
Natural Compounds Targeting the Autophagy Pathway in the Treatment of Colorectal Cancer


Abstract

1. Introduction
2. Epidemiology of CRC
3. Autophagy in CRC
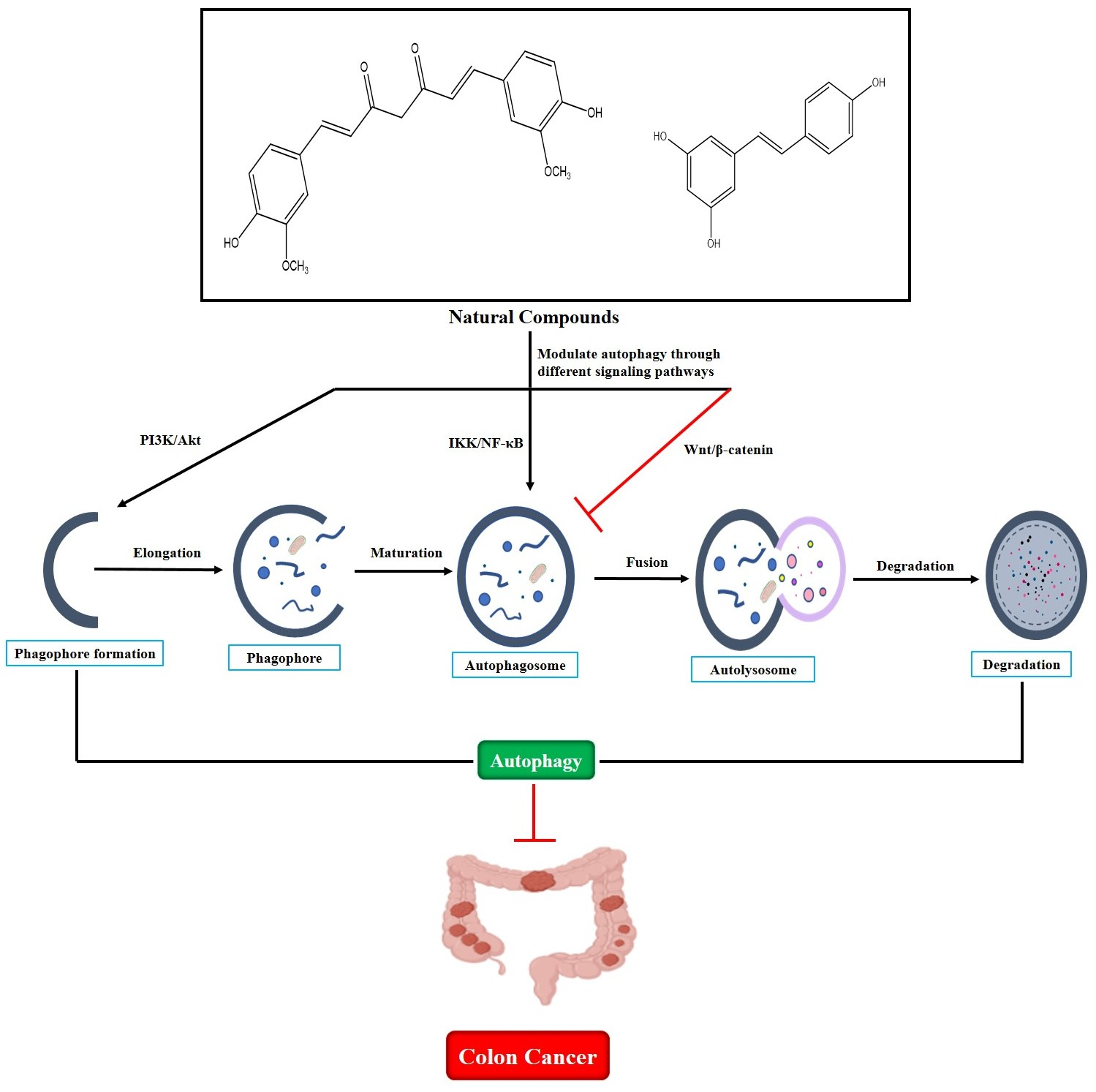
3.1. Overview of Autophagy
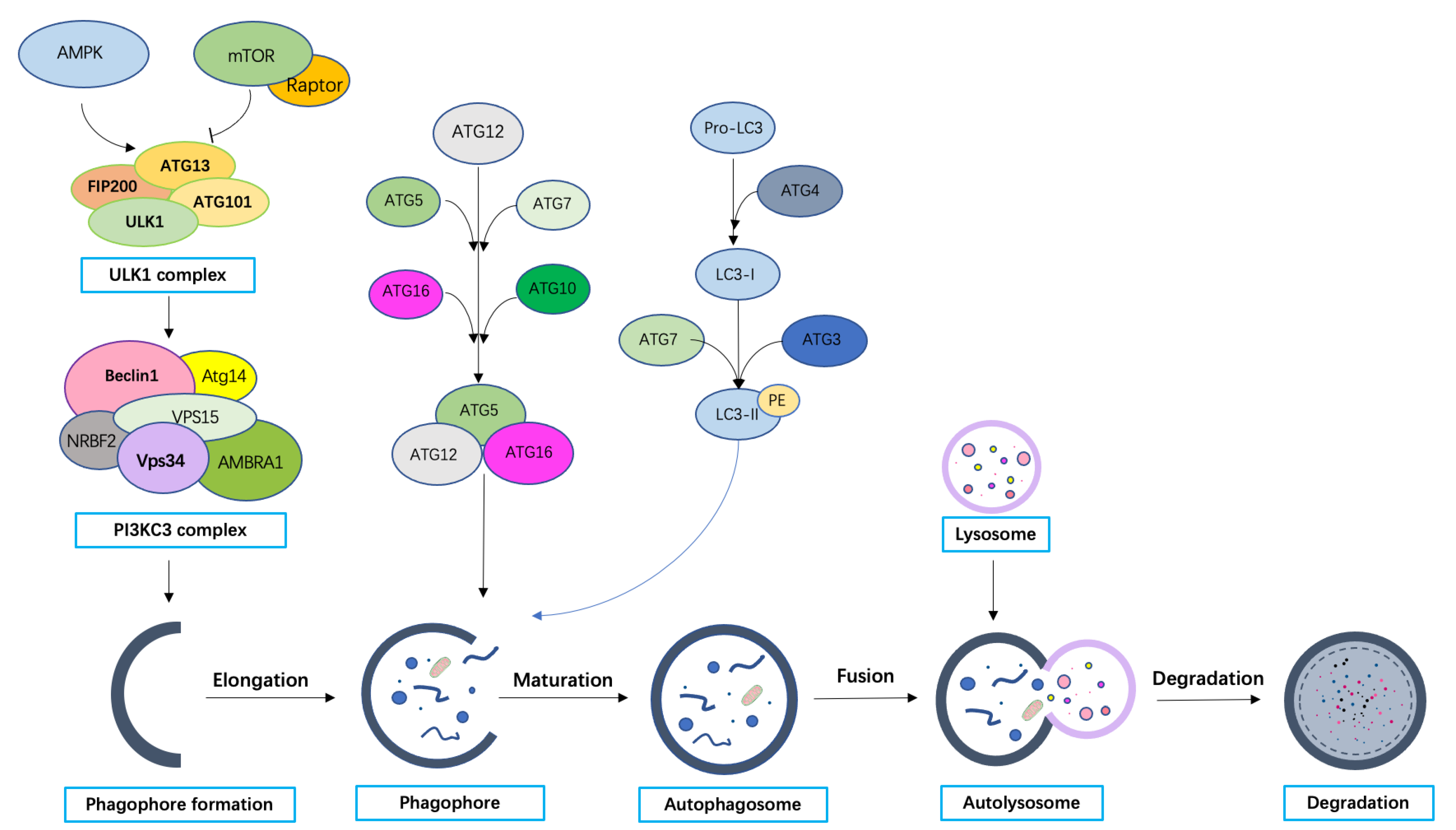
3.2. Molecular Machinery of Autophagy
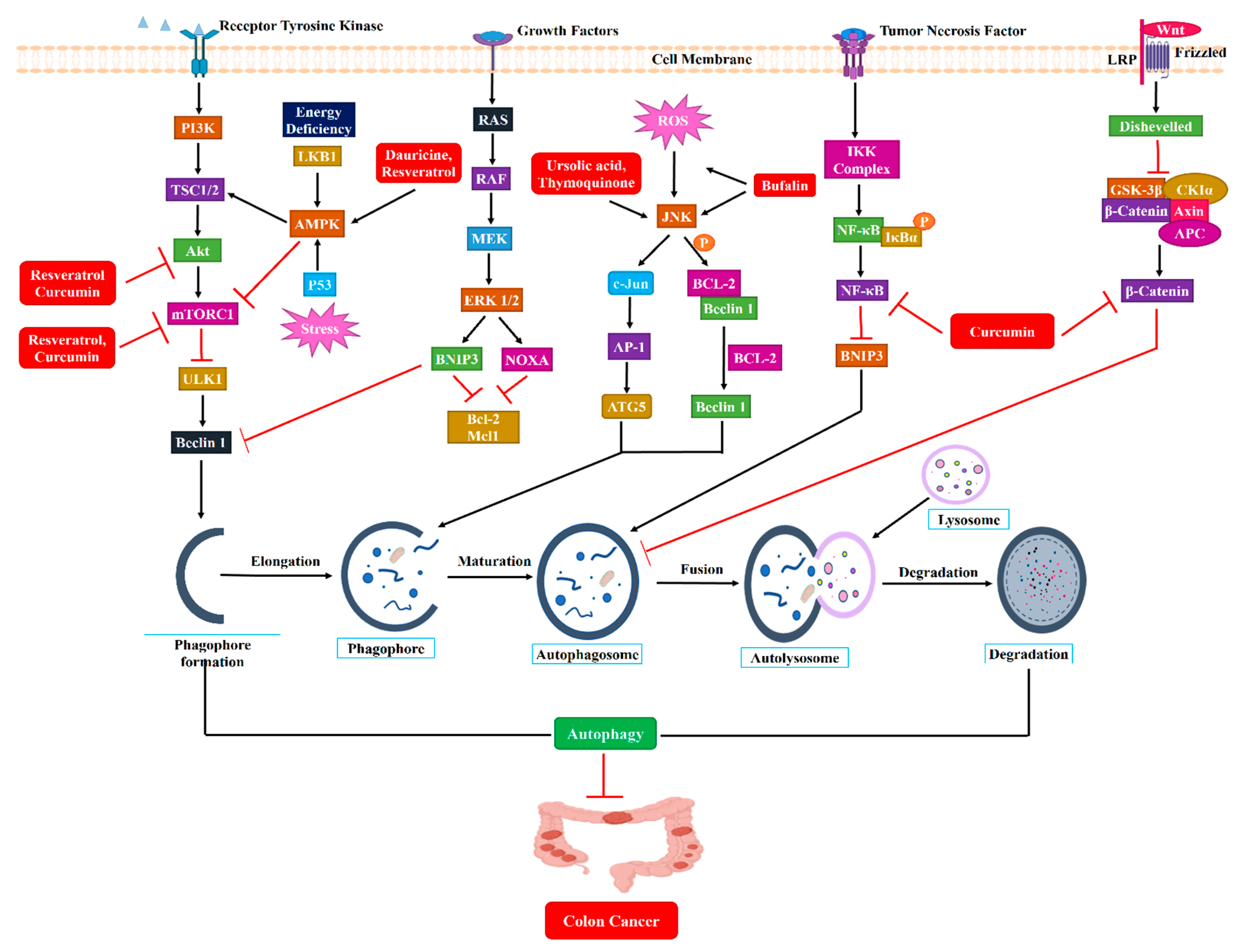
3.3. Dual Role of Autophagy in CRC Development
3.3.1. Tumor Inhibiting Function of Autophagy
3.3.2. Tumor Promoting Function of Autophagy
3.3.3. Autophagy and Chemotherapy Resistance in CRC
3.3.4. Autophagy and Gut Microbiota in CRC
3.3.5. Autophagy and Immunotherapy in CRC
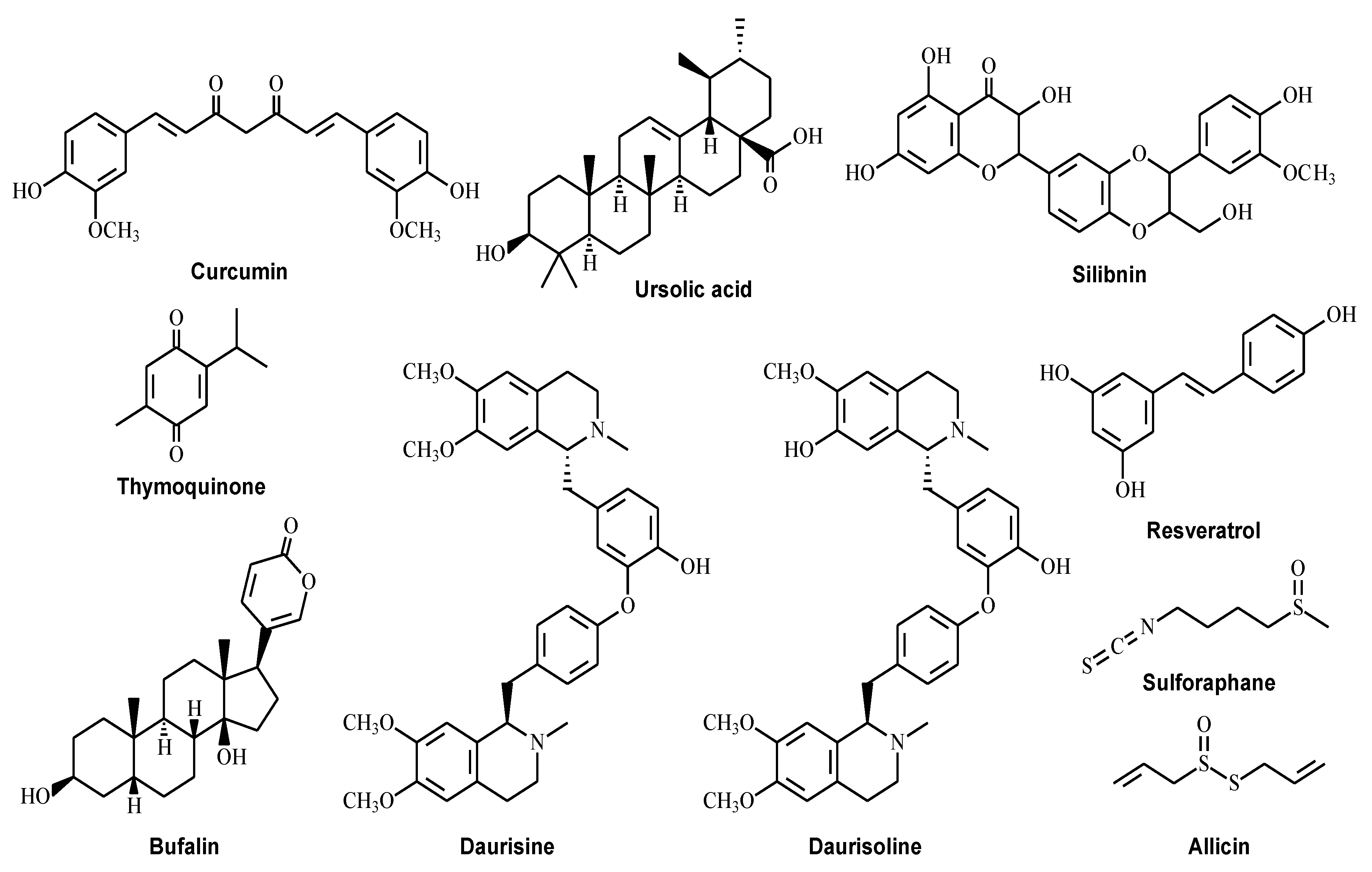
4. Natural Compounds That Modulate Autophagy as Potential CRC Treatments
4.1. Curcumin
4.2. Ursolic Acid
4.3. Silibinin
4.4. Thymoquinone
4.5. Dauricine and Daurisoline
4.6. Bufalin
4.7. Resveratrol
4.8. Sulforaphane
4.9. Allicin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CRC Cell Line | Effect on Autophagy | Mechanism of Action | IC50 | References |
|---|---|---|---|---|---|
| Curcumin | HT-29 HCT-15 HCT116 | Activator | Inducing autophagy-related death by inhibiting the AKT/mTOR pathway or activating the ERK1/2 pathway Induction of autophagy via suppression of mTOR and activation of transcription factor EB (TFEB), and also cause cancer cell cycle accumulation at the G2/M phase | 40 μM | [135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150] |
| Ursolic Acid | HCT-15 | Activator | Inducing autophagy which is activated by the CaMKK-AMPK-mTOR pathway. Accumulation of LC3 and p62 levels and activation of caspase-independent cancer cell apoptosis through the JNK pathway | 37.2 μM | [151,152,153,154,155,156,157,158] |
| Silibinin | SW480 LoVo HT-29 HCT116 | Activator | Activation of autophagy via oxidative stress-mediated BNIP3-dependent manner or the mTOR inhibition manner Inhibition of cancer cell proliferation through extracellular signal-regulated kinase 1/2 (ERK1/2), AKT phosphorylation, and NF-κB inactivation manner | 83 μM | [159,160,161,162,163,164,165,166] |
| Thymoquinone | LoVo HCT116 | Inhibitor | Sensitize the chemotherapy by inhibiting NF-κB and MEK signaling Inhibition of the late stage of autophagy and cause the accumulation of LC3 and p62 levels | 51.73 μM (HCT-116) 99.46 μM (HT29) | [167,168,169,170,171,172,173,174] |
| Dauricine Daurisoline | HCT116 HCT8 SW480 SW620 | Inhibitor | Impairing autophagy at the autophagosome maturation stage due to p62 accumulation and the increased ratio of GFP-LC3/RFP-LC3 Suppression of colon cancer invasion via CaMKKβ-AMPK-mTOR signaling cascades pathway and NF-κB pathways | Both are >20 μM in HCT8 cell line | [175,176,177,178,179,180] |
| Bufalin | HT-29 Caco-2 HCT116 | Activator | Inhibition of cancer cell proliferation by suppressing NF-κB and PI3K/AKT signaling pathways Activation of autophagy and apoptosis through inhibiting the phosphorylation of Akt and mTOR, while phosphorylated ERK1/2 | 12.823 ± 1.792 nM (HCT-116) 26.303 ± 2.498 nM (SW620) | [181,182,183,184,185] |
| Resveratrol | DLD1 HCT15 LoVo HCT116 HT-29 COLO 201 | Activator | Activation of autophagic cell death through MALAT1 regulated WNT/β-catenin signaling pathway Induction of autophagy via ROS production and mTOR inhibition | 170 μM | [186,187,188,189,190,191,192,193,194,195,196,197,198,199] |
| Sulforaphane | SW480, DLD1 HCT116 HT-29 | Activator | Induction of autophagic cell death by modulating Wnt/β-catenin and ERK/Nrf2 signaling pathways | 18.82 μM (DLD-1) 15.73 μM (HCT-116) | [200,201,202,203,204,205,206,207,208,209] |
| Allicin | HCT-116, LS174T HT-29 Caco-2 | Activator | Activation of autophagy through ROS accumulation and the AKT/mTOR inhibition Inducing CRC cell apoptosis through regulating Nrf2 mediated cytochrome c and NF-κB releasing process | 64.7 μM (SW620) 80 μM (HCT-116) | [210,211,212,213,214,215,216] |
5. Clinical Study of Autophagy-Modulating Natural Products in Colon Cancer
6. Discussion and Limitations
7. Conclusions and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kuipers, E.J.; Rösch, T.; Bretthauer, M. Colorectal cancer screening—Optimizing current strategies and new directions. Nat. Rev. Clin. Oncol. 2013, 10, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Carballal, S.; Rodríguez-Alcalde, D.; Moreira, L.; Hernández, L.; Rodríguez, L.; Rodríguez-Moranta, F.; Gonzalo, V.; Bujanda, L.; Bessa, X.; Poves, C. Colorectal cancer risk factors in patients with serrated polyposis syndrome: A large multicentre study. Gut 2016, 65, 1829–1837. [Google Scholar] [CrossRef]
- Simmonds, P.; Best, L.; George, S.; Baughan, C.; Buchanan, R.; Davis, C.; Fentiman, I.; Gosney, M.; Northover, J.; Williams, C. Surgery for colorectal cancer in elderly patients: A systematic review. Lancet 2000, 356, 968–974. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Mayer, R.J. Systemic treatment of colorectal cancer. Gastroenterology 2008, 134, 1296–1310.e1. [Google Scholar] [CrossRef] [PubMed]
- Chau, I.; Cunningham, D. Chemotherapy in colorectal cancer: New options and new challenges. Br. Med. Bull. 2002, 64, 159–180. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Heinemann, V.; Douillard, J.; Ducreux, M.; Peeters, M. Targeted therapy in metastatic colorectal cancer–an example of personalised medicine in action. Cancer Treat. Rev. 2013, 39, 592–601. [Google Scholar] [CrossRef]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Qin, Z.-H. Autophagy: Biology and Diseases; Springer: Singapore, 2019. [Google Scholar]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuk, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007, 21, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- White, E.; DiPaola, R.S. The Double-Edged Sword of Autophagy Modulation in CancerAutophagy in Cancer Therapy. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, metabolism, and cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Levine, B. Bcl-2 inhibition of autophagy: A new route to cancer? Cancer Res. 2006, 66, 2885–2888. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Fu, L.-l.; Cheng, Y.; Liu, B. Beclin-1: Autophagic regulator and therapeutic target in cancer. Int. J. Biochem. Cell Biol. 2013, 45, 921–924. [Google Scholar] [CrossRef]
- WHO Colorectal Cancer Awareness Month 2022. World Health Organization. Available online: https://www.iarc.who.int/featured-news/colorectal-cancer-awareness-month-2022/ (accessed on 24 May 2022).
- Tran, K.B.; Lang, J.J.; Compton, K.; Xu, R.; Acheson, A.R.; Henrikson, H.J.; Kocarnik, J.M.; Penberthy, L.; Aali, A.; Abbas, Q. The global burden of cancer attributable to risk factors, 2010–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 563–591. [Google Scholar] [CrossRef]
- Henrikson, N.B.; Webber, E.M.; Goddard, K.A.; Scrol, A.; Piper, M.; Williams, M.S.; Zallen, D.T.; Calonge, N.; Ganiats, T.G.; Janssens, A. Family history and the natural history of colorectal cancer: Systematic review. Genet. Med. 2015, 17, 702–712. [Google Scholar] [CrossRef]
- Shussman, N.; Wexner, S.D. Colorectal polyps and polyposis syndromes. Gastroenterol. Rep. 2014, 2, 1–15. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Allen, J.I.; Axilbund, J.E.; Boland, C.R.; Burke, C.A.; Burt, R.W.; Church, J.M.; Dominitz, J.A.; Johnson, D.A.; Kaltenbach, T. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi-society Task Force on colorectal cancer. Gastroenterology 2014, 147, 502–526. [Google Scholar] [CrossRef]
- Lynch, H.T.; Smyrk, T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome): An updated review. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1996, 78, 1149–1167. [Google Scholar] [CrossRef]
- Moreira, L.; Balaguer, F.; Lindor, N.; De la Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; Le Marchand, L.; Gallinger, S.; Newcomb, P.A. Identification of Lynch syndrome among patients with colorectal cancer. Jama 2012, 308, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.N.; Dove, W.F. APC and its modifiers in colon cancer. In Apc Proteins; Näthke, I.S., McCartney, B.M., Eds.; Springer: New York, NY, USA, 2009; pp. 85–106. [Google Scholar]
- Tenesa, A.; Campbell, H.; Barnetson, R.; Porteous, M.; Dunlop, M.; Farrington, S. Association of MUTYH and colorectal cancer. Br. J. Cancer 2006, 95, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Bardhan, K.; Liu, K. Epigenetics and colorectal cancer pathogenesis. Cancers 2013, 5, 676–713. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef]
- Nassar, D.; Blanpain, C. Cancer stem cells: Basic concepts and therapeutic implications. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 47–76. [Google Scholar] [CrossRef]
- Devoto, L.; Celentano, V.; Cohen, R.; Khan, J.; Chand, M. Colorectal cancer surgery in the very elderly patient: A systematic review of laparoscopic versus open colorectal resection. Int. J. Color. Dis. 2017, 32, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Guillou, P.J.; Quirke, P.; Thorpe, H.; Walker, J.; Jayne, D.G.; Smith, A.M.; Heath, R.M.; Brown, J.M. Short-term endpoints of conventional versus laparoscopic-assisted surgery in patients with colorectal cancer (MRC CLASICC trial): Multicentre, randomised controlled trial. Lancet 2005, 365, 1718–1726. [Google Scholar] [CrossRef]
- Gustafsson, U.O.; Hausel, J.; Thorell, A.; Ljungqvist, O.; Soop, M.; Nygren, J.; Group, E.R.A.S.S. Adherence to the enhanced recovery after surgery protocol and outcomes after colorectal cancer surgery. Arch. Surg. 2011, 146, 571–577. [Google Scholar] [CrossRef]
- Bondeven, P.; Hagemann-Madsen, R.; Laurberg, S.; Pedersen, B.G. Extent and completeness of mesorectal excision evaluated by postoperative magnetic resonance imaging. J. Br. Surg. 2013, 100, 1357–1367. [Google Scholar] [CrossRef]
- Audisio, R.A.; Papamichael, D. Treatment of colorectal cancer in older patients. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Heemskerk-Gerritsen, B.A.; Rookus, M.A.; Aalfs, C.M.; Ausems, M.G.; Collée, J.M.; Jansen, L.; Kets, C.M.; Keymeulen, K.B.; Koppert, L.B.; Meijers-Heijboer, H.E. Improved overall survival after contralateral risk-reducing mastectomy in BRCA1/2 mutation carriers with a history of unilateral breast cancer: A prospective analysis. Int. J. Cancer 2015, 136, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Bennouna, J.; Sastre, J.; Arnold, D.; Österlund, P.; Greil, R.; Van Cutsem, E.; von Moos, R.; Viéitez, J.M.; Bouché, O.; Borg, C. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): A randomised phase 3 trial. Lancet Oncol. 2013, 14, 29–37. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef]
- Ramesh, J.; Ronsard, L.; Gao, A.; Venugopal, B. Autophagy intertwines with different diseases—Recent strategies for therapeutic approaches. Diseases 2019, 7, 15. [Google Scholar] [CrossRef]
- Spaink, H.; Spaink, H. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2016, 12, 1–222. [Google Scholar]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 105–137. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-M.; Jung, C.H.; Seo, M.; Otto, N.M.; Grunwald, D.; Kim, K.H.; Moriarity, B.; Kim, Y.-M.; Starker, C.; Nho, R.S. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 2016, 12, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.-I.; Natsume, T.; Takehana, K.; Yamada, N. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Bakula, D.; Müller, A.J.; Zuleger, T.; Takacs, Z.; Franz-Wachtel, M.; Thost, A.-K.; Brigger, D.; Tschan, M.P.; Frickey, T.; Robenek, H. WIPI3 and WIPI4 β-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat. Commun. 2017, 8, 15637. [Google Scholar] [CrossRef]
- Thurston, T.L.; Ryzhakov, G.; Bloor, S.; Von Muhlinen, N.; Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar] [CrossRef]
- Weidberg, H.; Elazar, Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci. Signal. 2011, 4, pe39. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Mizushima, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Deosaran, E.; Larsen, K.B.; Hua, R.; Sargent, G.; Wang, Y.; Kim, S.; Lamark, T.; Jauregui, M.; Law, K.; Lippincott-Schwartz, J. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 2013, 126, 939–952. [Google Scholar] [CrossRef] [PubMed]
- von Muhlinen, N.; Akutsu, M.; Ravenhill, B.J.; Foeglein, Á.; Bloor, S.; Rutherford, T.J.; Freund, S.M.; Komander, D.; Randow, F. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol. Cell 2012, 48, 329–342. [Google Scholar] [CrossRef]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Young, A.R.; Chan, E.Y.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef] [PubMed]
- Orsi, A.; Razi, M.; Dooley, H.; Robinson, D.; Weston, A.; Collinson, L.; Tooze, S. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef]
- Reggiori, F.; Ungermann, C. Autophagosome maturation and fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Yim, W.W.-Y.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future DirectionsTargeting Autophagy in Cancer. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef]
- Koukourakis, M.; Giatromanolaki, A.; Sivridis, E.; Pitiakoudis, M.; Gatter, K.; Harris, A. Beclin 1 over-and underexpression in colorectal cancer: Distinct patterns relate to prognosis and tumour hypoxia. Br. J. Cancer 2010, 103, 1209–1214. [Google Scholar] [CrossRef]
- Shen, Y.; Li, D.-D.; Wang, L.-L.; Deng, R.; Zhu, X.-F. Decreased expression of autophagy-related proteins in malignant epithelial ovarian cancer. Autophagy 2008, 4, 1067–1068. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Aita, V.M.; Liang, X.H.; Murty, V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.C.; Levine, B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar] [CrossRef]
- Zhu, J.; Cai, Y.; Xu, K.; Ren, X.; Sun, J.; Lu, S.; Chen, J.; Xu, P. Beclin1 overexpression suppresses tumor cell proliferation and survival via an autophagy-dependent pathway in human synovial sarcoma cells. Oncol. Rep. 2018, 40, 1927–1936. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, J.; Sui, Y.; Jin, L.; Yang, Y.; Lin, S.; Shi, H. Beclin1 overexpression inhibitis proliferation, invasion and migration of CaSki cervical cancer cells. Asian Pac. J. Cancer Prev. 2011, 12, 1269–1273. [Google Scholar]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef]
- Qu, X.; Zou, Z.; Sun, Q.; Luby-Phelps, K.; Cheng, P.; Hogan, R.N.; Gilpin, C.; Levine, B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007, 128, 931–946. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ghoorun, R.A.; Fan, X.; Wu, P.; Bai, Y.; Li, J.; Chen, H.; Wang, L.; Wang, J. High expression of Beclin-1 predicts favorable prognosis for patients with colorectal cancer. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 98–106. [Google Scholar] [CrossRef]
- Chen, N.; Karantza-Wadsworth, V. Role and regulation of autophagy in cancer. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 1516–1523. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Zhang, C.; Yi, H.; Wu, C.; Wang, J.; Liu, Y.; Tan, J.; Wen, J. Downregulation of Beclin1 and impairment of autophagy in a small population of colorectal cancer. Dig. Dis. Sci. 2013, 58, 2887–2894. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.-P.; Budina, A.; Balaburski, G.; Bergenstock, M.K.; Murphy, M. Autophagy in tumor suppression and cancer therapy. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 71–100. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zhao, Z.; Yang, Y.; O’connell, D.; Zhang, X.; Oh, S.; Ma, B.; Lee, J.-H.; Zhang, T.; Varghese, B. Truncating mutation in the autophagy gene UVRAG confers oncogenic properties and chemosensitivity in colorectal cancers. Nat. Commun. 2015, 6, 7839. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Saeed, O.; Lopez-Beltran, A.; Fisher, K.W.; Scarpelli, M.; Montironi, R.; Cimadamore, A.; Massari, F.; Santoni, M.; Cheng, L. RAS genes in colorectal carcinoma: Pathogenesis, testing guidelines and treatment implications. J. Clin. Pathol. 2019, 72, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Tsuchihara, K.; Fujii, S.; Sugiyama, M.; Goya, T.; Atomi, Y.; Ueno, T.; Ochiai, A.; Esumi, H. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res. 2007, 67, 9677–9684. [Google Scholar] [CrossRef]
- Yang, A.; Rajeshkumar, N.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy Is Critical for Pancreatic Tumor Growth and Progression in Tumors with p53 AlterationsAutophagy Is Critical for Pancreatic Tumor Growth. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.; Nguyen, V.H.; Salazar, M.A.; Wong, P.; Diamond, D.J.; Yim, J.H.; Melstrom, L.G. Inhibition of autophagy amplifies baicalein-induced apoptosis in human colorectal cancer. Mol. Ther. Oncolytics 2020, 19, 1–7. [Google Scholar] [CrossRef]
- Sakitani, K.; Hirata, Y.; Hikiba, Y.; Hayakawa, Y.; Ihara, S.; Suzuki, H.; Suzuki, N.; Serizawa, T.; Kinoshita, H.; Sakamoto, K. Inhibition of autophagy exerts anti-colon cancer effects via apoptosis induced by p53 activation and ER stress. BMC Cancer 2015, 15, 795. [Google Scholar] [CrossRef] [PubMed]
- Scherr, A.-L.; Jassowicz, A.; Pató, A.; Elssner, C.; Ismail, L.; Schmitt, N.; Hoffmeister, P.; Neukirch, L.; Gdynia, G.; Goeppert, B. Knockdown of Atg7 induces nuclear-LC3 dependent apoptosis and augments chemotherapy in colorectal cancer cells. Int. J. Mol. Sci. 2020, 21, 1099. [Google Scholar] [CrossRef]
- Yang, X.; Yu, D.-D.; Yan, F.; Jing, Y.-Y.; Han, Z.-P.; Sun, K.; Liang, L.; Hou, J.; Wei, L.-X. The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 2015, 5, 14. [Google Scholar] [CrossRef]
- Ohmori, H.; Luo, Y.; Fujii, K.; Sasahira, T.; Shimomoto, T.; Denda, A.; Kuniyasu, H. Dietary linoleic acid and glucose enhances azoxymethane-induced colon cancer and metastases via the expression of high-mobility group box 1. Pathobiology 2010, 77, 210–217. [Google Scholar] [CrossRef]
- Süren, D.; Yıldırım, M.; Demirpençe, Ö.; Kaya, V.; Alikanoğlu, A.S.; Bülbüller, N.; Yıldız, M.; Sezer, C. The role of high mobility group box 1 (HMGB1) in colorectal cancer. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2014, 20, 530. [Google Scholar]
- Shteingauz, A.; Porat, Y.; Voloshin, T.; Schneiderman, R.S.; Munster, M.; Zeevi, E.; Kaynan, N.; Gotlib, K.; Giladi, M.; Kirson, E.D. AMPK-dependent autophagy upregulation serves as a survival mechanism in response to Tumor Treating Fields (TTFields). Cell Death Dis. 2018, 9, 1074. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Tsuno, N.H.; Okaji, Y.; Shuno, Y.; Sasaki, K.; Hongo, K.; Sunami, E.; Kitayama, J.; Takahashi, K.; Nagawa, H. Inhibition of autophagy potentiates sulforaphane-induced apoptosis in human colon cancer cells. Ann. Surg. Oncol. 2010, 17, 592–602. [Google Scholar] [CrossRef]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.-L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef]
- Xia, H.; Wang, W.; Crespo, J.; Kryczek, I.; Li, W.; Wei, S.; Bian, Z.; Maj, T.; He, M.; Liu, R.J. Suppression of FIP200 and autophagy by tumor-derived lactate promotes naive T cell apoptosis and affects tumor immunity. Sci. Immunol. 2017, 2, eaan4631. [Google Scholar] [CrossRef]
- Kim, J.-H.; Kim, H.Y.; Lee, Y.-K.; Yoon, Y.-S.; Xu, W.G.; Yoon, J.-K.; Choi, S.-E.; Ko, Y.-G.; Kim, M.-J.; Lee, S.-J. Involvement of mitophagy in oncogenic K-Ras-induced transformation: Overcoming a cellular energy deficit from glucose deficiency. Autophagy 2011, 7, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Lu, Y.; Zhang, Q.; Qu, X. Heterozygous deletion of ATG5 in Apc Min/+ mice promotes intestinal adenoma growth and enhances the antitumor efficacy of interferon-gamma. Cancer Biol. Ther. 2015, 16, 383–391. [Google Scholar] [CrossRef]
- Cho, D.-H.; Jo, Y.K.; Kim, S.C.; Park, I.J.; Kim, J.C. Down-regulated expression of ATG5 in colorectal cancer. Anticancer. Res. 2012, 32, 4091–4096. [Google Scholar] [PubMed]
- An, C.H.; Kim, M.S.; Yoo, N.J.; Park, S.W.; Lee, S.H. Mutational and expressional analyses of ATG5, an autophagy-related gene, in gastrointestinal cancers. Pathol.-Res. Pract. 2011, 207, 433–437. [Google Scholar] [CrossRef]
- Zheng, S.; Zhong, Y.-F.; Tan, D.-M.; Xu, Y.; Chen, H.-X.; Wang, D. miR-183-5p enhances the radioresistance of colorectal cancer by directly targeting ATG5. J. Biosci. 2019, 44, 92. [Google Scholar] [CrossRef]
- Levy, J.; Cacheux, W.; Bara, M.A.; L’Hermitte, A.; Lepage, P.; Fraudeau, M.; Trentesaux, C.; Lemarchand, J.; Durand, A.; Crain, A.-M. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat. Cell Biol. 2015, 17, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Zou, Z. Targeting autophagy to overcome drug resistance: Further developments. J. Hematol. Oncol. 2020, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Takeuchi, T.; Kuwano, H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann. Surg. Oncol. 2009, 16, 761–771. [Google Scholar] [CrossRef]
- Sasaki, K.; Tsuno, N.H.; Sunami, E.; Tsurita, G.; Kawai, K.; Okaji, Y.; Nishikawa, T.; Shuno, Y.; Hongo, K.; Hiyoshi, M. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010, 10, 370. [Google Scholar] [CrossRef]
- Xu, R.; Ji, Z.; Xu, C.; Zhu, J. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: A systematic review and meta-analysis. Medicine 2018, 97, e12912. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Huang, T.-C.; Shieh, T.-M.; Wu, C.-H.; Lin, L.-C.; Hsia, S.-M. Isoliquiritigenin induces autophagy and inhibits ovarian cancer cell growth. Int. J. Mol. Sci. 2017, 18, 2025. [Google Scholar] [CrossRef]
- Paillas, S.; Causse, A.; Marzi, L.; De Medina, P.; Poirot, M.; Denis, V.; Vezzio-Vie, N.; Espert, L.; Arzouk, H.; Coquelle, A. MAPK14/p38α confers irinotecan resistance to TP53-defective cells by inducing survival autophagy. Autophagy 2012, 8, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Morcillo, M.; Valero, M.; Callejas-Valera, J.; Arias-Gonzalez, L.; Melgar-Rojas, P.; Galán-Moya, E.; García-Gil, E.; García-Cano, J.; Sánchez-Prieto, R. P38MAPK is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluorouracil: Implication in resistance. Oncogene 2012, 31, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Yu, J. Gut microbiota in colorectal cancer: Mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 690–704. [Google Scholar] [CrossRef]
- Yang, Y.; Du, L.; Shi, D.; Kong, C.; Liu, J.; Liu, G.; Li, X.; Ma, Y. Dysbiosis of human gut microbiome in young-onset colorectal cancer. Nat. Commun. 2021, 12, 6757. [Google Scholar] [CrossRef] [PubMed]
- Laqueur, G.; McDaniel, E.; Matsumoto, H. Tumor induction in germfree rats with methylazoxymethanol (MAM) and synthetic MAM acetate. J. Natl. Cancer Inst. 1967, 39, 355–371. [Google Scholar]
- Onoue, M.; Kado, S.; Sakaitani, Y.; Uchida, K.; Morotomi, M. Specific species of intestinal bacteria influence the induction of aberrant crypt foci by 1, 2-dimethylhydrazine in rats. Cancer Lett. 1997, 113, 179–186. [Google Scholar] [CrossRef]
- Wong, S.H.; Zhao, L.; Zhang, X.; Nakatsu, G.; Han, J.; Xu, W.; Xiao, X.; Kwong, T.N.; Tsoi, H.; Wu, W.K. Gavage of fecal samples from patients with colorectal cancer promotes intestinal carcinogenesis in germ-free and conventional mice. Gastroenterology 2017, 153, 1621–1633.e6. [Google Scholar] [CrossRef]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z. Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; yi Liang, Q.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Wang, K.; Kim, M.K.; Di Caro, G.; Wong, J.; Shalapour, S.; Wan, J.; Zhang, W.; Zhong, Z.; Sanchez-Lopez, E.; Wu, L.-W. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity 2014, 41, 1052–1063. [Google Scholar] [CrossRef]
- Kirchberger, S.; Royston, D.J.; Boulard, O.; Thornton, E.; Franchini, F.; Szabady, R.L.; Harrison, O.; Powrie, F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 2013, 210, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.P.; Mahalingam, D. Immunotherapy in colorectal cancer: For the select few or all? J. Gastrointest. Oncol. 2018, 9, 170–179. [Google Scholar] [CrossRef]
- Boland, P.M.; Ma, W.W. Immunotherapy for colorectal cancer. Cancers 2017, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 1–30. [Google Scholar] [CrossRef]
- Mokarram, P.; Albokashy, M.; Zarghooni, M.; Moosavi, M.A.; Sepehri, Z.; Chen, Q.M.; Hudecki, A.; Sargazi, A.; Alizadeh, J.; Moghadam, A.R. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy 2017, 13, 781–819. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhao, Y.-L.; Deng, X.; Yang, S.; Mao, Y.; Li, Z.; Jiang, P.; Zhao, X.; Wei, Y. Chloroquine inhibits colon cancer cell growth in vitro and tumor growth in vivo via induction of apoptosis. Cancer Investig. 2009, 27, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; De Vera, M.E.; Buchser, W.J.; de Vivar Chavez, A.R.; Loughran, P.; Stolz, D.B.; Basse, P.; Wang, T.; Van Houten, B.; Zeh, H.J. Inhibiting Systemic Autophagy during Interleukin 2 Immunotherapy Promotes Long-term Tumor RegressionInhibition of IL-2–Induced Autophagy Prolongs Survival. Cancer Res. 2012, 72, 2791–2801. [Google Scholar] [CrossRef]
- Chen, D.; Xie, J.; Fiskesund, R.; Dong, W.; Liang, X.; Lv, J.; Jin, X.; Liu, J.; Mo, S.; Zhang, T. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun. 2018, 9, 873. [Google Scholar] [CrossRef]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.; Killingsworth, M.C.; Lee, C.S. The significance of autophagy in colorectal cancer pathogenesis and implications for therapy. J. Clin. Pathol. 2014, 67, 854–858. [Google Scholar] [CrossRef]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef]
- He, K.; Zheng, X.; Li, M.; Zhang, L.; Yu, J. mTOR inhibitors induce apoptosis in colon cancer cells via CHOP-dependent DR5 induction on 4E-BP1 dephosphorylation. Oncogene 2016, 35, 148–157. [Google Scholar] [CrossRef]
- Faller, W.J.; Jackson, T.J.; Knight, J.R.; Ridgway, R.A.; Jamieson, T.; Karim, S.A.; Jones, C.; Radulescu, S.; Huels, D.J.; Myant, K.B. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 2015, 517, 497–500. [Google Scholar] [CrossRef]
- Deng, S.; Shanmugam, M.K.; Kumar, A.P.; Yap, C.T.; Sethi, G.; Bishayee, A. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer 2019, 125, 1228–1246. [Google Scholar] [CrossRef]
- Menon, V.P.; Sudheer, A.R. Antioxidant and anti-inflammatory properties of curcumin. In The Molecular Targets and Therapeutic Uses of Curcumin in Health and Disease; Springer: Boston, MA, USA, 2007; pp. 105–125. [Google Scholar]
- Lin, J.-K.; Pan, M.-H.; Lin-Shiau, S.-Y. Recent studies on the biofunctions and biotransformations of curcumin. Biofactors 2000, 13, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Lima, C.F.; Pereira-Wilson, C.; Rattan, S.I. Curcumin induces heme oxygenase-1 in normal human skin fibroblasts through redox signaling: Relevance for anti-aging intervention. Mol. Nutr. Food Res. 2011, 55, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Tamaddoni, A.; Mohammadi, E.; Sedaghat, F.; Qujeq, D.; As’Habi, A. The anticancer effects of curcumin via targeting the mammalian target of rapamycin complex 1 (mTORC1) signaling pathway. Pharmacol. Res. 2020, 156, 104798. [Google Scholar] [CrossRef]
- Olivera, A.; Moore, T.W.; Hu, F.; Brown, A.P.; Sun, A.; Liotta, D.C.; Snyder, J.P.; Yoon, Y.; Shim, H.; Marcus, A.I. Inhibition of the NF-κB signaling pathway by the curcumin analog, 3, 5-Bis (2-pyridinylmethylidene)-4-piperidone (EF31): Anti-inflammatory and anti-cancer properties. Int. Immunopharmacol. 2012, 12, 368–377. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Eckert, R.L. Curcumin suppresses AP1 transcription factor-dependent differentiation and activates apoptosis in human epidermal keratinocytes. J. Biol. Chem. 2007, 282, 6707–6715. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Takada, Y.; Kondo, S.; Sawaya, R.; Aggarwal, B.B.; Kondo, Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: Role of Akt and extracellular signal-regulated kinase signaling pathways. Mol. Pharmacol. 2007, 72, 29–39. [Google Scholar] [CrossRef]
- Hanif, R.; Qiao, L.; Shiff, S.J.; Rigas, B. Curcumin, a natural plant phenolic food additive, inhibits cell proliferation and induces cell cycle changes in colon adenocarcinoma cell lines by a prostaglandin-independent pathway. J. Lab. Clin. Med. 1997, 130, 576–584. [Google Scholar] [CrossRef]
- Zhu, J.; Zhao, B.; Xiong, P.; Wang, C.; Zhang, J.; Tian, X.; Huang, Y. Curcumin induces autophagy via inhibition of yes-associated protein (YAP) in human colon cancer cells. Med. Sci. Monit. 2018, 24, 7035–7042. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kim, N.-Y.; Suh, Y.-A.; Lee, C. Involvement of ROS in curcumin-induced autophagic cell death. Korean J. Physiol. Pharmacol. 2011, 15, 1–7. [Google Scholar] [CrossRef]
- Zhang, P.; Lai, Z.-L.; Chen, H.-F.; Zhang, M.; Wang, A.; Jia, T.; Sun, W.-Q.; Zhu, X.-M.; Chen, X.-F.; Zhao, Z. Curcumin synergizes with 5-fluorouracil by impairing AMPK/ULK1-dependent autophagy, AKT activity and enhancing apoptosis in colon cancer cells with tumor growth inhibition in xenograft mice. J. Exp. Clin. Cancer Res. 2017, 36, 190. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Xu, J.; Lu, Y.; Jiang, J.; Wang, L.; Shen, H.-M.; Xia, D. Curcumin targets the TFEB-lysosome pathway for induction of autophagy. Oncotarget 2016, 7, 75659–75671. [Google Scholar] [CrossRef]
- Zhou, T.; Ye, L.; Bai, Y.; Sun, A.; Cox, B.; Liu, D.; Li, Y.; Liotta, D.; Snyder, J.P.; Fu, H. Autophagy and apoptosis in hepatocellular carcinoma induced by EF25-(GSH) 2: A novel curcumin analog. PLoS ONE 2014, 9, e107876. [Google Scholar] [CrossRef] [PubMed]
- Basile, V.; Belluti, S.; Ferrari, E.; Gozzoli, C.; Ganassi, S.; Quaglino, D.; Saladini, M.; Imbriano, C. bis-Dehydroxy-Curcumin triggers mitochondrial-associated cell death in human colon cancer cells through ER-stress induced autophagy. PLoS ONE 2013, 8, e53664. [Google Scholar] [CrossRef]
- Mao, X.; Zhang, X.; Zheng, X.; Chen, Y.; Xuan, Z.; Huang, P. Curcumin suppresses LGR5 (+) colorectal cancer stem cells by inducing autophagy and via repressing TFAP2A-mediated ECM pathway. J. Nat. Med. 2021, 75, 590–601. [Google Scholar] [CrossRef]
- Kantara, C.; O’Connell, M.; Sarkar, S.; Moya, S.; Ullrich, R.; Singh, P. Curcumin Promotes Autophagic Survival of a Subset of Colon Cancer Stem Cells, Which Are Ablated by DCLK1-siRNAEffects of DCLK1-siRNA±Curcumin on Cancer Stem Cells. Cancer Res. 2014, 74, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Murakami, A.; Ohigashi, H. Ursolic acid: An anti-and pro-inflammatory triterpenoid. Mol. Nutr. Food Res. 2008, 52, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.-W.; Dai, Y.-C.; Xue, J.-P.; Wang, J.-C.; Lin, F.-P.; Guo, Y.-H. In vitro and in vivo anticancer activity evaluation of ursolic acid derivatives. Eur. J. Med. Chem. 2011, 46, 2652–2661. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Wang, X.; Song, Z.; Zhang, H.; Zhou, S.; Zhao, J.; Wang, H. A phase I trial to evaluate the multiple-dose safety and antitumor activity of ursolic acid liposomes in subjects with advanced solid tumors. BioMed Res. Int. 2015, 2015, 809714. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, Y.; Zhang, R.; Tu, X.; Gong, X. Ursolic acid induces autophagy in U87MG cells via ROS-dependent endoplasmic reticulum stress. Chem. Biol. Interact. 2014, 218, 28–41. [Google Scholar] [CrossRef]
- Xavier, C.P.; Lima, C.F.; Pedro, D.F.; Wilson, J.M.; Kristiansen, K.; Pereira-Wilson, C. Ursolic acid induces cell death and modulates autophagy through JNK pathway in apoptosis-resistant colorectal cancer cells. J. Nutr. Biochem. 2013, 24, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Leng, S.; Hao, Y.; Du, D.; Xie, S.; Hong, L.; Gu, H.; Zhu, X.; Zhang, J.; Fan, D.; Kung, H.F. Ursolic acid promotes cancer cell death by inducing Atg5-dependent autophagy. Int. J. Cancer 2013, 133, 2781–2790. [Google Scholar] [CrossRef]
- Luo, J.; Hu, Y.L.; Wang, H. Ursolic acid inhibits breast cancer growth by inhibiting proliferation, inducing autophagy and apoptosis, and suppressing inflammatory responses via the PI3K/AKT and NF-κB signaling pathways in vitro. Exp. Ther. Med. 2017, 14, 3623–3631. [Google Scholar] [CrossRef]
- Jung, J.; Seo, J.; Kim, J.; Kim, J.H. Ursolic acid causes cell death in PC-12 cells by inducing apoptosis and impairing autophagy. Anticancer. Res. 2018, 38, 847–853. [Google Scholar] [PubMed]
- Marmouzi, I.; Bouyahya, A.; Ezzat, S.M.; El Jemli, M.; Kharbach, M. The food plant Silybum marianum (L.) Gaertn.: Phytochemistry, Ethnopharmacology and clinical evidence. J. Ethnopharmacol. 2021, 265, 113303. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Wang, W.; Jin, X.; Wang, Z.; Ji, Z.; Meng, G. Silibinin, a natural flavonoid, induces autophagy via ROS-dependent mitochondrial dysfunction and loss of ATP involving BNIP3 in human MCF7 breast cancer cells. Oncol. Rep. 2015, 33, 2711–2718. [Google Scholar] [CrossRef]
- Wang, C.; He, C.; Lu, S.; Wang, X.; Wang, L.; Liang, S.; Wang, X.; Piao, M.; Cui, J.; Chi, G. Autophagy activated by silibinin contributes to glioma cell death via induction of oxidative stress-mediated BNIP3-dependent nuclear translocation of AIF. Cell Death Dis. 2020, 11, 630. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.-L.; Tay, V.; Guo, S.-Z.; Ren, J.; Shu, M.-G. Silibinin induced human glioblastoma cell apoptosis concomitant with autophagy through simultaneous inhibition of mTOR and YAP. BioMed Res. Int. 2018, 2018, 6165192. [Google Scholar] [CrossRef]
- Raina, K.; Agarwal, C.; Agarwal, R. Effect of silibinin in human colorectal cancer cells: Targeting the activation of NF-κB signaling. Mol. Carcinog. 2013, 52, 195–206. [Google Scholar] [CrossRef]
- Singh, R.P.; Gu, M.; Agarwal, R. Silibinin inhibits colorectal cancer growth by inhibiting tumor cell proliferation and angiogenesis. Cancer Res. 2008, 68, 2043–2050. [Google Scholar] [CrossRef]
- Raina, K.; Agarwal, C.; Wadhwa, R.; Serkova, N.J.; Agarwal, R. Energy deprivation by silibinin in colorectal cancer cells: A double-edged sword targeting both apoptotic and autophagic machineries. Autophagy 2013, 9, 697–713. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Khader, M.; Eckl, P.M. Thymoquinone: An emerging natural drug with a wide range of medical applications. Iran. J. Basic Med. Sci. 2014, 17, 950–957. [Google Scholar]
- Gali-Muhtasib, H.; Ocker, M.; Kuester, D.; Krueger, S.; El-Hajj, Z.; Diestel, A.; Evert, M.; El-Najjar, N.; Peters, B.; Jurjus, A. Thymoquinone reduces mouse colon tumor cell invasion and inhibits tumor growth in murine colon cancer models. J. Cell. Mol. Med. 2008, 12, 330–342. [Google Scholar] [CrossRef]
- Ballout, F.; Monzer, A.; Fatfat, M.; El Ouweini, H.; Jaffa, M.A.; Abdel-Samad, R.; Darwiche, N.; Abou-Kheir, W.; Gali-Muhtasib, H. Thymoquinone induces apoptosis and DNA damage in 5-Fluorouracil-resistant colorectal cancer stem/progenitor cells. Oncotarget 2020, 11, 2959–2972. [Google Scholar] [CrossRef]
- Ndreshkjana, B.; Çapci, A.; Klein, V.; Chanvorachote, P.; Muenzner, J.K.; Huebner, K.; Steinmann, S.; Erlenbach-Wuensch, K.; Geppert, C.I.; Agaimy, A. Combination of 5-fluorouracil and thymoquinone targets stem cell gene signature in colorectal cancer cells. Cell Death Dis. 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed]
- Kensara, O.A.; El-Shemi, A.G.; Mohamed, A.M.; Refaat, B.; Idris, S.; Ahmad, J. Thymoquinone subdues tumor growth and potentiates the chemopreventive effect of 5-fluorouracil on the early stages of colorectal carcinogenesis in rats. Drug Des. Dev. Ther. 2016, 10, 2239. [Google Scholar]
- Zhang, L.; Bai, Y.; Yang, Y. Thymoquinone chemosensitizes colon cancer cells through inhibition of NF-κB. Oncol. Lett. 2016, 12, 2840–2845. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-C.; Lee, N.-H.; Hsu, H.-H.; Ho, T.-J.; Tu, C.-C.; Hsieh, D.J.-Y.; Lin, Y.-M.; Chen, L.-M.; Kuo, W.-W.; Huang, C.-Y. Thymoquinone induces caspase-independent, autophagic cell death in CPT-11-resistant lovo colon cancer via mitochondrial dysfunction and activation of JNK and p38. J. Agric. Food Chem. 2015, 63, 1540–1546. [Google Scholar] [CrossRef] [PubMed]
- Racoma, I.O.; Meisen, W.H.; Wang, Q.-E.; Kaur, B.; Wani, A.A. Thymoquinone inhibits autophagy and induces cathepsin-mediated, caspase-independent cell death in glioblastoma cells. PLoS ONE 2013, 8, e72882. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Wang, S.-F.; Cai, C.-Z.; Tan, J.-Q.; Li, M.; Lu, J.-J.; Chen, X.-P.; Wang, Y.-T.; Zheng, W.; Lu, J.-H. Natural autophagy blockers, dauricine (DAC) and daurisoline (DAS), sensitize cancer cells to camptothecin-induced toxicity. Oncotarget 2017, 8, 77673–77684. [Google Scholar] [CrossRef]
- Yang, Z.; Li, C.; Wang, X.; Zhai, C.; Yi, Z.; Wang, L.; Liu, B.; Du, B.; Wu, H.; Guo, X. Dauricine induces apoptosis, inhibits proliferation and invasion through inhibiting NF-κB signaling pathway in colon cancer cells. J. Cell. Physiol. 2010, 225, 266–275. [Google Scholar] [CrossRef]
- Law, B.Y.K.; Chan, W.K.; Xu, S.W.; Wang, J.R.; Bai, L.P.; Liu, L.; Wong, V.K.W. Natural small-molecule enhancers of autophagy induce autophagic cell death in apoptosis-defective cells. Sci. Rep. 2014, 4, 5510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ren, Y.; Qiu, J. Dauricine inhibits viability and induces cell cycle arrest and apoptosis via inhibiting the PI3K/Akt signaling pathway in renal cell carcinoma cells. Mol. Med. Rep. 2018, 17, 7403–7408. [Google Scholar] [CrossRef] [PubMed]
- Law, B.Y.; Mok, S.W.; Chen, J.; Michelangeli, F.; Jiang, Z.-H.; Han, Y.; Qu, Y.Q.; Qiu, A.C.; Xu, S.-W.; Xue, W.-W. N-desmethyldauricine induces autophagic cell death in apoptosis-defective cells via Ca2+ mobilization. Front. Pharmacol. 2017, 8, 388. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Qu, Y.Q.; Zheng, Z.; Law, B.Y.K.; Mok, S.W.F.; Jiang, Z.-H.; Wong, V.K.W.; Bai, L.-P. Novel dauricine derivatives suppress cancer via autophagy-dependent cell death. Bioorganic Chem. 2019, 83, 450–460. [Google Scholar] [CrossRef]
- Sun, X.; Ng, T.T.; Sham, K.W.; Zhang, L.; Chan, M.T.; Wu, W.K.; Cheng, C.H. Bufalin, a traditional Chinese medicine compound, prevents tumor formation in two murine models of colorectal cancer. Cancer Prev. Res. 2019, 12, 653–666. [Google Scholar] [CrossRef]
- Xie, C.-M.; Chan, W.Y.; Yu, S.; Zhao, J.; Cheng, C.H. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free. Radic. Biol. Med. 2011, 51, 1365–1375. [Google Scholar] [CrossRef]
- Qi, H.Y.; Qu, X.J.; Liu, J.; Hou, K.Z.; Fan, Y.B.; Che, X.F.; Liu, Y.P. Bufalin induces protective autophagy by Cbl-b regulating mTOR and ERK signaling pathways in gastric cancer cells. Cell Biol. Int. 2019, 43, 33–43. [Google Scholar] [CrossRef]
- Wang, J.; Chen, C.; Wang, S.; Zhang, Y.; Yin, P.; Gao, Z.; Xu, J.; Feng, D.; Zuo, Q.; Zhao, R. Bufalin inhibits HCT116 colon cancer cells and its orthotopic xenograft tumor in mice model through genes related to apoptotic and PTEN/AKT pathways. Gastroenterol. Res. Pract. 2015, 2015, 457193. [Google Scholar] [CrossRef]
- Dai, X.Y.; Zhou, B.F.; Xie, Y.Y.; Lou, J.; Li, K.Q. Bufalin and 5-fluorouracil synergistically induce apoptosis in colorectal cancer cells. Oncol. Lett. 2018, 15, 8019–8026. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef]
- Rauf, A.; Imran, M.; Butt, M.S.; Nadeem, M.; Peters, D.G.; Mubarak, M.S. Resveratrol as an anti-cancer agent: A review. Crit. Rev. Food Sci. Nutr. 2018, 58, 1428–1447. [Google Scholar] [CrossRef] [PubMed]
- Athar, M.; Back, J.H.; Tang, X.; Kim, K.H.; Kopelovich, L.; Bickers, D.R.; Kim, A.L. Resveratrol: A review of preclinical studies for human cancer prevention. Toxicol. Appl. Pharmacol. 2007, 224, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, G.; Jin, G.; Yao, K.; Zhao, Z.; Bie, L.; Guo, Y.; Li, N.; Deng, W.; Chen, X. Resveratrol suppresses colon cancer growth by targeting the AKT/STAT3 signaling pathway. Int. J. Mol. Med. 2019, 43, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Liu, X.; Fu, X.; Zhang, L.; Sui, H.; Zhou, L.; Sun, J.; Cai, J.; Qin, J.; Ren, J. Resveratrol inhibits invasion and metastasis of colorectal cancer cells via MALAT1 mediated Wnt/β-catenin signal pathway. PLoS ONE 2013, 8, e78700. [Google Scholar] [CrossRef]
- Patel, K.R.; Brown, V.A.; Jones, D.J.; Britton, R.G.; Hemingway, D.; Miller, A.S.; West, K.P.; Booth, T.D.; Perloff, M.; Crowell, J.A. Clinical Pharmacology of Resveratrol and Its Metabolites in Colorectal Cancer PatientsResveratrol in Colorectal Cancer Patients. Cancer Res. 2010, 70, 7392–7399. [Google Scholar] [CrossRef]
- Andreadi, C.; Britton, R.G.; Patel, K.R.; Brown, K. Resveratrol-sulfates provide an intracellular reservoir for generation of parent resveratrol, which induces autophagy in cancer cells. Autophagy 2014, 10, 524–525. [Google Scholar] [CrossRef]
- Honari, M.; Shafabakhsh, R.; Reiter, R.J.; Mirzaei, H.; Asemi, Z. Resveratrol is a promising agent for colorectal cancer prevention and treatment: Focus on molecular mechanisms. Cancer Cell Int. 2019, 19, 180. [Google Scholar] [CrossRef]
- Tian, Y.; Song, W.; Li, D.; Cai, L.; Zhao, Y. Resveratrol as a natural regulator of autophagy for prevention and treatment of cancer. OncoTargets Ther. 2019, 12, 8601–8609. [Google Scholar] [CrossRef]
- Miki, H.; Uehara, N.; Kimura, A.; Sasaki, T.; Yuri, T.; Yoshizawa, K.; Tsubura, A. Resveratrol induces apoptosis via ROS-triggered autophagy in human colon cancer cells. Int. J. Oncol. 2012, 40, 1020–1028. [Google Scholar] [CrossRef]
- Park, D.; Jeong, H.; Lee, M.N.; Koh, A.; Kwon, O.; Yang, Y.R.; Noh, J.; Suh, P.-G.; Park, H.; Ryu, S.H. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci. Rep. 2016, 6, 21772. [Google Scholar] [CrossRef]
- Selvaraj, S.; Sun, Y.; Sukumaran, P.; Singh, B.B. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol. Carcinog. 2016, 55, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhang, S.; Zhou, J.; Li, X. Effect of resveratrol on drug resistance in colon cancer chemotherapy. RSC Adv. 2019, 9, 2572–2580. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.H.; Heo, E.Y.; Song, J.H.; Kwon, B.E.; Lee, J.Y.; Park, Y.; Kim, J.; Chang, S.-Y.; Chin, Y.-W.; Jeon, S.-M.; et al. Trans-scirpusin A showed antitumor effects via autophagy activation and apoptosis induction of colorectal cancer cells. Oncotarget 2017, 8, 41401–41411. [Google Scholar] [CrossRef]
- Guerrero-Beltrán, C.E.; Calderón-Oliver, M.; Pedraza-Chaverri, J.; Chirino, Y.I. Protective effect of sulforaphane against oxidative stress: Recent advances. Exp. Toxicol. Pathol. 2012, 64, 503–508. [Google Scholar] [CrossRef]
- Clarke, J.D.; Dashwood, R.H.; Ho, E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008, 269, 291–304. [Google Scholar] [CrossRef]
- Frydoonfar, H.; McGrath, D.; Spigelman, A. Sulforaphane inhibits growth of a colon cancer cell line. Color. Dis. 2004, 6, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Bernkopf, D.B.; Daum, G.; Brückner, M.; Behrens, J. Sulforaphane inhibits growth and blocks Wnt/β-catenin signaling of colorectal cancer cells. Oncotarget 2018, 9, 33982–33994. [Google Scholar] [CrossRef]
- Gwon, Y.; Oh, J.; Kim, J.-S. Sulforaphane induces colorectal cancer cell proliferation through Nrf2 activation in a p53-dependent manner. Appl. Biol. Chem. 2020, 63, 86. [Google Scholar] [CrossRef]
- Herman-Antosiewicz, A.; Johnson, D.E.; Singh, S.V. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res. 2006, 66, 5828–5835. [Google Scholar] [CrossRef]
- Hao, Q.; Wang, M.; Sun, N.X.; Zhu, C.; Lin, Y.M.; Li, C.; Liu, F.; Zhu, W.W. Sulforaphane suppresses carcinogenesis of colorectal cancer through the ERK/Nrf2-UDP glucuronosyltransferase 1A metabolic axis activation. Oncol. Rep. 2020, 43, 1067–1080. [Google Scholar] [CrossRef]
- Jo, C.; Kim, S.; Cho, S.-J.; Choi, K.J.; Yun, S.-M.; Koh, Y.H.; Johnson, G.V.; Park, S.I. Sulforaphane induces autophagy through ERK activation in neuronal cells. FEBS Lett. 2014, 588, 3081–3088. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Ren, Y.; Yang, L.; Jia, A.; Hu, Y.; Zhao, Y.; Zhao, W.; Yu, B.; Zhao, W.; Zhang, J.; et al. Inhibiting autophagy enhances sulforaphane-induced apoptosis via targeting NRF2 in esophageal squamous cell carcinoma. Acta Pharm. Sin. B 2021, 11, 1246–1260. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, S.; Muhammad, J.S.; Maghazachi, A.A.; Hamid, Q. Autophagy: A versatile player in the progression of colorectal cancer and drug resistance. Front. Oncol. 2022, 3175. [Google Scholar] [CrossRef]
- Rahman, M.S. Allicin and other functional active components in garlic: Health benefits and bioavailability. Int. J. Food Prop. 2007, 10, 245–268. [Google Scholar] [CrossRef]
- Li, X.; Ni, J.; Tang, Y.; Wang, X.; Tang, H.; Li, H.; Zhang, S.; Shen, X. Allicin inhibits mouse colorectal tumorigenesis through suppressing the activation of STAT3 signaling pathway. Nat. Prod. Res. 2019, 33, 2722–2725. [Google Scholar] [CrossRef]
- Bat-Chen, W.; Golan, T.; Peri, I.; Ludmer, Z.; Schwartz, B. Allicin purified from fresh garlic cloves induces apoptosis in colon cancer cells via Nrf2. Nutr. Cancer 2010, 62, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhao, J.; Zhao, M.; Wang, K. Allicin activates autophagic cell death to alleviate the malignant development of thyroid cancer. Exp. Ther. Med. 2018, 15, 3537–3543. [Google Scholar] [CrossRef]
- Chu, Y.-L.; Ho, C.-T.; Chung, J.-G.; Rajasekaran, R.; Sheen, L.-Y. Allicin induces p53-mediated autophagy in Hep G2 human liver cancer cells. J. Agric. Food Chem. 2012, 60, 8363–8371. [Google Scholar] [CrossRef]
- Huang, W.; Wu, S.F.; Xu, S.T.; Ma, Y.C.; Wang, R.; Jin, S.; Zhou, S. Allicin enhances the radiosensitivity of colorectal cancer cells via inhibition of NF-κB signaling pathway. J. Food Sci. 2020, 85, 1924–1931. [Google Scholar] [CrossRef] [PubMed]
- Țigu, A.B.; Toma, V.-A.; Moț, A.C.; Jurj, A.; Moldovan, C.S.; Fischer-Fodor, E.; Berindan-Neagoe, I.; Pârvu, M. The synergistic antitumor effect of 5-fluorouracil combined with allicin against lung and colorectal carcinoma cells. Molecules 2020, 25, 1947. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Yang, P.; Shen, Y.; Bei, W.; Zhang, Y.; Ge, Y.; Newman, R.A.; Cohen, L.; Liu, L.; Thornton, B. Pilot study of huachansu in patients with hepatocellular carcinoma, nonsmall-cell lung cancer, or pancreatic cancer. Cancer 2009, 115, 5309–5318. [Google Scholar] [CrossRef] [PubMed]
- De, A.; De, A.; Papasian, C.; Hentges, S.; Banerjee, S.; Haque, I.; Banerjee, S.K. Emblica officinalis extract induces autophagy and inhibits human ovarian cancer cell proliferation, angiogenesis, growth of mouse xenograft tumors. PLoS ONE 2013, 8, e72748. [Google Scholar] [CrossRef]
- Winawer, S.J.; Fletcher, R.H.; Miller, L.; Godlee, F.; Stolar, M.; Mulrow, C.; Woolf, S.; Glick, S.; Ganiats, T.; Bond, J. Colorectal cancer screening: Clinical guidelines and rationale. Gastroenterology 1997, 112, 594–642. [Google Scholar] [CrossRef]
- Horita, Y.; Yamada, Y.; Kato, K.; Hirashima, Y.; Akiyoshi, K.; Nagashima, K.; Nakajima, T.; Hamaguchi, T.; Shimada, Y. Phase II clinical trial of second-line FOLFIRI plus bevacizumab for patients with metastatic colorectal cancer: AVASIRI trial. Int. J. Clin. Oncol. 2012, 17, 604–609. [Google Scholar] [CrossRef]
- Lee, R.M.; Cardona, K.; Russell, M.C. Historical perspective: Two decades of progress in treating metastatic colorectal cancer. J. Surg. Oncol. 2019, 119, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-M.; Yang, Z.-J.; Xie, Q.; Zhang, Z.-K.; Zhang, H.; Ma, J.-Y. Natural products for treating colorectal cancer: A mechanistic review. Biomed. Pharmacother. 2019, 117, 109142. [Google Scholar] [CrossRef]
- Cunningham, D.; Pyrhönen, S.; James, R.D.; Punt, C.J.; Hickish, T.F.; Heikkila, R.; Johannesen, T.B.; Starkhammar, H.; Topham, C.A.; Awad, L. Randomised trial of irinotecan plus supportive care versus supportive care alone after fluorouracil failure for patients with metastatic colorectal cancer. Lancet 1998, 352, 1413–1418. [Google Scholar] [CrossRef]
- Tanaka, S.; Haruma, K.; Yoshihara, M.; Kajiyama, G.; Kira, K.; Amagase, H.; Chayama, K. Aged garlic extract has potential suppressive effect on colorectal adenomas in humans. J. Nutr. 2006, 136, 821S–826S. [Google Scholar] [CrossRef]
- Hsieh, C. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer. Res 2001, 21, 2895–2900. [Google Scholar]
- Boocock, D.J.; Faust, G.E.; Patel, K.R.; Schinas, A.M.; Brown, V.A.; Ducharme, M.P.; Booth, T.D.; Crowell, J.A.; Perloff, M.; Gescher, A.J. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1246–1252. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.S.; Garland, L.L.; Hsu, C.-H.; Vining, D.R.; Chew, W.M.; Miller, J.A.; Perloff, M.; Crowell, J.A.; Alberts, D.S. Resveratrol modulates drug-and carcinogen-metabolizing enzymes in a healthy volunteer study. Cancer Prev. Res. 2010, 3, 1168–1175. [Google Scholar] [CrossRef]
- Popat, R.; Plesner, T.; Davies, F.; Cook, G.; Cook, M.; Elliott, P.; Jacobson, E.; Gumbleton, T.; Oakervee, H.; Cavenagh, J. A phase 2 study of SRT501 (resveratrol) with bortezomib for patients with relapsed and or refractory multiple myeloma. Br. J. Haematol. 2012, 160, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Britton, R.G.; Kovoor, C.; Brown, K. Direct molecular targets of resveratrol: Identifying key interactions to unlock complex mechanisms. Ann. N. Y. Acad. Sci. 2015, 1348, 124–133. [Google Scholar] [CrossRef]
- Grigalunas, M.; Brakmann, S.; Waldmann, H. Chemical evolution of natural product structure. J. Am. Chem. Soc. 2022, 144, 3314–3329. [Google Scholar] [CrossRef] [PubMed]
- Song, J.X.; Sun, Y.R.; Peluso, I.; Zeng, Y.; Yu, X.; Lu, J.H.; Xu, Z.; Wang, M.Z.; Liu, L.F.; Huang, Y.Y.; et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 2016, 12, 1372–1389. [Google Scholar] [CrossRef]
- Wu, Y.C.; Wu, W.K.K.; Li, Y.; Yu, L.; Li, Z.J.; Wong, C.C.M.; Li, H.T.; Sung, J.J.Y.; Cho, C.H. Inhibition of macroautophagy by bafilomycin A1 lowers proliferation and induces apoptosis in colon cancer cells. Biochem. Biophys. Res. Commun. 2009, 382, 451–456. [Google Scholar] [CrossRef]
- Zhang, J.; Roberts, T.M.; Shivdasani, R.A. Targeting PI3K signaling as a therapeutic approach for colorectal cancer. Gastroenterology 2011, 141, 50–61. [Google Scholar] [CrossRef]



| Natural Compounds | Condition/Disease | Clinical Trial Number | Phase | Outcomes | Status |
|---|---|---|---|---|---|
| Curcumin and Avatin/FOLFIRI | Colorectal cancer | NCT02439385 | 2 | Improves survival rates, safety, and tolerability | Completed |
| Curcumin and Irinotecan | Advanced colorectal cancer | NCT01859858 | 1 | Evaluates the effect of curcumin on the pharmacokinetics of irinotecan | Completed |
| Curcumin C3 tablet | Colorectal cancer | NCT01333917 | 1 | Measures gene expression, RNA levels, and apoptosis activity of curcumin | Completed |
| Oral complex C3 curcumin + chemotherapy | Colorectal cancer | NCT01490996 | 1 | Evaluates the efficacy of curcumin in terms of disease response and survival | Completed |
| Curcumin | Colorectal cancer | NCT00027495 | 1 | Determines the tolerable dose of curcumin that can aid in the prevention of colon cancer in healthy men and women. | Completed |
| Curcumin | Familial adenomatous polyposis | NCT00927485 | Not applicable | Determines the safety and effectiveness of curcumin in reducing intestinal adenomas by counting the number of polyps in the duodenum, colon, and ileum. | Completed |
| Curcumin | Familial adenomatous polyposis | NCT00641147 | 2 | Measures the efficacy and safety of curcumin in patients with familial adenomatous polyposis. | Completed |
| Curcuminoids | Colorectal cancer | NCT00027495 | 1 | Evaluates the pharmacokinetics profile and the safest dose of curcumin for preventing colon cancer in healthy people. | Completed |
| Resveratrol | Colorectal cancer | NCT00433576 | 1 | Inhibits tumor cell growth by inhibiting some of the enzymes required for cell proliferation. | Completed |
| SRT501 (Resveratrol) | Neoplasms, Colorectal cancer | NCT00920803 | 1 | Determines the safety, tolerability, pharmacodynamics, and pharmacokinetic profile of SRT501 in blood and both normal and malignant metastatic tissue samples. | Completed |
| FOLFOX (Oxaliplatin, 5 Fluorouracil, Lederfolin) | Colon Cancer | NCT00646607 | 3 | Evaluates the overall survival, toxicity, and adverse events | Completed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Y.-X.; Mamun, A.A.; Lyu, A.-P.; Zhang, H.-J. Natural Compounds Targeting the Autophagy Pathway in the Treatment of Colorectal Cancer. Int. J. Mol. Sci. 2023, 24, 7310. https://doi.org/10.3390/ijms24087310
Du Y-X, Mamun AA, Lyu A-P, Zhang H-J. Natural Compounds Targeting the Autophagy Pathway in the Treatment of Colorectal Cancer. International Journal of Molecular Sciences. 2023; 24(8):7310. https://doi.org/10.3390/ijms24087310
Chicago/Turabian StyleDu, Yin-Xiao, Abdullah Al Mamun, Ai-Ping Lyu, and Hong-Jie Zhang. 2023. "Natural Compounds Targeting the Autophagy Pathway in the Treatment of Colorectal Cancer" International Journal of Molecular Sciences 24, no. 8: 7310. https://doi.org/10.3390/ijms24087310
APA StyleDu, Y.-X., Mamun, A. A., Lyu, A.-P., & Zhang, H.-J. (2023). Natural Compounds Targeting the Autophagy Pathway in the Treatment of Colorectal Cancer. International Journal of Molecular Sciences, 24(8), 7310. https://doi.org/10.3390/ijms24087310