dual Inhibitors of α-Amylase and α-Glucosidase Activity: An in-Silico and in-Vitro Approach. Med. Chem. Res. 2021, 30, 2184–2202. [Google Scholar] [CrossRef]
- Ahmad, P.; Alvi, S.S.; Iqbal, J.; Khan, M.S. Target-Based Virtual and Biochemical Screening Against HMG-CoA Reductase Reveals Allium sativum-Derived Organosulfur Compound N-Acetyl Cysteine as a Cardioprotective Agent. Rev. Bras. Farmacogn. 2022, 32, 962–973. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal Chemical Properties of Successful Central Nervous System Drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef]
- Hitchcock, S.A.; Pennington, L.D. Structure-Brain Exposure Relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef]
- Prasanna, S.; Doerksen, R. Topological Polar Surface Area: A Useful Descriptor in 2D-QSAR. Curr. Med. Chem. 2008, 16, 21–41. [Google Scholar] [CrossRef]
- Ma, X.L.; Chen, C.; Yang, J. Predictive Model of Blood-Brain Barrier Penetration of Organic Compounds. Acta Pharmacol. Sin. 2005, 26, 500–512. [Google Scholar] [CrossRef]
- Leão, R.P.; Cruz, J.V.; da Costa, G.V.; Cruz, J.N.; Ferreira, E.F.B.; Silva, R.C.; de Lima, L.R.; Borges, R.S.; Dos Santos, G.B.; Santos, C.B.R. Identification of New Rofecoxib-Based Cyclooxygenase-2 Inhibitors: A Bioinformatics Approach. Pharmaceuticals 2020, 13, 209. [Google Scholar] [CrossRef]
- Bittermann, K.; Goss, K.U. Predicting Apparent Passive Permeability of Caco-2 and MDCK Cell-Monolayers: A Mechanistic Model. PLoS ONE 2017, 12, e0190319. [Google Scholar] [CrossRef]
- Volpe, D.A. Variability in Caco-2 and MDCK Cell-Based Intestinal Permeability Assays. J. Pharm. Sci. 2008, 97, 712–725. [Google Scholar] [CrossRef]
- Yamashita, S.; Furubayashi, T.; Kataoka, M.; Sakane, T.; Sezaki, H.; Tokuda, H. Optimized Conditions for Prediction of Intestinal Drug Permeability Using Caco-2 Cells. Eur. J. Pharm. Sci. 2000, 10, 195–204. [Google Scholar] [CrossRef]
- Lundborg, M.; Wennberg, C.L.; Narangifard, A.; Lindahl, E.; Norlén, L. Predicting Drug Permeability through Skin Using Molecular Dynamics Simulation. J. Control. Release 2018, 283, 269–279. [Google Scholar] [CrossRef]
- Chen, C.-P.; Chen, C.-C.; Huang, C.-W.; Chang, Y.-C. Evaluating Molecular Properties Involved in Transport of Small Molecules in Stratum Corneum: A Quantitative Structure-Activity Relationship for Skin Permeability. Molecules 2018, 23, 911. [Google Scholar] [CrossRef]
- Roberts, J.A.; Pea, F.; Lipman, J. The Clinical Relevance of Plasma Protein Binding Changes. Clin. Pharmacokinet. 2013, 52, 1–8. [Google Scholar] [CrossRef]
- Gurevich, K.G. Effect of Blood Protein Concentrations on Drug-Dosing Regimes: Practical Guidance. Theor. Biol. Med. Model. 2013, 10, 20. [Google Scholar] [CrossRef]
- Kim, J.E.; Cho, H.J.; Kim, J.S.; Shim, C.K.; Chung, S.J.; Oak, M.H.; Yoon, I.S.; Kim, D.D. The Limited Intestinal Absorption via Paracellular Pathway Is Responsible for the Low Oral Bioavailability of Doxorubicin. Xenobiotica 2013, 43, 579–591. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Le, J.; Abraham, M.H.; Hersey, A.; Eddershaw, P.J.; Luscombe, C.N.; Boutina, D.; Beck, G.; Sherborne, B.; Cooper, I.; et al. Evaluation of Human Intestinal Absorption Data and Subsequent Derivation of a Quantitative Structure—Activity Relationship (QSAR) with the Abraham Descriptors. J. Pharm. Sci. 2001, 90, 749–784. [Google Scholar] [CrossRef]
- Palanissami, G.; Paul, S.F.D. RAGE and Its Ligands: Molecular Interplay Between Glycation, Inflammation, and Hallmarks of Cancer—A Review. Horm. Cancer 2018, 9, 295–325. [Google Scholar] [CrossRef] [PubMed]
- Nabi, R.; Alvi, S.S.; Saeed, M.; Ahmad, S.; Khan, M.S. Glycation and HMG-CoA Reductase Inhibitors: Implication in Diabetes and Associated Complications. Curr. Diabetes Rev. 2019, 15, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Nabi, R.; Alvi, S.S.; Shah, A.; Chaturvedi, C.P.; Iqbal, D.; Ahmad, S.; Khan, M.S. Modulatory Role of HMG-CoA Reductase Inhibitors and Ezetimibe on LDL-AGEs-Induced ROS Generation and RAGE-Associated Signalling in HEK-293 Cells. Life Sci. 2019, 235, 116823. [Google Scholar] [CrossRef] [PubMed]
- Alvi, S.S.; Nabi, R.; Khan, M.S.; Akhter, F.; Ahmad, S.; Khan, M.S. Glycyrrhizic Acid Scavenges Reactive Carbonyl Species and Attenuates Glycation-Induced Multiple Protein Modification: An In Vitro and In Silico Study. Oxid. Med. Cell. Longev. 2021, 2021, 7086951. [Google Scholar] [CrossRef]
- Nabi, R.; Alvi, S.S.; Shah, M.S.; Ahmad, S.; Faisal, M.; Alatar, A.A.; Khan, M.S. A Biochemical & Biophysical Study on In-Vitro Anti-Glycating Potential of Iridin against D-Ribose Modified BSA. Arch. Biochem. Biophys. 2020, 686, 108373. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Teh, L.K.; Bertilsson, L. Pharmacogenomics of CYP2D6: Molecular Genetics, Interethnic Differences and Clinical Importance. Drug Metab. Pharmacokinet. 2012, 27, 55–67. [Google Scholar] [CrossRef]
- Walko, C.M.; McLeod, H. Use of CYP2D6 Genotyping in Practice: Tamoxifen Dose Adjustment. Pharmacogenomics 2012, 13, 691–697. [Google Scholar] [CrossRef]
- Neves Cruz, J.; Santana De Oliveira, M.; Gomes Silva, S.; Pedro Da Silva Souza Filho, A.; Santiago Pereira, D.; Lima E Lima, A.H.; De Aguiar Andrade, E.H. Insight into the Interaction Mechanism of Nicotine, NNK, and NNN with Cytochrome P450 2A13 Based on Molecular Dynamics Simulation. J. Chem. Inf. Model. 2020, 60, 766–776. [Google Scholar] [CrossRef]
- Wang, G.; Liu, Z.; Li, M.; Li, Y.; Alvi, S.S.; Ansari, I.A.; Salman Khan, M. Ginkgolide B Mediated Alleviation of Inflammatory Cascades and Altered Lipid Metabolism in HUVECs via Targeting PCSK-9 Expression and Functionality. Biomed Res. Int. 2019, 2019, 7284767. [Google Scholar] [CrossRef]
- Alvi, S.S.; Iqbal, D.; Ahmad, S.; Khan, M.S. Molecular Rationale Delineating the Role of Lycopene as a Potent HMG-CoA Reductase Inhibitor: In Vitro and in Silico Study. Nat. Prod. Res. 2016, 30, 2111–2114. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Wang, P.; Yang, C.; Li, S. Reduced Pim-1 Expression Increases Chemotherapeutic Drug Sensitivity in Human Androgen-independent Prostate Cancer Cells by Inducing Apoptosis. Exp. Ther. Med. 2019, 18, 2731–2738. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, B.; Li, X.; Lu, D.; Yang, L.; Chen, L.; Li, Y.; Cheng, L.; Lv, F.; Zhang, P.; et al. ATF4/CEMIP/PKCα Promotes Anoikis Resistance by Enhancing Protective Autophagy in Prostate Cancer Cells. Cell Death Dis. 2022, 13, 46. [Google Scholar] [CrossRef]
- Waiz, M.; Alvi, S.S.; Khan, M.S. Potential Dual Inhibitors of PCSK-9 and HMG-R from Natural Sources in Cardiovascular Risk Management. EXCLI J. 2022, 21, 47–76. [Google Scholar] [CrossRef]
- Jahan, F.; Alvi, S.S.; Islam, M.H. Berberis Aristata and Its Secondary Metabolites: Insights into Nutraceutical and Therapeutical Applications. Pharmacol. Res.—Mod. Chin. Med. 2022, 5, 100184. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in Cancer Therapy: The Bright Side of the Moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Hashim, A.; Alvi, S.S.; Ansari, I.A.; Salman Khan, M. Phyllanthus Virgatus Forst Extract and It’s Partially Purified Fraction Ameliorates Oxidative Stress and Retino-Nephropathic Architecture in Streptozotocin-Induced Diabetic Rats. Pak. J. Pharm. Sci. 2019, 32, 2697–2708. [Google Scholar] [CrossRef]
- Nabi, R.; Alvi, S.S.; Khan, R.H.; Ahmad, S.; Ahmad, S.; Khan, M.S. Antiglycation Study of HMG-R Inhibitors and Tocotrienol against Glycated BSA and LDL: A Comparative Study. Int. J. Biol. Macromol. 2018, 116, 983–992. [Google Scholar] [CrossRef]
- Zhang, J.H.; Xu, M. DNA Fragmentation in Apoptosis. Cell Res. 2000, 10, 205–211. [Google Scholar] [CrossRef]
- Estandarte, A.K.; Botchway, S.; Lynch, C.; Yusuf, M.; Robinson, I. The Use of DAPI Fluorescence Lifetime Imaging for Investigating Chromatin Condensation in Human Chromosomes. Sci. Rep. 2016, 6, 31417. [Google Scholar] [CrossRef]
- Begue, F.; Tanaka, S.; Mouktadi, Z.; Rondeau, P.; Veeren, B.; Diotel, N.; Tran-Dinh, A.; Robert, T.; Vélia, E.; Mavingui, P.; et al. Altered High-Density Lipoprotein Composition and Functions during Severe COVID-19. Sci. Rep. 2021, 11, 2291. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Wang, Z.; Cheng, Y.; Ren, J.; Wu, B.; Li, W.; Wang, X.; Su, X.; Liu, Z. Computational Study of Novel Natural Inhibitors Targeting Aminopeptidase N(CD13). Aging 2020, 12, 8523–8535. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.S.; De Fabritiis, G. DeepSite: Protein-Binding Site Predictor Using 3D-Convolutional Neural Networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef] [PubMed]
- Alvi, S.S.; Ansari, I.A.; Ahmad, M.K.; Iqbal, J.; Khan, M.S. Lycopene Amends LPS Induced Oxidative Stress and Hypertriglyceridemia via Modulating PCSK-9 Expression and Apo-CIII Mediated Lipoprotein Lipase Activity. Biomed. Pharmacother. 2017, 96, 1082–1093. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | p-TSA (mM) | Solvent | Time (h) | Temp. | Yield (%) |
|---|---|---|---|---|---|
| 1 | 0 | EtOH | 16 | RT | trace |
| 2 | 0.25 | EtOH | 12 | RT | 55 |
| 3 | 0.25 | EtOH | 10 | RT | 55 |
| 4 | 0.5 | EtOH | 10 | RT | 62 |
| 5 | 0.5 | EtOH | 6 | RT | 70 |
| 6 | 0.5 | EtOH | 8 | RT | 95 |
| 7 | 0.7 | EtOH | 10 | RT | 60 |
| 8 | 0.7 | EtOH | 8 | RT | 65 |
| 9 | 0.5 | MeOH | 8 | RT | 65 |
| 10 | 0.5 | EtOH:H2O (2:1) | 8 | RT | trace |
| 11 | 0.8 | EtOH: H2O (2:1) | 8 | RT | trace |
| 12 | 0.5 | EtOAc | 8 | RT | ND |
| 13 | 0.5 | Acetone | 8 | RT | ND |
| 14 | 0.5 | Dime. sulfoxide | 8 | RT | ND |
| 15 | 0.5 | Isopropanol | 8 | RT | trace |
| Entry | DOX | Arylamine | Time (h) | SOX | Melting Point (°C) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 |  1a |  2a | 8 |  4a | 244 | 95 |
| 2 |  1a |  2b | 8 |  4b | 243 | 79 |
| 3 |  1a |  2c | 6 |  4c | 245 | 83 |
| 4 |  1b |  2c | 9 |  4d | 248 | 84 |
| 5 |  1b |  2b | 12 |  4e | 239 | 74 |
| 6 |  1b |  2a | 8 |  4f | 236 | 81 |
| 7 |  1c |  2c | 8 |  4g | 246 | 84 |
| 8 |  1c |  2b | 8 |  4h | 241 | 95 |
| S. No. | Compounds | CD44 (PDB ID: 1UUH) | EGFR (PDB ID: 1M17) | AKR1D1 (PDB ID: 3CQA) | HER-2 (PDB ID: 3PP0) |
|---|---|---|---|---|---|
| 1 | 4a | −7.4 | −8.7 | −10.1 | −7.8 |
| 2 | 4b | −7.0 | −8.3 | −9.3 | −6.8 |
| 3 | 4c | −7.1 | −8.0 | −9.4 | −6.6 |
| 4 | 4d | −7.2 | −8.2 | −8.1 | −6.8 |
| 5 | 4e | −6.7 | −8.3 | −7.6 | −7.1 |
| 6 | 4f | −7.2 | −8.2 | −7.1 | −7.4 |
| 7 | 4g | −6.5 | −8.0 | −9.9 | −7.7 |
| 8 | 4h | −6.8 | −8.4 | −9.8 | −7.3 |
| 9 | * Doxorubicin | −7.0 | −10.1 | −10.4 | −7.7 |
| Complex | Binding Energy | Interacting Residues |
|---|---|---|
| EGFR-4a | −6.55 Kcal/mol | Leu694, Val702, Ala720, Lys721, Leu768, Met769, Gly772, Cys773, Arg817, Asn818, Leu820, Thr830, Asp831 |
| EGFR-Doxo | −10.11 Kcal/mol | Leu694, Phe699, Ala719, Lys721, Glu738, Met742, Val762, Leu764, Thr766, Gln767, Leu768, Met769, Pro770, Gly772, Arg817, Asn818, Leu820, Thr830, Asp831, Phe832 |
| CD44-4a | −6.65 Kcal/mol | Arg29, Phe34, Phe56, Asn120, Thr130, Ser131, Val132, Thr133, Asp134, Pro136, Ser158 |
| CD44-Doxo | −5.83 Kcal/mol | Arg29, Asn57, Asn120, Ala121, Ser122, Ala123, Pro124, Asn128, Thr130, Ser131, Val132, Thr133, Asp134, Leu135, Pro136 |
| AKR1D1-4a | −8.73 Kcal/mol | Gly24, Thr25, Tyr26, Ser27, Glu28, Tyr58, Lys87, Tyr219, Ser220, Pro221, Ser225, Tyr230, Ile271, Pro272, Gly273, Val309 |
| AKR1D1-Doxo | −10.01 Kcal/mol | Gly24, Tyr26, Ile57, Tyr58, Lys87, Trp89, Val121, Glu120, Tyr132, Asn170, Gln193, Tyr219, Ser220, Pro221, Thr224, Trp230, Ile271, Lys273, Val309, Leu311, Met313, Trp314, Phe322 |
| HER-2-4a | −7.27 Kcal/mol | Gly77, Ser78, Lue726, Gyl729, Val734, Ala751, Leu800, Met801, Gly804, Cys805, Asp808, Asp850, Leu852, Arg859, Thr862, Asp863 |
| HER-2-Doxo | −8.94 Kcal/mol | Lys724, Val725, Leu726, Val734, Ala751, Thr798, Leu800, Met801, Pro802, Tyr803, Gly804, Cys805, Asp808, Arg849, Asn850, Leu852, Thr862 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asif, M.; Alvi, S.S.; Azaz, T.; Khan, A.R.; Tiwari, B.; Hafeez, B.B.; Nasibullah, M. Novel Functionalized Spiro [Indoline-3,5′-pyrroline]-2,2′dione Derivatives: Synthesis, Characterization, Drug-Likeness, ADME, and Anticancer Potential. Int. J. Mol. Sci. 2023, 24, 7336. https://doi.org/10.3390/ijms24087336
Asif M, Alvi SS, Azaz T, Khan AR, Tiwari B, Hafeez BB, Nasibullah M. Novel Functionalized Spiro [Indoline-3,5′-pyrroline]-2,2′dione Derivatives: Synthesis, Characterization, Drug-Likeness, ADME, and Anticancer Potential. International Journal of Molecular Sciences. 2023; 24(8):7336. https://doi.org/10.3390/ijms24087336
Chicago/Turabian StyleAsif, Mohd, Sahir Sultan Alvi, Tazeen Azaz, Abdul Rahman Khan, Bhoopendra Tiwari, Bilal Bin Hafeez, and Malik Nasibullah. 2023. "Novel Functionalized Spiro [Indoline-3,5′-pyrroline]-2,2′dione Derivatives: Synthesis, Characterization, Drug-Likeness, ADME, and Anticancer Potential" International Journal of Molecular Sciences 24, no. 8: 7336. https://doi.org/10.3390/ijms24087336
APA StyleAsif, M., Alvi, S. S., Azaz, T., Khan, A. R., Tiwari, B., Hafeez, B. B., & Nasibullah, M. (2023). Novel Functionalized Spiro [Indoline-3,5′-pyrroline]-2,2′dione Derivatives: Synthesis, Characterization, Drug-Likeness, ADME, and Anticancer Potential. International Journal of Molecular Sciences, 24(8), 7336. https://doi.org/10.3390/ijms24087336